芳族杂环通过地球丰富的无过渡金属的催化剂的甲硅烷基化的制作方法
相关申请的交叉引用
本申请要求于2014年8月6日提交的第62/033,975号、于2014年12月19日提交的第62/094,381号和于2015年4月2日提交的第62/141,905号美国专利申请的优先权,出于所有目的,其内容通过引用并入本文。
政府权利
本发明是在美国政府支持下,在美国国家科学基金会资助的拨款号che1212767下做出的。政府在本发明中具有某些权利。
本发明涉及用于使用氢氧化物(尤其氢氧化钾)和硅烷试剂使芳族基质(包括杂芳族基质)甲硅烷基化的方法。
背景
使有机部分甲硅烷基化的能力已在近年来吸引了极大的关注,这归因于甲硅烷基化材料本身或作为用于例如在农用化学、药学和电子材料应用中所使用的其他重要材料的中间体的实用性。此外,用有机硅烷使多核芳族化合物官能化的能力提供了利用这些材料的令人感兴趣的性质的机会。
以往,芳族化合物的甲硅烷基化已经通过涉及用热的方法、用光化学的方法或以其他方式衍生的自由基源的自由基过程来实现。芳族化合物已知与氢化硅在以下情况下反应:在气相中在500℃-850℃下、在液相中在自生的压力下在350℃-500℃下、在过氧化物的存在下在135℃下在气相缩合下、以及使用电放电反应。此类反应条件不适合于非挥发性材料或热敏材料。
目前,杂芳族c-si键构建的最常见方法包括杂芳基锂试剂或杂芳基镁试剂与硅亲电体的相交(interception)。然而,此方法常常在范围方面受限,并且要求通过使用自燃的有机金属物质以化学计量的量使杂芳烃预官能化(prefunctionalization)。对于c-si键构建,强有力的杂芳族官能化策略例如minisci型自由基取代和傅-克反应(friedel–craftsreaction)已经具有受限制的用途,这是由于产生对应的甲硅烷基自由基和三配位硅正离子(silyliumion)的困难。
最近,过渡金属介导的芳族c-h甲硅烷基化已被描述,其中所描述的不同体系基于例如co、rh、ir、fe、ru、os、ni、pd和pt催化剂。但是某些电子应用,甚至低水平的此类残余物的存在可能不利地影响甲硅烷基化材料的性能。相似地,在某些药学应用或电子应用中,对残余的过渡金属的限制是相当严格的,并且完全避免它们的能力在合成后的处理(work-up)期间提供益处。
本发明利用本文中引用的发现来避免与先前已知的方法有关的问题中的至少一些。
概述
本公开内容提供关于芳族基质的丁醇盐催化的甲硅烷基化的新的信息,以及可以使得koh(氢氧化钾)作为在本反应中的催化剂是可操作的最近的发现。与早期发现相反的是,现在已经发现,koh对于杂芳族物质与氢硅烷(hydrosilane)在某些条件下的直接甲硅烷基化可以是有效的催化剂。现在看来,通过修改反应条件,此koh催化剂体系可以与每种基质一起使用,在所述每种基质中叔丁醇钾(或其他“强碱”)先前被示出是有效的,但其中koh先前被示出是不能实行的,例如如在美国专利申请序列号14/043,929和国际申请号pct/us2013/062963(两者均于2013年10月2日提交)中所描述的。koh的使用提供重要的实际益处,例如较低的成本和毒性、更易操作、以及有利的反应建立和纯化。另外,其提供在使用较强的碱(包括醇盐)的反应中未见到的选择性。
本说明书还公开了先前未明确地描述的从叔丁醇钾方面描述的另外的实施方案,本说明书示出这些方法的多用性的更完整的一组实施例。
本发明的多个实施方案提供用于使有机化合物甲硅烷基化的化学体系,每种体系包含(a)至少一种有机硅烷和(b)至少一种强碱的混合物或基本上由(a)至少一种有机硅烷和(b)至少一种强碱的混合物组成,所述强碱的定义现在还包括koh,当优选地在大体上无过渡金属化合物的情况下进行时,所述体系还对于使芳族前体甲硅烷基化是可操作的。体系还包含至少一种有机芳族基质。
其他实施方案提供方法,每种方法包括使有机芳族基质与包含(a)至少一种有机硅烷和(b)至少一种强碱或基本上由(a)至少一种有机硅烷和(b)至少一种强碱组成的混合物在足以使基质甲硅烷基化的条件下接触,所述强碱的定义现在还包括koh。在一些实施方案中,所述混合物和所述基质优选地但不必要地大体上无过渡金属化合物。
附图简述
当结合附图阅读时,进一步地理解本申请。出于阐明主题的目的,在附图中示出了主题的示例性实施方案;然而,本文公开的主题不限于所公开的具体的方法、装置和体系。此外,附图不一定按比例绘制。在附图中:
图1a和图1b图示通过本文描述的方法可用的反应的一些的实例。
图2图示吲哚类的碱催化的甲硅烷基化的范围。在这些实例中,ko-t-bu被用作示例性的碱。[si]–h=et3sih、et2sih2、etme2sih、phme2sih或n-bu3sih。mom,甲氧基甲基;sem,2-[(三甲基甲硅烷基)乙氧基]甲基。
图3图示含n-、含o-以及含s-的杂芳烃的碱催化的甲硅烷基化的范围。在这些实例中,ko-t-bu被用作示例性的碱。详见实施例6.9.1至实施例6.9.51。[si]–h=et3sih、et2sih2、etme2sih、phme2sih或n-bu3sih。
图4a-4e示出碱催化的c-h甲硅烷基化的某些合成应用。在这些实例中,ko-t-bu被用作示例性的碱。图4a示出142g的c2-甲硅烷基化的吲哚构建单元2a的制备的示意图。图4b图示杂芳基硅烷在交叉偶联和苯并噻吩的c7处的缩甲醛c-h硼基化(borylation)中的某些应用。图4c图示选择性前体至高级材料和聚合物的某些具体合成。图4d图示被用于制备后期化学选择性和区域选择性修饰的活性药物成分的本发明方法的选择性实例。图4e示出通过氧定向的sp2(oxygen-directedsp2)和固有的(innate)苄型sp3c–h甲硅烷基化对芳烃的官能化的实例。详见实施例6.7.1至实施例6.7.4。[si]=et3si;i-pr,异丙基;dba,二亚苄基丙酮(dibenzylideneacetone);bpin,4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷;tmeda,四甲基乙二胺;edot,3,4-亚乙二氧基噻吩。
图5a/b示出在3当量的et3sih和不同的koh载量下,在45℃下,1-甲基吲哚的甲硅烷基化的转化率相对于时间数据(时间以分钟计并且转化率以百分比计)。图5a示出作为时间的函数的总转化率,并且图5b示出作为时间的函数的c2:c3的比率。顶部曲线(正方形)是关于20mol%koh,并且底部曲线是关于5mol%。
图6示出在3当量的et3sih下,在65℃下,1-甲基吲哚的甲硅烷基化的koh催化剂载量数据。
图7示出对用koh催化体系甲硅烷基化的代表性基质进行测试的结果。条件a:起始材料(0.5mmol,1当量);koh(0.1mmol,5.6mg,20mol%);siet3h(1.5mmol,3当量,240μl),在thf(0.5ml)中在65℃下。条件b:起始材料(0.5mmol,1当量);koh(0.1mmol,5.6mg,20mol%);siet3h(0.6mmol,1.2当量,96μl),在thf(0.5ml)中在45℃下。
说明性实施方案详述
本发明建立于一系列反应上,这些反应中的每一个都依赖于有机硅烷和强碱的简单混合物,所述强碱的定义现在还包括氢氧化物,尤其koh,它们一起形成能够使液相中的芳族分子尤其杂芳基化合物在不存在过渡金属催化剂、uv辐射或电放电(包括等离子体放电)的情况下甲硅烷基化的原位体系(insitusystem)(活性物质的结构和性质是尚未知的)。这些反应与在开发用于制备对药学和电子应用重要的产物的实用方法中的重要进展有关。重要的是,该反应是令人感兴趣的,因为其仅产生了环境友好的硅酸盐(environmentallybenignsilicate)和二氢(dihydrogen)作为副产物并且可以避免如在为了这个目的文献中提出的几乎所有的其他方法中将观察到的有毒金属废物流。通过这些体系中的至少一些所呈现出的显著容易性和区域专一性为这些领域中的化学工作者的工具箱中提供有用的工具。
本公开内容包括先前在美国专利申请序列号14/043,929和国际申请号pct/us2013/062963(两者均于2013年10月2日提交)中提供的一些信息,以及先前未明确地描述的从叔丁醇钾方面描述的新的另外的实施方案,本公开内容示出这些方法的多用性的一组更完整的实施方案。本公开内容还提供与可以使得koh(氢氧化钾)和其他氢氧化物在本文反应中作为催化剂是可操作的最近发现有关的数据。与早期发现相反的是,现在已经发现,koh对于杂芳族物质与氢硅烷在某些条件下的直接甲硅烷基化可以是有效的催化剂。尽管本文提供的许多实例在叔丁醇盐、氢化物等等的方面被描述,但这些实例还可以被扩展到包括其中koh是有效的催化剂的那些,并且关于前者所描述的实施方案还扩展到使用后者的那些。相似地,关于叔丁醇盐体系的可操作性(例如对于官能团的耐受(tolerance))的评论明确地意图还反映koh体系的可操作性。
本文所描述的甲硅烷基化反应在温和的条件下、在氢受体、配体或添加剂不存在下进行,并且在任选地无溶剂条件下可扩展到大于100克。难以用贵金属催化剂活化的基质种类以良好的收率并且以优良的区域选择性被甲硅烷基化。衍生的杂芳基硅烷产物容易地参与使得用于杂芳族的加工(heteroaromaticelaboration)的新的合成策略成为可能的多用性转化,并且凭其自身因素而在药学应用和材料科学应用中是有用的。
本发明可以通过参照结合附图和实施例的以下描述而更容易地理解,其全部形成本公开内容的一部分。应理解,本发明不限于本文中描述的或示出的具体的产物、方法、条件或参数,并且本文中使用的术语仅用实例的方式用于描述具体实施方案的目的且不意图是任何要求保护的发明的限制。相似地,除非另有具体说明,否则关于用于改进的可能的作用机理或模式或理由的任何描述意味着仅是说明性的,并且本文的发明将不被用于改进的任何此类建议的作用机理或模式或理由的正确性或不正确性约束。贯穿本文,意识到的是,该描述指的是组合物以及制造和使用所述组合物的方法。换言之,如果本公开内容描述或要求保护与组合物或者制造或使用组合物的方法相关联的特征或实施方案,那么应意识到,这种描述或权利要求意图使这些特征或实施方案延伸至这些上下文中的每个中的实施方案(即组合物、制造方法和使用方法)。
在本公开内容中,单数形式“一(a)”、“一(an)”和“该(the)”包括复数指代物,并且提到的具体数值的指代包括至少该具体值,除非上下文另有清楚地指示。因此,例如,对“一种材料”的指代是对本领域技术人员已知的此类材料及其等同物中的至少一种的指代,等等。
当值通过使用描述符“约”被表示为近似值时,应理解,该具体值形成另一个实施方案。一般而言,术语“约”的使用指示近似值,该近似值可以取决于将通过公开的主题获得的所寻求的期望性能而变化并且将在其中使用该主题的具体上下文中基于该主题的功能来理解。本领域技术人员将能够按常规解释该术语。在某些情况下,用于具体值的有效数字的数目可以是确定词语“约”的程度的一个非限制性方法。在其他情况下,在一系列值中使用的等级可以用于确定预期范围,该预期范围对于每个值的术语“约”是可用的。如果存在,那么所有的范围是包括端点的并且是可组合的。换言之,对以范围陈述的值的指代包括在该范围内的每个值。
应意识到,为了清楚起见,在本文中的单独的实施方式的上下文中描述的本发明的某些特征也可以在单一的实施方案中被组合地提供。换言之,除非明显地不可相容或具体地排除的,否则每个单个的实施方案被视为是与任何其他实施方案可组合的并且这种组合被认为是另一个实施方案。相反地,为了简洁起见,单一的实施方案的上下文中描述的本发明的各种特征也可以被单独提供或以任何子组合来提供。最后,虽然实施方案可以被描述为一系列步骤的一部分或更多通用结构的一部分,但是每个所述步骤也可以被认为是与其他实施方案可组合的自身独立的实施方案。
过渡术语“包含”、“基本上由......组成”和“由......组成”意图包含它们的在专利行话中的被普遍接受的含义;即,(i)与“包括”、“含有”或“以......为特征”同义的“包含”是包括端点的或开放式的并且不排除另外的、未叙述的要素或方法步骤;(ii)“由......组成”排除未在权利要求中指定的任何要素、步骤或成分;并且(iii)“基本上由......组成”将权利要求的范围限制于指定的材料或步骤以及“不实质上影响要求保护的发明的基本特征和新颖特征的材料或步骤”。在措辞“包含”(或其等同物)的方面所描述的实施方案也提供在“由......组成”和“基本上由......组成”的方面独立地描述的那些作为实施方案。对于在“基本上由......组成”的方面提供的那些实施方案,基本特征和新颖特征是用于使芳族有机部分甲硅烷基化的方法(或在此类方法中使用的体系或从其衍生的组合物)的简便的可操作性。在提供包括使用基本上由基质、有机硅烷(可选择地被称为氢硅烷)、和强碱(强碱的定义现在还包括氢氧化物,尤其koh)组成的混合物的体系和方法的那些实施方案中,其指的是以下的事实:此体系操作以使基质以对应于本文所描述的那些的速率,在如本文所描述的可比较的条件下甲硅烷基化而没有另外的(例如过渡金属)催化剂或等离子体或紫外辐射源。虽然可以存在某种水平的过渡金属,但它们对于方法的可操作性是不需要的,并且可以被认为是为了该反应的目的的旁观者。事实上,进行的大量实验和分析排除通过外来的过渡金属残余物的催化(参见实施例3.1至实施例3.3)。相似地,虽然其他先前的甲硅烷基化反应已经采用了等离子体或紫外辐照来操作,但本发明不需要这些能源。这些能源的另外的存在不应当被认为替代引起本文方法的可操作性的基础。
当呈现出清单时,除非另有指示,否则应理解,该清单的每个单个的要素以及该清单的每个组合是单独的实施方案。例如,作为“a、b、或c”呈现的实施方案的清单应被解释为包括实施方案“a”、“b”、“c”、“a或b”、“a或c”、“b或c”、或“a、b、或c”。相似地,诸如c1-3的指定包括作为单独的实施方案的c1、c2、c3、c1-2、c2-3、c1,3,以及c1-3。
贯穿本说明书,词语被赋予它们的正常含义,如相关领域技术人员将理解的。然而,为了避免误解,某些术语的含义将被特别地定义或澄清。
如本文使用的术语“烷基”指的是直链的、支链的或环状的饱和烃基团,其通常但不一定含有1个至约24个碳原子,优选地含有1个至约12个碳原子,例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、辛基、癸基、及类似基团,以及环烷基例如环戊基、环己基及类似基团。通常,但再次不一定地,本文的烷基含有1至约12个碳原子。术语“低级烷基”意指1个至6个碳原子的烷基,并且特定术语“环烷基”意指通常具有4个至8个、优选5个至7个碳原子的环状的烷基。术语“被取代的烷基”指的是被一个或更多个取代基基团取代的烷基,并且术语“含杂原子的烷基”和“杂烷基”指的是其中至少一个碳原子被杂原子代替的烷基。如果没有另外指明,那么术语“烷基”和“低级烷基”分别包括直链的、支链的、环状的、未被取代的、被取代的和/或含杂原子的烷基和低级烷基。
如本文使用的术语“亚烷基”指的是双官能的直链的、支链的或环状的烷基,其中“烷基”如上文所定义。
如本文使用的术语“烯基”指的是含有至少一个双键的2个至约24个碳原子的直链的、支链的或环状的烃基,例如乙烯基、正丙烯基、异丙烯基、正丁烯基、异丁烯基、辛烯基、癸烯基、十四烯基、十六烯基、二十烯基、二十四烯基、及类似基团。本文中优选的烯基含有2个至约12个碳原子。术语“低级烯基”意指2个至6个碳原子的烯基,并且特定术语“环烯基”意指优选地具有5个至8个碳原子的环状的烯基。术语“被取代的烯基”指的是被一个或更多个取代基基团取代的烯基,并且术语“含杂原子的烯基”和“杂烯基”指的是其中至少一个碳原子被杂原子代替的烯基。如果没有另外指明,那么术语“烯基”和“低级烯基”分别包括直链的、支链的、环状的、未被取代的、被取代的和/或含杂原子的烯基和低级烯基。
如本文使用的术语“亚烯基”指的是双官能的直链的、支链的或环状的烯基,其中“烯基”如上文所定义。
如本文使用的术语“炔基”指的是含有至少一个三键的2个至约24个碳原子的直链的或支链的烃基,例如乙炔基、正丙炔基、及类似基团。本文中优选的炔基含有2个至约12个碳原子。术语“低级炔基”意指2个至6个碳原子的炔基。术语“被取代的炔基”指的是被一个或更多个取代基基团取代的炔基,并且术语“含杂原子的炔基”和“杂炔基”指的是其中至少一个碳原子被杂原子代替的炔基。如果没有另外指明,那么术语“炔基”和“低级炔基”分别包括直链的、支链的、未被取代的、被取代的和/或含杂原子的炔基和低级炔基。
如本文使用的术语“烷氧基”意指通过单个的末端醚键结合的烷基;即,“烷氧基”可以表示为-o-烷基,其中烷基是如上文定义的。“低级烷氧基”意指含有1个至6个碳原子的烷氧基。类似地,“烯氧基”和“低级烯氧基”分别指的是通过单个末端醚键结合的烯基和低级烯基,并且“炔氧基”和“低级炔氧基”分别是指通过单个末端醚键结合的炔基和低级炔基。
术语“芳族的”指的是满足芳香性的hückel4n+2规则的环部分,并且包括芳基(即,碳环的)结构和杂芳基(也被称为杂芳族的)结构二者,这些结构包括芳基、芳烷基、烷芳基、杂芳基、杂芳烷基、或烷杂芳基的部分、或其预聚合的(pre-polymeric)(例如单体的、二聚合的)类似物、低聚物类似物或聚合物类似物。虽然包括koh的方法和体系的描述关于杂芳族基质被提供,其中其可操作性是优选的,然而合理地认为,它们还对于芳基基质起作用。
如本文使用的术语“芳基”,并且除非另有指定,否则指的是含有单个芳环或稠合在一起、直接连接或间接连接(使得不同的芳环被结合到共用基团例如亚甲基部分或亚乙基部分)的多个芳环的芳族取代基或结构。除非另有修饰,否则术语“芳基”指的是碳环结构。优选的芳基含有5个至24个碳原子,并且特别优选的芳基含有5个至14个碳原子。示例性芳基含有一个芳环或两个稠合或连接的芳环,例如苯基、萘基、联苯、二苯醚、二苯胺、二苯甲酮等。“被取代的芳基”是指被一个或更多个取代基基团取代的芳基部分,并且术语“含杂原子的芳基”和“杂芳基”是指其中至少一个碳原子被杂原子替换的芳基取代基,如下文将进一步详细描述。
如本文使用的术语“芳氧基”指的是通过单独的指的是通过单个、末端醚键结合的芳基,其中“芳基”是如上文定义的。“芳氧基”基团可以表示为-o-芳基,其中芳基是如上文定义的。优选的芳氧基含有5个至24个碳原子,并且特别优选的芳氧基含有5个至14个碳原子。芳氧基的实例包括但不限于苯氧基、邻-卤代-苯氧基、间-卤代-苯氧基、对-卤代-苯氧基、邻-甲氧基-苯氧基、间-甲氧基-苯氧基、对-甲氧基-苯氧基、2,4-二甲氧基-苯氧基、3,4,5-三甲氧基-苯氧基、及类似基团。
术语“烷芳基”指的是具有烷基取代基的芳基,并且术语“芳烷基”指的是具有芳基取代基的烷基,其中“芳基”和“烷基”是如上文定义的。优选的烷芳基和芳烷基含有6个至24个碳原子,并且特别优选的烷芳基和芳烷基含有6个至16个碳原子。烷芳基包括,例如,对-甲基苯基、2,4-二甲基苯基、对-环己基苯基、2,7-二甲基萘基、7-环辛基萘基、3-乙基-环戊-1,4-二烯、及类似基团。芳烷基的实例包括但不限于苄基、2-苯基-乙基、3-苯基-丙基、4-苯基-丁基、5-苯基-戊基、4-苯基环己基、4-苄基环己基、4-苯基环己基甲基、4-苄基环己基甲基、及类似基团。术语“烷芳氧基”和“芳烷氧基”指的是式-or的取代基,其中r分别是如刚刚定义的烷芳基或芳烷基。
术语“酰基”指的是具有式-(co)-烷基、-(co)-芳基、或-(co)-芳烷基的取代基,并且术语“酰氧基”指的是具有式-o(co)-烷基、-o(co)-芳基、或-o(co)-芳烷基的取代基,其中“烷基”、“芳基”和“芳烷基”如上文所定义。
术语“环状的”和“环”指的是脂环族基团或芳族基团,这些基团可以是或可以不是被取代的和/或含杂原子的,并且可以是单环的、双环的或多环的。术语“脂环族的”在常规的意义上用于指代脂肪族的环状部分,这与芳族的环状部分相对,并且可以是单环的、双环的或多环的。术语“脂环族的”指的是其中双键不被包含在环结构内的结构。
术语“卤素(halo)”、“卤化物”和“卤素(halogen)”在常规的意义上用于指代氯、溴、氟或碘取代基。
“烃基”指的是含有1个至约30个碳原子、优选1个至约24个碳原子、最优选1个至约12个碳原子的一价烃基基团,包括直链的、支链的、环状的、饱和的和不饱和的物质,例如烷基、烯基、芳基、及类似基团。术语“低级烃基”意指1个至6个碳原子、优选1个至4个碳原子的烃基,并且术语“亚烃基”意指含有1个至约30个碳原子、优选1个至约24个碳原子、最优选1个至约12个碳原子的二价烃基部分,包括直链的、支链的、环状的、饱和的和不饱和的物质。术语“低级亚烃基”意指1个至6个碳原子的亚烃基。“被取代的烃基”指的是被一个或更多个取代基基团取代的烃基,并且术语“含杂原子的烃基”和“杂烃基”指的是其中至少一个碳原子被杂原子代替的烃基。相似地,“被取代的亚烃基”指的是被一个或更多个取代基基团取代的亚烃基,并且术语“含杂原子的亚烃基”和“亚杂烃基”指的是其中至少一个碳原子被杂原子代替的亚烃基。除非另外指明,否则术语“烃基”和“亚烃基”应分别被解释为包括被取代的和/或含杂原子的烃基部分和亚烃基部分。
如在“含杂原子的烃基”中的术语“含杂原子的”指的是其中一个或更多个碳原子被除碳之外的原子例如氮、氧、硫、磷或硅、通常是氮、氧或硫代替的烃分子或烃基分子碎片。相似地,术语“杂烷基”指的是含杂原子的烷基取代基,术语“杂环的”指的是含杂原子的环状取代基,术语“杂芳基”和“杂芳族的”分别指的是含杂原子的“芳基”和“芳族的”取代基及类似基团。应注意,“杂环”基团或化合物可以是或可以不是芳族的,并且进一步地,“杂环”可以是如上文关于术语“芳基”描述的单环、双环或多环。杂烷基的实例包括烷氧基芳基、烷基硫烷基(alkylsulfanyl)取代的烷基、n-烷基化的氨基烷基等。杂芳基取代基的非限制性实例包括吡咯基、吡咯烷基、吡啶基、喹啉基、吲哚基、嘧啶基、咪唑基、1,2,4-三唑基、四唑基,等等,并且含杂原子的脂环族基团的实例是吡咯烷基、吗啉基、哌嗪基、哌啶基,等等。
如本文使用的,术语“基质”或“有机基质”意图表示离散的小分子(有时描述为“有机化合物”)和含有此类“芳族部分”的低聚物和聚合物二者。术语“芳族部分”意图指的是具有所指示的芳族结构的至少一种的化合物、预聚合物(pre-polymer)(即,能够聚合的单体化合物)、低聚物或聚合物的那些部分。当作为结构示出时,该部分至少含有被示出的部分,以及含有另外的官能团、取代基或二者,包括但不限于本文中描述为“fn”的官能团。
如在上文提到的定义中的某些中提到的,如在“被取代的烃基”、“被取代的烷基”、“被取代的芳基”及类似物中的“被取代的”意指在烃基、烷基、芳基、杂芳基或其他部分中结合至碳(或其他)原子的至少一个氢原子被一个或更多个非氢取代基代替。此类取代基的实例包括不限于:本文被称为“fn”的官能团,例如卤素(例如f、cl、br、i)、羟基、氢硫基、c1-c24烷氧基、c2-c24烯氧基、c2-c24炔氧基、c5-c24芳氧基、c6-c24芳烷氧基、c6-c24烷芳氧基、酰基(包括c1-c24烷基羰基(-co-烷基)和c6-c24芳基羰基(-co-芳基))、酰氧基(-o-酰基,包括c2-c24烷基羰基氧基(-o-co-烷基)和c6-c24芳基羰基氧基(-o-co-芳基))、c2-c24烷氧基羰基((co)-o-烷基)、c6-c24芳氧基羰基(-(co)-o-芳基)、卤代羰基(-co)-x,其中x是卤素)、c2-c24烷基碳酸根(-o-(co)-o-烷基)、c6-c24芳基碳酸根(-o-(co)-o-芳基)、羧基(-cooh)、羧酸根(-coo-)、氨基甲酰基(-(co)-nh2)、单(c1-c24烷基)取代的氨基甲酰基(-(co)nh(c1-c24烷基))、二(c1-c24烷基)取代的氨基甲酰基(-(co)-n(c1-c24烷基)2)、单(c1-c24卤代烷基)取代的氨基甲酰基(-(co)-nh(c1-c24烷基))、二(c1-c24卤代烷基)取代的氨基甲酰基(-(co)-n(c1-c24烷基)2)、单(c5-c24芳基)取代的氨基甲酰基(-(co)-nh-芳基)、二(c5-c24芳基)取代的氨基甲酰基(-(co)-n(c5-c24芳基)2)、二-n-(c1-c24烷基)取代的氨基甲酰基、n-(c5-c24芳基)取代的氨基甲酰基、硫代氨基甲酰基(-(cs)-nh2)、单(c1-c24烷基)取代的硫代氨基甲酰基(-(co)-nh(c1-c24烷基))、二(c1-c24烷基)取代的硫代氨基甲酰基(-(co)-n(c1-c24烷基)2)、单(c5-c24芳基)取代的硫代氨基甲酰基(-(co)-nh-芳基)、二(c5-c24芳基)取代的硫代氨基甲酰基(-(co)-n(c5-c24芳基)2)、二-n-(c1-c24烷基)取代的硫代氨基甲酰基、n-(c5-c24芳基)取代的硫代氨基甲酰基、脲基(-nh-(co)-nh2)、氰基(-c≡n)、氰酸根(-o-c=n)、硫代氰酸根(-s-c=n)、甲酰基(-(co)-h)、硫代甲酰基(-(cs)-h)、氨基(-nh2)、单(c1-c24烷基)取代的氨基、二(c1-c24烷基)取代的氨基、单(c5-c24芳基)取代的氨基、二(c5-c24芳基)取代的氨基、c1-c24烷基酰氨基(-nh-(co)-烷基)、c6-c24芳基酰氨基(-nh-(co)-芳基)、亚氨基(-cr=nh,其中r=氢、c1-c24烷基、c5-c24芳基、c6-c24烷芳基、c6-c24芳烷基等等)、c2-c20烷基亚氨基(-cr=n(烷基),其中r=氢、c1-c24烷基、c5-c24芳基、c6-c24烷芳基、c6-c24芳烷基等等)、芳基亚氨基(-cr=n(芳基),其中r=氢、c1-c20烷基、c5-c24芳基、c6-c24烷芳基、c6-c24芳烷基等等)、硝基(-no2)、亚硝基(-no)、磺基(-so2oh)、磺酸根(so2o-)、c1-c24烷基硫烷基(-s-烷基;还被称为“烷基硫代”)、c5-c24芳基硫烷基(-s-芳基;还被称为“芳基硫代”)、c1-c24烷基亚磺酰基(-(so)-烷基)、c5-c24芳基亚磺酰基(-(so)-芳基)、c1-c24烷基磺酰基(-so2-烷基)、c1-c24单烷基氨基磺酰基(-so2-n(h)烷基)、c1-c24二烷基氨基磺酰基-so2-n(烷基)2、c5-c24芳基磺酰基(-so2-芳基)、硼基(-bh2)、二羟硼基(-b(oh)2)、硼酸根(boronato)(-b(or)2,其中r是烷基或其他烃基)、膦酰基(-p(o)(oh)2)、膦酸根(-p(o)(o)2)、次磷酸根(p(o)(o-))、二氧膦基(-po2)、以及膦(-ph2);以及烃基部分,c1-c24烷基(优选地c1-cl2烷基、更优选地c1-c6烷基)、c2-c24烯基(优选地c2-c12烯基、更优选地c2-c6烯基)、c2-c24炔基(优选地c2-c12炔基、更优选地c2-c6炔基)、c5-c24芳基(优选地c5-c24芳基)、c6-c24烷芳基(优选地c6-c16烷芳基)、以及c6-c24芳烷基(优选地c6-c16芳烷基)。在这些取代基结构内,“烷基”、“亚烷基”、“烯基”、“亚烯基”、“炔基”、“亚炔基”、“烷氧基”、“芳族的”、“芳基”、“芳氧基”、“烷芳基”和“芳烷基”部分可以任选地是氟化的或全氟化的。此外,提到的醇、醛、胺、羧酸、酮或其他相似的反应性官能团还包括它们的被保护的类似物。例如,提到的羟基或醇还包括其中羟基被以下保护的那些取代基:乙酰基(ac)、苯甲酰基(bz)、苄基(bn、bnl)、β-甲氧基乙氧基甲醚(mem)、二甲氧基三苯甲基、[双-(4-甲氧基苯基)苯基甲基](dmt)、甲氧基甲醚(mom)、甲氧基三苯甲基[(4-甲氧基苯基)二苯基甲基、mmt)、对-甲氧基苄基醚(pmb)、甲基硫代甲醚、新戊酰基(piv)、四氢吡喃基(thp)、四氢呋喃(thf)、三苯甲基(三苯基甲基、tr)、硅醚(最常用的硅醚包括三甲基甲硅烷基(tms)、叔丁基二甲基甲硅烷基(tbdms)、三异丙基甲硅烷氧基甲基(tom)、和三异丙基甲硅烷基(tips)醚)、乙氧基乙基醚(ee)。提到的胺还包括其中胺被以下保护的那些取代基:boc甘氨酸、苄氧羰基(cbz)、对-甲氧基苄基羰基(moz或meoz)、叔丁基氧基羰基(boc)、9-芴基甲氧基羰基(fmoc)、乙酰基(ac)、苯甲酰基(bz)、苄基(bn)、氨基甲酸酯、对-甲氧基苄基(pmb)、3,4-二甲氧基苄基(dmpm)、对-甲氧基苯基(pmp)、甲苯磺酰基(ts)基团、或磺酰胺(nosyl和nps)基团。提到的含有羰基的取代基还包括其中羰基被以下保护的那些取代基:缩醛基或缩酮基、缩羰酯基、或代森基(diathanegroup)。提到的含有羧酸或羧酸酯基团的取代基还包括其中羧酸或羧酸酯基团被以下保护的那些取代基:其甲酯、苄酯、叔丁酯、2,6-二取代的苯酚(例如2,6-二甲基苯酚、2,6-二异丙基苯酚、2,6-二叔丁基苯酚)的酯、甲硅烷基酯、原酸酯或噁唑啉。优选的取代基是在本文被确定为不影响或很少影响甲硅烷基化化学的取代基,例如包括包含以下的那些取代基:烷基;烷氧化物、芳氧化物、芳烷基烷氧化物、被保护的羰基;任选地被f、cl、-cf3取代的芳基;环氧化合物;n-烷基氮丙啶类;顺式和反式烯烃;乙炔类;吡啶类、伯胺、仲胺以及叔胺;膦类;以及氢氧化物。
如在“官能化的烃基”、“官能化的烷基”、“官能化的烯烃”、“官能化的环烯烃”及类似物中的“官能化的”意指在烃基、烷基、芳基、杂芳基、烯烃、环烯烃或其他部分中结合至碳(或其他)原子的至少一个氢原子被一个或更多个官能团例如在这里和上文描述的那些代替。术语“官能团”意指包括适合于本文描述的用途的任何官能物质。特别地,如本文使用的,官能团将必需具备与基质表面上的相应的官能团反应或结合的能力。
此外,如果特定的基团允许,那么前面提到的官能团可以被一个或更多个另外的官能团或被一个或更多个烃基部分例如上文具体地列举的那些进一步地取代。类似地,上文提到的烃基部分可以被一个或更多个官能团或另外的烃基部分例如具体地列举的那些进一步地取代。
“任选的”或“任选地”意指后续描述的情况可以或可以不发生,使得该描述包括其中该情况发生的例子和其中该情况不发生的例子。例如,措辞“任选地被取代的”意指非氢取代基可以或可以不存在于给定的原子上,并且,因此,该描述包括其中存在非氢取代基的结构和其中不存在非氢取代基的结构。
如本文使用的,术语“甲硅烷基化”指的是碳-硅键的形成,通常是在先前被碳-氢键、通常是未活化的c-h键占据的位置形成。在本文描述的条件下,用c-si键直接地代替c-h键的能力被认为是无先例的(unprecedented)。
本发明包括与用于使芳族化合物和芳族部分甲硅烷基化的化学体系和方法相关的实施方案。具体的实施方案提供用于使芳族化合物和芳族部分甲硅烷基化的化学体系,每个体系包含(a)至少一种有机硅烷和(b)至少一种强碱的混合物,所述强碱的定义现在还包括氢氧化物,尤其koh,所述体系优选地但不必要地大体上无过渡金属化合物。
意识到,在不同的条件下(主要是在较高的温度下)提供芳族化合物和芳族部分的甲硅烷基化的体系和反应还能够使芳族基质内的c-o键、c-n键、c-s键裂解。该后者的还原裂解特征是于2013年10月2日提交的名称为“transition-metal-freereductivecleavageofaromaticc-o,c-n,andc-sbondsbyactivatedsilanes”的共同未决的美国专利申请第14/043,917号的主题,其为了所有目的也通过引用以其整体并入。体系和方法通过其操作的机理尚未被理解,例如,甲硅烷基化是否是裂解反应的中间步骤或共产物(co-product)或副产物(某些观察提示不是),但确实表现为每个分支的相对贡献可以通过反应条件来操纵。例如,如果其他因素是相似的或相等的并且在某些例外情况下,那么似乎较高的温度和较长的反应时间相对于甲硅烷基化反应(其在相对较温和的温度下发生)倾向于有利于c-o键、c-n键、c-s键的裂解。相似地,氢和氢供体分子的不存在(甚至在较高的温度下)和强碱的亚化学计量的量的使用(所述强碱的定义现在还包括氢氧化物,尤其koh)(相对于有机硅烷),似乎有利于甲硅烷基化反应并且不利于c-x裂解。
对于杂芳族化合物的至少甲硅烷基化的初步的机理研究表明涉及自由基物质,尽管连续的机理(acontinuumofmechanism)可以是可操作的。元素性甲硅烷基自由基产生-取代机理(elementarysilylradicalgeneration-substitutionmechanism)似乎是不可能的,这是由于与缺电子杂芳烃例如吡啶的差的反应性(例如,实施6.9.49至实施例6.9.51)。此外,甲硅烷基化的速率在含硫杂芳烃中比在含氧杂芳烃中更快,并且在含氧杂芳烃中比在含氮杂芳烃中更快,如在内部竞争研究(参见例如实施例7.1)中观察到的,该研究提供对于亲电取代和minisci型反应的补充的反应性。这些观察结果指明不同于已知杂芳族c-h官能化反应的潜在的机理。
如本文使用的,术语“大体上无过渡金属化合物”意图反映出,在本文所述的相对温和的条件下该体系对于其使芳族化合物和芳族部分甲硅烷基化的预期目的是有效的,即使在没有任何外源的(即,有意地加入的或以其他方式加入的)过渡金属催化剂时。虽然某些实施方案提供了包括能够催化甲硅烷基化反应的那些的过渡金属可以以与此催化活性通常相关联的水平存在于本文描述的体系或方法内,但是不要求此类金属(作为催化剂或旁观者化合物(spectatorcompound))的存在并且在许多情况下不是合意的。据此,在优选的实施方案中,体系和方法是“大体上无过渡金属化合物”。除非另有声明,否则那么,术语“大体上无过渡金属化合物”定义成反映出,甲硅烷基化体系内的过渡金属的总水平独立地或在有机基质的存在下小于约5ppm,如在下文实施例3.3中描述的由icp-ms测量的。另外的实施方案还提供了过渡金属的浓度小于约10wt%、5wt%、1wt%、100ppm、50ppm、30ppm、25ppm、20ppm、15ppm、10ppm、或5ppm至约1ppm或0ppm。如本文使用的,术语“过渡金属”定义成包括co、rh、ir、fe、ru、os、ni、pd、pt、cu、或其组合。在另外的具体的独立的实施方案中,如由icp-ms测量的ni的浓度小于25ppm、小于10ppm、小于5ppm或小于1ppm。
这些体系通常包含基于烃或醚的溶剂,或体系可以在没有溶剂下操作。如本文描述的,溶剂例如苯、甲苯、均三甲苯、和四氢呋喃类(包括2-甲基四氢呋喃)已被示出很好地起作用。在某些实施方案中,反应在纯的基质(neatsubstrate)中进行。
虽然可以不必限制该体系向水和氧气的暴露,但是,在某些实施方案中,化学体系和方法在大体上无水、无氧气、或无水和氧气二者的环境中进行。在其他实施方案中,存在空气和/或水。除非另有指定,否则术语“大体上无水”指的是水的水平小于约500ppm且“大体上无氧气”指的是氧气的水平对应于小于1托的分压。如果声明的话,那么另外的独立的实施方案可以提供:“大体上无水”指的是水的水平小于1.5%、1%、0.5%、1000ppm、500ppm、250ppm、100ppm、50ppm、10ppm、或1ppm并且“大体上无氧气”指的是对应于小于50托、10托、5托、1托、500毫托、250毫托、100毫托、50毫托、或10毫托的分压的氧气水平。在本文描述的一般程序中,做出有意的努力以排除水和氧气两者,除非另外指定的。
如为了描述体系和方法在本文所使用的,术语“有机硅烷”或“氢硅烷”可以可交换地被使用并且指的是具有至少一个硅-氢(si-h)键的化合物或试剂。有机硅烷还可以含有硅-碳键、硅-氧键、硅-氮键或其组合,并且可以是单体的或被包含在低聚物框架或聚合物框架内,包括被链接至异质支持结构或均质支持结构。在某些实施方案中,这些有机硅烷可以包含至少一种式(i)或式(ii)的化合物:
(r)4-msi(h)m(i)r—[—sih(r)-o—]n—r(ii)
其中:m是1、2、或3,优选是1或2;
n在从约5至约500、从约10至约100或从约25至约50的范围内;并且
每个r独立地是任选地被取代的c1-12烷基或杂烷基、任选地被取代的c5-20芳基或c4-20杂芳基、任选地被取代的c6-30烷芳基或c4-30杂烷芳基、任选地被取代的c6-30芳烷基或c4-30杂芳烷基、任选地被取代的-o-c1-12烷基或杂烷基、任选地被取代的-o-c5-20芳基或–o-c4-20杂芳基、任选地被取代的-o-c6-30烷芳基或–o-c4-30杂烷芳基、或任选地被取代的-o-c6-30芳烷基或–o-c4-30杂芳烷基,并且如果被取代,那么取代基可以是膦酸根、磷酰基、磷烷基(phosphanyl)、膦基、磺酸根、c1-c20烷基硫烷基、c5-c20芳基硫烷基、c1-c20烷基磺酰基、c5-c20芳基磺酰基、c1-c20烷基亚磺酰基、c5-c20芳基亚磺酰基、磺酰氨基、氨基、酰氨基、亚氨基、硝基、亚硝基、羟基、c1-c20烷氧基、c5-c20芳氧基、c2-c20烷氧基羰基、c5-c20芳氧基羰基、羧基、羧酸根、巯基、甲酰基、c1-c20硫酯、氰基、氰酸根、硫代氰酸根、异氰酸酯、硫代异氰酸酯、氨基甲酰基、环氧基、苯乙烯基、甲硅烷基、甲硅烷氧基(silyloxy)、硅烷基、硅氧氮烷基(siloxazanyl)、硼酸根、硼基、或卤素、或含金属的基团或含准金属的基团,其中所述准金属是sn或ge,其中所述取代基可以任选地向包括氧化铝、二氧化硅、或碳的不可溶的或微溶的支持介质提供链接(tether)。示例性的、非限制性的有机硅烷包括(r)3sih,其中r是c1-6烷基,特别地是三乙基硅烷和三丁基硅烷、混合的芳基烷基硅烷例如phme2sih,以及聚合物材料例如聚甲基氢硅氧烷(pmhs)。一般结构(r)2sih2的有机硅烷的使用也良好地工作,并且提供偶联反应或桥接反应的机会。
如本文使用的,术语“强碱”指的是尤其在但不仅仅在非水介质中对氢原子具有强亲合力的化合物。在具体的独立的实施方案中,至少一种强碱包括碱金属或碱性金属氢化物或醇盐。然后应认识到,该定义不严格地限于经典的共轭酸碱模型—因为氢化物的共轭酸将是二氢。这种“强亲合力”的一个测量可以是该强碱(如果与水反应的话)将反应以实质上从其完全形成氢氧化物。其他“强碱”可以被视为包括烷基锂化合物或酰胺离子,例如双(三甲基甲硅烷基)氨基钾。再次,这些描述先前已经被用于描述醇盐、烷基(例如,烷基锂化合物)、酰胺离子、氢化物、以及其他非常强的碱。在先前公开内容的上下文中,这些描述在被描述为“超强碱(superbase)”的材料的上下文中被使用。现在发现的是,在本发明的范围内,术语“强碱”还可以被认为涵盖氢氧化物,特别地koh(氢氧化钾)。
有用的醇盐包括包含c1-12直链的或支链的烷基部分或c5-10芳族部分或c4-10杂芳族部分的那些,例如甲醇盐、乙醇盐、丙醇盐、丁醇盐、2-乙基-己醇盐、或苯甲醇盐。这些中的每种似乎给出类似的反应性。另外,抗衡阳离子的选择也影响化学体系的活性的效力,使得钾是优选的。更具体地,甲醇钾、乙醇钾和叔丁醇钾和2-乙基-己基醇铯已被示出在这种作用中是有效的。通过比较,et3sih与叔丁醇锂或钠的反应提供很少或不提供反应性,这表明抗衡离子在活性甲硅烷基化物质的产生中并且可能地在基质醚的活化中或在两者中起重要作用。相似地,在足以充当钾螯合剂的18-冠-6的存在下进行与叔丁醇钾的反应导致该反应的几乎完全抑制。
氢氧化物例如氢氧化钾(koh)在本发明的方法中,现在首次被认为是碱的有用的来源。该氢氧化物,koh,可以原位形成,例如通过游离态的金属(例如钾)与水的反应原位形成,但在优选的实施方案中,氢氧化物(例如koh)按原样有意地被添加并且优选地无水地添加(即,在水不存在下)。似乎先前描述的反应的条件不产生足以使其以此容量(capacity)工作的koh。
虽然有机硅烷和强碱(所述强碱的定义现在还包括氢氧化物,尤其koh)的相对量不被认为是特别重要的,只要二者均以充分的量存在,但在某些实施方案中,有机硅烷和至少一种强碱(所述强碱的定义现在还包括氢氧化物,尤其koh)以相对于彼此的在从约20:1至约1:1的范围内的摩尔比共同地存在。在其他实施方案中,这些比率可以是约5:1至约1:1、从约3:1至约1:1或从约3:2至约1:1的级别。甲硅烷基化反应似乎也有利于其中碱相对于基质、尤其是更有活性的体系为0.01:1至0.9:1的亚化学计量的那些条件。另外的实施方案提供碱相对于基质以从约0.01:1至约0.6或从约0.1:1至约0.6的比率存在。参见例如表6。
另外的实施方案提供还包含基于n的化合物(优选是基于n的螯合剂)的体系,包括,例如,任选地被取代的四烷基乙二胺(例如,四甲基乙二胺)、任选地被取代的1,10-菲咯啉衍生物、任选地被取代的2,2’-联吡啶衍生物、和任选地被取代的4-二甲基氨基吡啶衍生物。参见,例如,实施例2和表2。
在这一点上,本发明已在能够使芳族化合物或芳族部分甲硅烷基化的化学体系方面被描述,但是还应明显的是,本发明还包括实施这些转化的方法。换言之,各种另外的实施方案包括其中使包含芳族部分的有机基质与上文描述的化学体系中的任何在足以使基质的至少一部分甲硅烷基化的条件下接触的那些方法。换言之,某些实施方案提供方法,每种方法包括:在足以使基质甲硅烷基化的条件下,使包含芳族部分的有机基质与(a)至少一种有机硅烷和(b)至少一种强碱(所述强碱的定义现在还包括氢氧化物,尤其koh)的混合物接触;其中所述混合物和所述基质优选地,但不必要地大体上无过渡金属化合物。这些实施方案通常在液相中进行,而没有uv辐照或电放电条件或等离子体放电条件。
在某些实施方案中,足以使有机基质甲硅烷基化的条件包括:在约10℃至约165℃的范围内的温度下,加热基质与(a)所述至少一种有机硅烷和(b)所述至少一种强碱(所述强碱的定义现在还包括氢氧化物,尤其koh)的混合物。在某些情况下,温度可以在从约20℃、约30℃、约40℃、约50℃、约60℃、或约80℃至约165℃、约150℃、约125℃、约100℃、或至约80℃的范围内被应用。实施例中描述的任何温度均可以被认为是独立的实施方案。典型的操作反应时间可以在从约2小时、从约4小时、从约6小时、或从约10小时至约28天、至约14天、至约7天、至约4天、至约3天、至约48小时、至约24小时、至约12小时,或至约6小时的范围中。
如上文所描述的,描述为与用于使芳族化合物和芳族部分甲硅烷基化的化学体系有关的那些特征也与使这些芳族化合物和芳族部分甲硅烷基化的方法有关。例如,在各种实施方案中,该方法提供了该体系大体上无水、无氧气、或无水和氧气二者。
在其他实施方案中,至少一种有机硅烷包括式(i)或式(ii)的有机硅烷:
(r)4-msi(h)m(i)r—[—sih(r)-o—]n—r(ii)
其中m是1、2、或3(优选地1或2);
n是10至100;并且
每个r独立地是任选地被取代的c1-12烷基或杂烷基、任选地被取代的c5-20芳基或c4-20杂芳基、任选地被取代的c6-30烷芳基或c4-30杂烷芳基、任选地被取代的c6-30芳烷基或杂芳烷基、任选地被取代的-o-c1-12烷基或杂烷基、任选地被取代的-o-c5-20芳基或–o-c4-20杂芳基、任选地被取代的-o-c6-30烷芳基或c4-30杂烷芳基、或任选地被取代的-o-c6-30芳烷基或–o-c4-30杂芳烷基,并且如果被取代,那么取代基可以是膦酸根、磷酰基、磷烷基、膦基、磺酸根、c1-c20烷基硫烷基、c5-c20芳基硫烷基、c1-c20烷基磺酰基、c5-c20芳基磺酰基、c1-c20烷基亚磺酰基、c5-c20芳基亚磺酰基、磺酰氨基、氨基、酰氨基、亚氨基、硝基、亚硝基、羟基、c1-c20烷氧基、c5-c20芳氧基、c2-c20烷氧基羰基、c5-c20芳氧基羰基、羧基、羧酸根、巯基、甲酰基、c1-c20硫酯、氰基、氰酸根、硫代氰酸根、异氰酸酯、硫代异氰酸酯、氨基甲酰基、环氧基、苯乙烯基、甲硅烷基、甲硅烷氧基、硅烷基、硅氧氮烷基、硼酸根、硼基、或卤素、或含金属的基团或含准金属的基团,其中所述准金属是sn或ge,其中所述取代基可以任选地向包括氧化铝、二氧化硅、或碳的不可溶的或微溶的支持介质提供链接。
在另外其他的实施方案中,有机硅烷是(r)3sih,其中r是c1-6烷基、优选地et3sih或et2mesih、或(r)2sih2。至少一种强碱可以包括碱金属或碱性金属的氢化物,如上文所描述的,例如,氢化钙或氢化钾。至少一种强碱可以包括碱金属或碱性金属的醇盐,如上文所描述的,例如,其中至少一种醇盐包含c1-12直链的或支链的烷基部分或c5-10芳基或c4-10杂芳基部分,优选地是甲醇盐、乙醇盐、丙醇盐、丁醇盐、或2-乙基-己醇盐。碱金属阳离子优选地是钾或铯。在最优选的实施方案中,所述有机硅烷是三乙基硅烷、三甲基硅烷、二乙基甲基硅烷、二乙基硅烷、二甲基硅烷、二甲基乙基硅烷、乙基二甲基硅烷、二甲基苯基硅烷、二乙基苯基硅烷并且强碱是叔丁醇钾。强碱现在可以包括氢氧化钾。其他组合或示例性的反应物在这点上提供另外的实施方案。
在某些实施方案中,有机硅烷(或单体等效物)和至少一种强碱(所述强碱的定义现在还包括氢氧化物,尤其koh)以相对于彼此的在从约20:1至约1:1的范围内的摩尔比共同地存在。在某些实施方案中,至少一种强碱(包括koh)和有机基质以相对于彼此的在从约0.01:1至约5:1的范围内的摩尔比共同地存在。优选地,碱相对于有机基质是亚化学计量的-即,以0.01:1至0.9:1的比率。即,方法可以被认为相对于本文所预期的强碱是催化性的。
此外,在方法的上下文中,术语“大体上无过渡金属化合物”具有与上文针对化学体系描述的相同的含义和相关实施方案;即,反映出该方法在无任何有意添加的过渡金属催化剂的存在下有效地进行。除非另有声明,否则当描述方法或体系时,术语定义为反映过渡金属的总水平小于约50ppm,如通过在下文实施例3.3中描述的由icp-ms测量的。另外的实施方案还提供了过渡金属的浓度小于相对于总体系的重量(即,相对于甲硅烷基化体系以及甲硅烷基化体系和有机基质二者)的约10wt%、5wt%、1wt%、100ppm、50ppm、30ppm、25ppm、20ppm、15ppm、10ppm、或5ppm至约1ppm或0ppm。如本文使用的,术语“过渡金属”至少定义成包括co、rh、ir、fe、ru、os、ni、pd、pt、cu、或其组合。在另外的独立的实施方案中,如由icp-ms测量的ni的浓度小于25ppm、小于10ppm、小于5ppm或小于1ppm。在此注意,化学体系的某些实施方案可以包含至少一种有机硅烷和强碱(所述强碱的定义现在还包括氢氧化物,尤其koh),应认识到,当考虑这些混合物组合中的每一个时,独立的实施方案提供了过渡金属的水平保持低于刚刚描述的水平。
另外的实施方案提供了该方法还包括使用亚化学计量的量(相对于基质)的包括以下的基于n的化合物(优选是基于n的螯合剂):例如,任选地被取代的四烷基乙二胺(例如,四甲基乙二胺)、任选地被取代的1,7-菲咯啉衍生物、任选地被取代的1,10-菲咯啉衍生物、任选地被取代的2,2'-联吡啶衍生物、和任选地被取代的4-二甲基氨基吡啶衍生物。
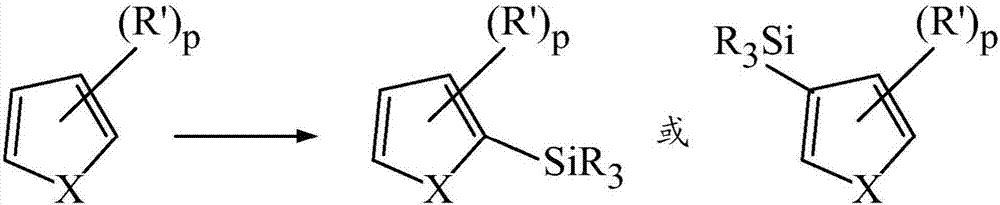
该方法相对于基质是相当灵活的,并且适应含有芳基和杂芳基部分二者的那些。包含芳基部分的示例性基质包括:包含任选地被取代的苯(包括均三甲苯和甲苯)、联苯、萘、蒽或高级多芳环结构(higherpolyaromaticringstructure)的那些。这些纯的烃基质通常要求比杂芳基体系更有力的条件以使环碳甲硅烷基化。参见实施例6.4。然而,使这些烃环结构官能化的能力是这些方法和体系的重要特征。
如果芳基部分或杂芳基部分包含α-甲基或亚甲基c-h键,如在任选地被取代的c1-6烷基中(如通过实施例中的甲苯、均三甲苯、1,2-二甲基吲哚或2,5-二甲基噻吩的甲基例示的),那么似乎反应进行以在比使环碳甲硅烷基化所需的温度更低的温度下形成α硅烷。如本文使用的,术语“α碳”指的是位于芳族部分的环外的第一个碳,并且如在“α甲基或亚甲基”中的“α”,意指在直接地附接至芳环的第一个环外的碳上的甲基或亚甲基。术语“α-硅烷”指的是结合至该α碳的硅烷。术语“α”被认为包括6元芳基芳族化合物的苄基碳。引起此类甲硅烷基化的方法在本发明的范围内。
包括具有环外的芳族c-x键的那些的其他的环外的环取代基,通常根据本文描述的方法来反应。术语“环外的”指的是o、n或s相对于芳环体系的位置。例如,术语“环外的”指的是这样的键,在该键中碳被包含在芳环体系内,但是相应的氧、氮或硫原子不被包含在芳环体系内,并且(在氮的情况下)反之亦然。例如苯酚、二甲基苯胺、1-甲基-1h-吡咯和苯硫酚分别地含有环外的芳族的c-o键、c-n键和c-s键。示例性有机基质包括但不限于任选地被取代的苯基醚类、苯胺类、苯硫醚类、萘基醚类、萘胺类、或萘硫醚类部分、n-烷基或n-芳基吡咯类、或其组合。
当x是o或n时,反应在含有环外的c-x键的碳的邻位或邻近处的碳处有利于环的甲硅烷基化。富电子体系或给电子基团或取代基通常表现为比缺电子体系或吸电子基团或取代基通常更具反应性;后者可能要求比前者更有力的条件,但是请注意,来源于较高温度的更有力的条件可能导致驱动c-x裂解分支(cleavagemanifold),参见,例如于2013年10月2日提交的名称为“transition-metal-freereductivecleavageofaromaticc-o,c-n,andc-sbondsbyactivatedsilanes”的共同提交的美国专利申请第14/043,917号。苯甲醚和2-甲氧基萘示出对邻位位置的特别偏好,并且这种选择性为包括此类基质的选择性邻位甲硅烷基化的实施方案提供基础。参见例如,实施例6.7.1和实施例6.7.4。
注意,这些化合物可以被看作聚合物或低聚物的代用品。例如,使二甲氧基苯、二苯基醚、以及3-甲氧基萘甲硅烷基化所呈现的能力提供能够支持使具有例如以下的连接基(linkage)的聚合物甲硅烷基化的能力:
包括例如亚苯基氧化物、萘氧化物、或亚烷基亚苯基氧化物的聚合物或共聚物,并且实现这些转化的方法被认为是在本公开内容的范围内。
令人关注地,并且对比之下,具有环外的芳族c-x键(其中x是s-烷基)的那些基质提供了不同的反应性,示出对使烷基而非芳环体系甲硅烷基化的倾向。参见,例如,实施例6.7.5。这种反应性模式为包括此类基质的β-甲硅烷基化的那些实施方案提供基础。
在某些实施方案中,该方法被应用于包含杂芳基部分的有机基质。非限制性的杂芳基部分包括那些任选地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、三唑、异噁唑、噁唑、噻唑、异噻唑、噁二唑、吡啶、哒嗪、嘧啶、吡嗪、三嗪酮、苯并呋喃、苯并吡咯、苯并噻吩、异苯并呋喃、异苯并吡咯、异苯并噻吩、吲哚、异吲哚、吲嗪、吲唑、氮杂吲哚、苯并异噁唑、苯并噁唑、喹啉、异喹啉、噌啉、喹唑啉、萘啶、2,3-二氢苯并呋喃、2,3-二氢苯并吡咯、2,3-二氢苯并噻吩、二苯并呋喃、呫吨、二苯并吡咯、二苯并噻吩。在更优选的实施方案中,所述基质包含包括以下的部分:任选地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、苯并呋喃、苯并吡咯、苯并噻吩、吲哚、氮杂吲哚、二苯并呋喃、呫吨、二苯并吡咯、或二苯并噻吩部分。独立的实施方案提供了该方法获得如本文描述的被取代的甲硅烷基化产物。
在其他的具体实施方案中,该方法关于包含以下部分的基质是可操作的:
其中x是n-r”、o、或s;
y是h、n(r”)2、o-r”、或s-r”;
p是0至4、0至3、0至2、或0至1;
r’是如上文所描述的官能团“fn”,或(r’)p包括稠合的脂环族部分、杂脂环族(例如亚甲基、亚乙基、或亚丙基连接的二醚)部分、芳基部分或杂芳基部分;并且
r”是胺保护基团或任选地被取代的烷基、芳基、杂芳基、烷芳基或烷杂芳基,优选是任选地被取代的c1-c6烷基、苯基、甲苯基、苄基或苯乙基。
示例性稠合的杂环部分包括例如以下基团:
在某些更具体的实施方案中,该方法关于包含以下部分的有机基质是可操作的:
其中x、y、r'、r”和p如上文所定义。注意,在每种情况下,指定
意图允许在任一个芳族环上的取代。
杂芳基部分似乎根据本发明的方法在比它们的芳基同族(cogener)更温和的条件下反应,使得在混合的芳基-杂芳基体系中,反应通常进行以优先地使杂芳基环甲硅烷基化。
另外,5元杂芳基部分似乎根据本发明的方法在比甚至6元杂芳基部分更温和的条件下反应。例如,如在实施例6.9.26至实施例9.9.29中示出的,1h-吡咯并吡啶被示出在分子的5元杂环部分中优先地甲硅烷基化。并且两个环都在比对于吡啶所发现的条件温和得多的条件下甲硅烷基化。
用包含5元杂芳基部分的基质的甲硅烷基化反应也提供显著清楚的且显然地可调节的区域选择性。包含含有o或n的5元杂芳基环的基质显然可以在c-2或c-3位甲硅烷基化,这取决于时间和温度,但较温和的条件似乎有利于在c-2位的取代。虽然不意图受任何特定理论的正确性或不正确性的束缚,但是似乎在c-2位的甲硅烷基化代表反应的动力学结果,而在c-3位的甲硅烷基化是热力学上有利的。虽然在“动力学”和“热力学”路径方面被描述,但不清楚的是,在c-3位的甲硅烷基化必然通过c-2中间体进行。事实上,使用其中c-2位被甲基基团封阻的1,2-二甲基吲哚和2,5-二甲基噻吩的实验,反应进行以优先地使α-甲基基团甲硅烷基化,而无c-3位中的甲硅烷基化的迹象。
除非另有声明,否则提到的在特定位的甲硅烷基化意图暗示产物的在该位置的大于约80%的区域选择性或区域专一性。但是其他实施方案提供了在该位置的区域专一性大于约50%、大于约75%、大于约90%或大于约95%。
甲硅烷基化反应还对于一系列官能团是明显地耐受的(参见例如实施例7.2)。通常,羰基是不被耐受的,但如果被保护为对应的缩醛或缩酮,则可以被使得是相容的。芳基–f、芳基–cl、芳基–cf3、环氧化物、n-烷基氮丙啶、顺式烯烃和反式烯烃、乙炔、吡啶、以及叔胺和膦部分全部与甲硅烷基化化学是相容的。甚至游离的oh和nh基团在某种程度上被耐受,这显然归因于杂原子的幸运的(fortuitous)甲硅烷基化的原位保护。相比之下,芳基–br、芳基–i、芳基–cn、以及芳基–no2的存在全部似乎使反应停止。此多用性对于将当前方法应用于例如生物碱天然产物合成和在早期的或用于高级中间体官能化的药学科学应用是重要的。
本发明方法的产物在农用化学、药学和电子应用的范围内是有用的,如在下文描述的。杂芳基硅烷衍生物,例如本文所描述的,被已知经历多种强有力的合成转化;许多代表性实例此处被证实(图4b)。再次,这些下游转化中的每个由于本发明的工艺是可达到的,并且因此这些下游步骤(当与本发明的甲硅烷基化结合时)被认为在本发明的范围内。
芳族硅烷的使用,例如本文描述的那些,对于制备联芳化合物/联芳族化合物是有用的合成子,例如,使用本领域中一般地认可的hiyama偶联方法。如技术人员所理解的,“联芳族的(biaromatic)”指的是通过单键连接的两个独立的芳族/杂芳族环体系-例如,双环呋喃(bifuran)、联苯、联吡啶、联噻吩、苯基-吡啶等等。技术人员将很好地能够使这些hiyama偶联方法的教导与此处呈现的那些教导在不过度实验下组合,以制备联芳化合物/联芳族化合物,并且此类制备被认为是在本发明的范围内。另外,ball和同事(ball等人,science2012年9月28日:第337卷,第6102期,第1644-1648页,针对其催化剂、方法和基质的教导内容通过引用将其并入本文)最近已描述了另一种方法,使用金催化剂来偶联三烷基硅烷,例如本文描述的那些,从而形成联芳化合物/联芳族化合物。再次,技术人员将能够再次在无过度实验情况下很好地结合包括在ball参考文献中教导或表明的至少第二芳基化合物的ball偶联的教导内容,以制备联芳化合物或联芳族化合物,并且此类方法和制备被认为是在本发明的范围内。在此类实施方案中,本发明的甲硅烷基化产物,无论是被分离还是被原位地生成,都在足以使甲硅烷基化产物与第二芳族化合物偶联以制备联芳产物或联芳族产物的条件(包括合适的过渡金属催化剂的存在)下进一步反应。如本文意图的,第二芳族化合物包含任选地被取代的芳族部分,该芳族部分包括任选地被取代的芳基部分和杂芳基部分,其中术语“任选地被取代的”、“芳族的”、“芳基”和“杂芳基”具有与已经在本文中描述的相同的定义。
这样的转化在本文被说明。例如,通过zhao和snieckus的方法的c2si定向的suzuki–miyaura交叉偶联,或经由杂芳基硅醇6的hiyama–denmark交叉偶联,提供2-芳基化的吲哚。苯并噻吩的不常见的直接c7官能化以给出硼酸酯7和8通过使用保护基策略从甲硅烷基化的前体4h来实现。参见实施例8.4.1和实施例8.4.2。此一般转化(即,使用本发明的甲硅烷基化以保护/脱保护某些有利的位置)被认为是在本发明的范围内。事实上,虽然实施例8.4.1和实施例8.4.2在吲哚类(并且引申开来,苯并呋喃类和噻吩类)的c2位的上下文中这样示出,但区域专一性地设置并且然后除去甲硅烷基的能力在化学家的工具箱中是有价值的新的工具。
使用熟知的fleming-tamao氧化方法,芳族硅烷的转化例如本文描述的那些也已知可转化成芳族的羟基化合物。技术人员将能够再次在无过度实验情况下很好地组合这些fleming-tamao氧化的教导内容与在此提出的那些教导内容,以制备羟基化的芳族化合物,并且此类方法和制备被认为是在本发明的范围内。在此类实施方案中,本发明的芳族的甲硅烷基化产物,无论是被分离还是被原位地生成,都在足以使甲硅烷基化产物转化为羟基化的芳族产物的条件(包括合适的过渡金属催化剂的存在)下进一步反应。在羟基化之后,芳族产物可以通过常规方法被转化成对应的烷基或芳基醚、烷基或芳基酯、卤化物(氯代、溴代、氟代、碘代)、硝酸酯、亚硝酸酯、或其他类似的官能团。芳基或杂芳基碘化物对于一系列偶联反应是尤其方便的前体(参见例如,如在nishihara,等人,tetrahedronletters,50(2009)4643-4646中描述的此类化合物与炔基硅烷的钯/铜催化的硅杂-sonogashira反应)。由其产生的所有此类转化和产物被认为是在本发明的范围内(当结合本发明的甲硅烷基化进行时)。
另外,本发明的为了提供α-碳取代基(或在环外的硫的情况中的β-甲硅烷基基团)而提供甲硅烷基化的能力也提供了那些产物可以用作用于peterson烯烃化反应的合成子。当α-亚甲基质子邻近于硅烷的硅时(“α硅效应”),已知的易于使该α-亚甲基质子去质子化以获得α-甲硅烷基负碳离子可以形成用于该烯烃化反应的方便的前体。技术人员将能够再次在无过度实验情况下很好地组合这些peterson烯烃化反应的教导内容与在此提出的那些教导内容,以用α烯烃代替α甲硅烷基基团,并且此类方法和制备被认为是在本发明的范围内。在此类实施方案中,本发明的芳族的甲硅烷基化产物,无论是被分离还是被原位地生成,都在足以使甲硅烷基化产物转化为芳族的α-烯烃产物的条件(包括合适的过渡金属催化剂的存在)下进一步反应。
另外的实施方案包括以下的实施方案:其中本发明的芳族甲硅烷基化产物,无论是被分离还是被原位生成,在足以使α甲硅烷基化产物转化成对应的羧酸的条件(包括合适的过渡金属催化剂的存在)下使用例如在mita,等人,organicletters,2012,第14卷,第13期,3462-3465中描述的方法进一步反应。技术人员将能够在再次无过度实验情况下很好地组合这些反应的教导内容与在此提出的那些教导内容,以制备羧基化的芳族化合物,并且此类方法和制备被认为是在本发明的范围内。
又另外的实施方案包括以下的实施方案:其中本发明的芳族甲硅烷基化产物,无论是被分离还是被原位地生成,在足以使芳族的甲硅烷基化产物转化成硼的卤化物和酯、卤化物(包括氯代、溴代、和碘代)、以及亚硝基的条件(包括合适的过渡金属催化剂的存在)下使用例如在zhao,等人,organicletters,2005,第7卷,第13期,2523-2526中描述的方法进一步反应。技术人员将能够在再次无过度实验情况下很好地组合这些反应的教导内容与在此提出的那些教导内容,以制备羧基化的芳族化合物,并且此类方法和制备被认为是在本发明的范围内。另外,如在zhao参考文献中所描述的,衍生自本发明的这些芳族的甲硅烷基化的前体还可以使用上文描述的suzuki-miyaura交叉偶联方案与芳族卤化物交叉偶联,以产生联芳族产物。
使被取代的噻吩和三聚噻吩(terthiophene)甲硅烷基化所呈现的能力还提供这些产物与全氟芳烃(perfluoroarene)的进一步反应,以提供交替的噻吩-全氟芳烃共聚物,如在wangy.和watsonm.,j.amer.chem.soc.,2006,128,2536-2537中所描述的。技术人员将能够在再次无过度实验情况下很好地组合wang和watson的教导与在此呈现的那些教导,以制备无过渡金属的交替的噻吩-全氟芳烃共聚物,并且此类方法和从其衍生的产物被认为是在本发明的范围内。
有机硅在高级材料的开发中已经广泛地被研究,这归因于硅的独特的物理的和化学的性质。在本上下文中,本公开内容提供在材料和药物上下文中有价值的化合物和转化的实例(参见例如,图4c和实施例8.8.1至实施例8.8.5)。在仅一个实例中,硅杂杂环9在一步中直接从可商购的未被官能化的杂芳烃通过无先例的双c-h官能化(包括分子间甲硅烷基化,随后是分子内甲硅烷基化)来制备。噻吩低聚体10的高收率双甲硅烷基化(bis-silylation)为至交替的共聚物的完全无过渡金属的催化路线提供起始材料。最后,3,4-亚乙二氧基噻吩单体的单选择性甲硅烷基化为聚噻吩衍生的材料的修饰提供可能的策略(图4c,11)。使噻吩类(包括edot)和三聚噻吩类甲硅烷基化的一般能力是本发明的许多重要的方面中的一个。
硅杂药物类似物已经从医药化学工作者获得许多注意力,因为与母体全碳化合物比较,它们可以提供改进的稳定性、溶解度以及药代动力学性质。此外,装入的有机硅官能性可以用作用于随后详细阐述的合成操作,这有助于库合成(librarysynthesis)并且使得能够进行构效关系研究(structure–activityrelationshipstudy)。结果,包含有机硅的小分子是药物科学中逐渐增长的兴趣,并且先导化合物(leadcompound)的直接甲硅烷基化因此将代表药物发现中新的且可能强有力的工具。为了评估用于此类后期c-h官能化应用的本文方法,使抗组胺药西那利定和抗血小板药物噻氯匹定经受代表性催化甲硅烷基化条件。反应在两种活性药物成分的情况下都平稳地进行,这产生56%–68%收率的具有优良的化学选择性和区域选择性的含si目标化合物12和13a–c(图4d)。哌啶类、苯胺、苄型c-h键和芳基氯部分全部被耐受,而没有任何观察到的副反应。氮杂类似物14的甲硅烷基化也很好地进行,这证明这些方法与具有可能的药物重要性的含吡啶的络合物分子的相容性。最后,在这些研究期间,在环境温度下,少量的sp2和sp3c–h甲硅烷基化副产物分别在甲氧基和甲基取代的吲哚类的情况下(即,15和16;图4e)被观察到。简单的芳烃类似地反应。苯甲醚的邻位甲硅烷基化和甲苯的无定向取代基的(directinggroup-free)c(sp3)–h甲硅烷基化被发现,这分别提供甲硅烷基化的衍生物17a和18a。四个另外的实施例被呈现,这提供具有优良的选择性的甲硅烷基芳烃(17b和17c)和苄基硅烷(18b和18c)。应特别注意的是2,6-卢剔啶的c(sp3)–h甲硅烷基化,这提供在缺电子体系中c-h甲硅烷基化的实例。令人关注地,甲氧基甲苯19和苄基醚21(两者均包含可能反应性sp2和sp3c–h键)在相反的选择性下被甲硅烷基化,以产生20和22。在22的情况下,反应引入si取代的手性中心。
以下实施方案的清单意图补充而非代替或取代之前的描述。
实施方案1.用于使包含芳族部分的有机基质甲硅烷基化的化学体系,所述体系包含(a)至少一种有机硅烷和(b)至少一种强碱的混合物或基本上由(a)至少一种有机硅烷和(b)至少一种强碱的混合物组成,所述强碱的定义现在还包括氢氧化物,尤其koh,所述体系优选地但不必要地大体上无过渡金属化合物,所述强碱足以实现有机部分的甲硅烷基化,而不用过渡金属催化剂、等离子体、或紫外辐射。
实施方案2.如实施方案1所述的体系,其中所述过渡金属化合物以相对于总体系的重量的小于10ppm而存在。
实施方案3.如实施方案1或2所述的化学体系,还包含任选地被取代的四烷基乙二胺(例如,四甲基乙二胺)、任选地被取代的1,7-菲咯啉衍生物、任选地被取代的1,10-菲咯啉衍生物、任选地被取代的2,2'-联吡啶衍生物、或任选地被取代的4-二甲基氨基吡啶衍生物。
实施方案4.如实施方案1至3中任一项所述的体系,所述体系大体上无水、无氧气、或无水和氧气二者,优选地无氧的且无水的。
实施方案5.如实施方案1至4中任一项所述的体系,其中至少一种有机硅烷包括式(i)或式(ii)的有机硅烷:
(r)4-msi(h)m(i)r—[—sih(r)-o—]n—r(ii)
其中:m是1、2、或3;n是10至100;并且每个r独立地是任选地被取代的c1-12烷基或杂烷基、任选地被取代的c5-20芳基或c4-20杂芳基、任选地被取代的c6-30烷芳基或杂烷芳基、任选地被取代的c5-30芳烷基或杂芳烷基、任选地被取代的-o-c1-12烷基或杂烷基、任选地被取代的-o-c5-20芳基或–o-c4-20杂芳基、任选地被取代的-o-c5-30烷芳基或杂烷芳基、或任选地被取代的-o-c5-30芳烷基或杂芳烷基,并且如果被取代,那么取代基可以是膦酸根、磷酰基、磷烷基、膦基、磺酸根、c1-c20烷基硫烷基、c5-c20芳基硫烷基、c1-c20烷基磺酰基、c5-c20芳基磺酰基、c1-c20烷基亚磺酰基、c5-c20芳基亚磺酰基、磺酰氨基、氨基、酰氨基、亚氨基、硝基、亚硝基、羟基、c1-c20烷氧基、c5-c20芳氧基、c2-c20烷氧基羰基、c5-c20芳氧基羰基、羧基、羧酸根、巯基、甲酰基、c1-c20硫酯、氰基、氰酸根、硫代氰酸根、异氰酸酯、硫代异氰酸酯、氨基甲酰基、环氧基、苯乙烯基、甲硅烷基、甲硅烷氧基、硅烷基、硅氧氮烷基、硼酸根、硼基、或卤素、或含金属的基团或含准金属的基团,其中所述准金属是sn或ge,其中所述取代基可以任选地向包括氧化铝、二氧化硅、或碳的不可溶的或微溶的支持介质提供链接。
实施方案6.如实施方案5所述的体系,其中所述有机硅烷是(r)3sih或(r)2sih2,其中r是芳基、芳烷基、或c1-6烷基。
实施方案7.如实施方案1至6中任一项所述的体系,其中所述至少一种强碱包括碱金属或碱性金属的氢化物或醇盐。
实施方案8.如实施方案1至7中任一项所述的体系,其中所述至少一种强碱包括碱金属或碱性金属的氢化物。
实施方案9.如实施方案8所述的体系,其中所述至少一种强碱包括氢化钙或氢化钾。
实施方案10.如实施方案1至7中任一项所述的体系,其中所述至少一种强碱包括碱金属或碱性金属的醇盐。
实施方案11.如实施方案10所述的体系,其中所述至少一种醇盐包含c1-12直链的或支链的烷基部分或c5-10芳族或杂芳族的部分。
实施方案12.如实施方案11所述的体系,其中所述至少一种醇盐包括甲醇盐、乙醇盐、丙醇盐、丁醇盐、或2-乙基-己醇盐。
实施方案13.如实施方案7至12中任一项所述的体系,其中所述碱金属或碱性金属的氢化物或醇盐碱(alkoxidebase)是钾或铯的醇盐。
实施方案14.如实施方案1至13中任一项所述的体系,其中所述有机硅烷是三乙基硅烷并且所述强碱是叔丁醇钾。
实施方案15.如实施方案1至7中任一项所述的体系,其中所述至少一种强碱包括氢氧化钾(koh)。
实施方案16.如实施方案1至15中任一项所述的体系,其中所述有机硅烷和所述至少一种强碱以相对于彼此的在从约20:1至约1:1的范围内的摩尔比共同地存在。
实施方案17.如实施方案1至15中任一项所述的体系,还包含有机芳族化合物,所述化合物是溶剂、基质、或溶剂和基质二者。
实施方案18.如实施方案17所述的体系,其中所述有机化合物包含任选地被取代的苯、联苯、萘或蒽的环结构。
实施方案19.如实施方案17或18所述的体系,其中所述有机芳族化合物包含杂芳基部分。
实施方案20.如实施方案19所述的体系,其中所述有机芳族化合物包含任选地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、三唑、异噁唑、噁唑、噻唑、异噻唑、噁二唑、吡啶、哒嗪、嘧啶、吡嗪、三嗪酮、苯并呋喃、苯并吡咯、苯并噻吩、异苯并呋喃、异苯并吡咯、异苯并噻吩、吲哚、异吲哚、吲嗪、吲唑、氮杂吲哚、苯并异噁唑、苯并噁唑、喹啉、异喹啉、噌啉、喹唑啉、萘啶、2,3-二氢苯并呋喃、2,3-二氢苯并吡咯、2,3-二氢苯并噻吩、二苯并呋喃、呫吨、二苯并吡咯、或二苯并噻吩部分。
实施方案21.如实施方案19或20所述的体系,其中所述有机芳族化合物包含任选地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、苯并呋喃、苯并吡咯、苯并噻吩、吲哚、氮杂吲哚、二苯并呋喃、呫吨、二苯并吡咯、二苯并噻吩或受阻的二苯并呋喃、二苯并吡咯或二苯并噻吩部分。
实施方案22.如实施方案17至21中任一项所述的体系,其中所述有机芳族化合物包含以下部分中的至少一种:
其中x是n-r”、o、或s;
y是h、n(r”)2、o-r”、或s-r”;
p是0至4、0至3、0至2、或0至1;
r’是如上文所描述的官能团“fn”,或(r’)p包括任选地被取代的稠合的脂环族部分、杂脂环族部分(例如亚甲基、亚乙基、或亚丙基连接的二醚)、芳基部分或杂芳基部分;并且
r”是胺保护基团或任选地被取代的烷基、芳基、杂芳基、烷芳基或烷杂芳基,优选是任选地被取代的c1-c6烷基、苯基、甲苯基、苄基或苯乙基。
实施方案23.如实施方案17至22中任一项所述的体系,其中所述基质包含以下部分中的至少一种:
其中x、y、r'、r”和p如上文所定义。注意,在每种情况下,指定
意图允许在任一个芳族环上的取代。
实施方案24.如实施方案17至22中任一项所述的方法的体系,其中所述芳族有机化合物包含至少一种α-甲基或亚甲基c-h键,所述方法导致α硅烷的形成。
实施方案25.一种使包含芳族部分的基质甲硅烷基化的方法,所述方法包括使一定量的有机基质与实施方案1至24中任一项所述的体系接触。
实施方案26.一种方法,包括:使包含芳族部分的有机基质与包含(a)至少一种有机硅烷和(b)至少一种强碱或基本上由(a)至少一种有机硅烷和(b)至少一种强碱组成的混合物在足以使所述基质甲硅烷基化的条件下接触,所述强碱的定义现在还包括氢氧化物,尤其koh;其中所述混合物和所述基质优选地但不必要地大体上无过渡金属化合物。
实施方案27.如实施方案26所述的方法,其中所述过渡金属化合物以相对于总体系的重量的小于10ppm而存在。
实施方案28.如实施方案26或27所述的方法,其中所述混合物还包含任选地被取代的四烷基乙二胺(例如,四甲基乙二胺)、任选地被取代的1,7-菲咯啉衍生物、任选地被取代的1,10-菲咯啉衍生物、任选地被取代的2,2'-联吡啶衍生物、或任选地被取代的4-二甲基氨基吡啶衍生物。
实施方案29.如实施方案26至28中任一项所述的方法,所述方法大体上无水、无氧气、或无水和氧气二者。
实施方案30.如实施方案26至29中任一项所述的方法,其中至少一种有机硅烷包括式(i)或式(ii)的有机硅烷:
(r)4-msi(h)m(i)r—[—sih(r)-o—]n—r(ii)
其中m是1、2、或3(优选地1或2);
n是10至100;并且
每个r独立地是任选地被取代的c1-12烷基或杂烷基、任选地被取代的c5-20芳基或c4-20杂芳基、任选地被取代的c6-30烷芳基或杂烷芳基、任选地被取代的c6-30芳烷基或杂芳烷基、任选地被取代的-o-c1-12烷基或杂烷基、任选地被取代的-o-c5-20芳基或-c4-20杂芳基、任选地被取代的-o-c6-30烷芳基或-o-c4-30杂烷芳基、或任选地被取代的-o-c6-30芳烷基或–o-c4-30杂芳烷基,并且如果被取代,那么取代基可以是膦酸根、磷酰基、磷烷基、膦基、磺酸根、c1-c20烷基硫烷基、c5-c20芳基硫烷基、c1-c20烷基磺酰基、c5-c20芳基磺酰基、c1-c20烷基亚磺酰基、c5-c20芳基亚磺酰基、磺酰氨基、氨基、酰氨基、亚氨基、硝基、亚硝基、羟基、c1-c20烷氧基、c5-c20芳氧基、c2-c20烷氧基羰基、c5-c20芳氧基羰基、羧基、羧酸根、巯基、甲酰基、c1-c20硫酯、氰基、氰酸根、硫代氰酸根、异氰酸酯、硫代异氰酸酯、氨基甲酰基、环氧基、苯乙烯基、甲硅烷基、甲硅烷氧基、硅烷基、硅氧氮烷基、硼酸根、硼基、或卤素、或含金属的基团或含准金属的基团,其中所述准金属是sn或ge,其中所述取代基可以任选地向包括氧化铝、二氧化硅、或碳的不可溶的或微溶的支持介质提供链接。
实施方案31.如实施方案26至30中任一项所述的方法,其中所述有机硅烷是(r)3sih,其中r独立地是c1-6烷基。
实施方案32.如实施方案26至31中任一项所述的方法,其中所述至少一种强碱包括碱金属或碱性金属的氢化物或醇盐。
实施方案33.如实施方案26至32中任一项所述的方法,其中所述至少一种强碱包括碱金属或碱性金属的氢化物。
实施方案34.如实施方案33所述的方法,其中所述至少一种强碱包括氢化钙或氢化钾。
实施方案35.如实施方案26至34中任一项所述的方法,其中所述至少一种强碱包括碱金属或碱性金属的醇盐。
实施方案36.如实施方案35所述的方法,其中所述至少一种醇盐包含c1-12直链的或支链的烷基部分或c5-10芳基或c4-10杂芳基部分。
实施方案37.如实施方案36所述的方法,其中所述至少一种醇盐包括甲醇盐、乙醇盐、丙醇盐、丁醇盐或2-乙基-己醇盐。
实施方案38.如实施方案32至37中任一项所述的方法,其中所述碱金属或碱性金属的氢化物或醇盐是钾或铯的醇盐。
实施方案39.如实施方案26至38中任一项所述的方法,其中所述有机硅烷是三乙基硅烷且所述强碱是叔丁醇钾。
实施方案40.如实施方案26所述的方法,其中所述有机硅烷是三乙基硅烷且所述强碱是氢氧化钾。
实施方案41.如实施方案26至29中任一项所述的方法,其中所述有机硅烷和所述至少一种强碱以相对于彼此的在从约20:1至约1:1的范围内的摩尔比共同地存在,所述强碱的定义现在包括氢氧化物,尤其koh。
实施方案42.如实施方案26至41中任一项所述的方法,其中所述至少一种强碱和所述基质以相对于彼此的在从约0.01:1至约5:1的范围内、优选地在从约0.01:1至约0.9:1的范围内的摩尔比共同地存在,所述强碱的定义现在包括氢氧化物,尤其koh。
实施方案43.如实施方案26至42中任一项所述的方法,其中所述有机基质包含任选地被取代的苯、联苯、萘或蒽的环结构。
实施方案44.如实施方案26至43中任一项所述的方法,其中所述有机基质包含环外的芳族c-x键,其中x是n、o或s。
实施方案45.如实施方案26至44中任一项所述的方法,其中所述有机基质包含环外的芳族的c-x键并且所述甲硅烷基化在环外的c-x键的邻位发生,其中x是n、o或s。
实施方案46.如实施方案26至45中任一项所述的方法,其中所述有机基质包含杂芳基部分。
实施方案47.如实施方案26至46中任一项所述的方法,其中所述基质包含任选地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、三唑、异噁唑、噁唑、噻唑、异噻唑、噁二唑、吡啶、哒嗪、嘧啶、吡嗪、三嗪酮、苯并呋喃、苯并吡咯、苯并噻吩、异苯并呋喃、异苯并吡咯、异苯并噻吩、吲哚、异吲哚、吲嗪、吲唑、氮杂吲哚、苯并异噁唑、苯并噁唑、喹啉、异喹啉、噌啉、喹唑啉、萘啶、2,3-二氢苯并呋喃、2,3-二氢苯并吡咯、2,3-二氢苯并噻吩、二苯并呋喃、呫吨、二苯并吡咯、或二苯并噻吩部分。
实施方案48.如实施方案26至47中任一项所述的方法,其中所述基质包括任选地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、苯并呋喃、苯并吡咯、苯并噻吩、吲哚、氮杂吲哚、二苯并呋喃、呫吨、二苯并吡咯、或二苯并噻吩。
实施方案49.如实施方案26至48中任一项所述的方法,其中所述有机芳族基质包含以下部分中的至少一种:
其中x是n-r”、o、或s;
y是h、n(r”)2、o-r”、或s-r”;
p是0至4、0至3、0至2、或0至1;
r’是如上所述的官能团“fn”,或(r’)p是任选地被取代的稠合的脂环族部分、杂脂环族部分、芳基部分或杂芳基部分;并且
r”是胺保护基团或任选地被取代的烷基、芳基、杂芳基、烷芳基或烷杂芳基,优选是任选地被取代的c1-c6烷基、苯基、甲苯基、苄基或苯乙基。
实施方案50.如实施方案26至48中任一项所述的方法,其中所述基质包含以下部分中的至少一种:
其中x、y、r'、r”和p如上文所定义。注意,在每种情况下,指定
意图允许在任一个芳族环上的取代。
实施方案51.如实施方案26至50中任一项所述的方法,其中所述有机基质包含以下结构的杂芳基部分:
并且所述甲硅烷基化在杂芳基环的c-2位发生。
实施方案52.如实施方案26至51中任一项所述的方法,其中所述有机基质包含以下结构的杂芳基部分:
并且所述甲硅烷基化在杂芳基环的c-3位发生。
实施方案53.如实施方案26至52中任一项所述的方法,其中芳族基质包含至少一种α-甲基或亚甲基c-h键,所述方法导致α硅烷的形成。
实施方案54.如实施方案26至53中任一项所述的方法,其中所述芳族基质是聚合的或是聚合物前体。
实施方案55.如实施方案26至54中任一项所述的方法,其中所述芳族的甲硅烷基化产物在足以使所述甲硅烷基化产物与第二芳族化合物偶联以制备联芳族产物的条件下进一步反应。
实施方案56.如实施方案26至54中任一项所述的方法,其中所述芳族的甲硅烷基化产物在足以使所述甲硅烷基化产物转化为芳族的羟基化的(被保护的或游离的羟基)、烷氧基化的(或芳氧基化的)、或烷基或芳基羧酸化的产物的条件下进一步反应。
实施方案57.如实施方案26至54中任一项所述的方法,其中所述芳族的甲硅烷基化产物在足以使所述甲硅烷基化产物转化为芳族的α-烯烃产物的条件下进一步反应。
实施方案58.如实施方案26至54中任一项所述的方法,其中所述芳族的甲硅烷基化产物在足以使所述甲硅烷基化产物转化为芳族卤化物(氯代、溴代、氟代、碘代)、硝酸酯、或亚硝酸酯的条件下进一步反应。
实施方案59.如实施方案26至54中任一项所述的方法,其中所述芳族的甲硅烷基化产物在足以使所述甲硅烷基化产物转化为芳族的α-羧酸或羧酸烷基酯或羧酸芳基酯的条件下进一步反应。
实施方案60.如实施方案26至54中任一项所述的方法,其中所述芳族的甲硅烷基化产物在足以使所述甲硅烷基化产物转化为芳族的硼卤化物或硼酸酯(boronicester)的条件下进一步反应。
实施方案61.如实施方案26至54中任一项所述的方法,其中所述甲硅烷基化的噻吩产物在足以使所述甲硅烷基化产物转化为交替的噻吩-全氟芳烃共聚物的条件下进一步反应。
实施例
提供以下实施例以例证在本公开内容内描述的概念中的某些。虽然每个实施例被认为是提供组合物、制备方法和用途的具体的单独的实施方案,但是无一实施例应当被认为限制本文描述的较一般的实施方案。
在以下实施例中,虽然已作出努力来确保关于所使用的数字(例如量、温度等等)的准确度,但是某些实验误差和偏差应当被考虑在内。除非另有指示,否则温度以摄氏度计,压力在或接近大气压。
实施例1:一般信息
除非另有指定,否则所有的反应都在干燥玻璃器皿(例如烘箱干燥的或火焰干燥的)中使用标准舒伦克线(schlenkline)技术在氩气气氛下或在真空气氛手套箱中在氮气气氛下进行。
溶剂通过在氩气下通过穿过活性氧化铝柱来干燥。反应进程通过薄层色谱法(tlc)分析、uhplc-lcms分析或gc-fid分析来监测。tlc使用e.merck硅胶60f254预涂覆的玻璃板(0.25mm)进行,并且通过紫外荧光猝灭、磷钼酸、或kmno4染色来可视化。silicyclesiliaflashp60academic硅胶(粒度40–63nm)被用于快速色谱法。
均三甲苯(纯度≥99.0%(gc))用钠/二苯甲酮回流,然后蒸馏。四氢呋喃通过穿过溶剂纯化柱来纯化,然后用钠-钾合金进一步蒸馏并且用氩气脱气。所有的其他溶剂通过穿过溶剂纯化柱来纯化并且用氩气进一步脱气。用于空气敏感实验的nmr溶剂用cah2干燥并且被真空转移或蒸馏至干燥的舒伦克瓶(schlenkflask)中,并且随后用氩气脱气。三乙基硅烷(99%)购自sigma-aldrich、用分子筛回流,并且接下来被蒸馏。然后,其在使用之前通过三个冷冻-泵-解冻(freeze-pump-thaw)循环来脱气并且随后传递穿过中性的氧化铝。氘代三乙基硅烷(97原子%d)购自sigma-aldrich并且在使用之前通过三个冷冻-泵-解冻循环来脱气并且其他可商购获得的液体试剂被类似地处理。苯基二甲基硅烷(≥98%)、乙基二甲基硅烷(98%)和二乙基硅烷(99%)购自sigma-aldrich并且用cah2蒸馏并且在使用之前通过三个冷冻-泵-解冻循环来脱气。其他可商购获得的液体试剂被类似地处理。1-甲基吲哚(≥97%)、苯并呋喃(99%)、硫茚(98%)、1-甲氧基萘(≥98%)、苯甲醚(99%)和硫代苯甲醚(99%)购自sigma-aldrich并且在使用之前被蒸馏。2-甲氧基萘从沸腾的et2o重结晶两次。1-苯基吡咯(99%)被溶解在et2o中并且被传递穿过活性氧化铝。将醚在真空中除去,并且将固体残余物从绝对etoh/水的3:1混合物中重结晶两次。1-苯基吡咯(99%)、二苯基醚(≥99%)、二苯并噻吩(≥99%)购自sigma-aldrich,并且按原样被使用。4-甲氧基吡啶(97%)和2,6-二甲氧基吡啶(98%)购自sigma-aldrich,通过中性的活性氧化铝传递若干次,并且在使用之前经受3个冷冻-泵-解冻循环。1-甲基-7-氮杂吲哚遵照cheve,g.等人,medchemcomm2012,3,788的程序来制备。升华级kot-bu(99.99%)购自sigma-aldrich并且在使用之前经受真空升华(30毫托,160℃)。二-4-(甲基)苯基醚、1-萘酚、2-萘酚、4-叔丁基苯甲醚、4-甲基苯甲醚、1,3-二苯氧基苯、2-甲氧基萘、和1.0m四丁基氟化铵thf溶液购自sigma-aldrich并且按原样使用。4-(甲氧基)二苯并呋喃、二-4-(叔丁基)苯基醚、萘基醚类、4-(苯基)苯基苯基醚、2-乙氧基萘、2-新戊氧基萘、2-叔丁氧基萘根据文献程序来合成。
杂芳族基质购自aldrich、tci、或acros,或根据文献程序例如(a)kong,a.;han,x.;lu,x.org.lett.2006,8,1339.(b)islam,s.;larrosa,i.chem.–eur.j.2013,19,15093.(c)huestis,m.p.;fagnou,k.org.lett.2009,11,1357.(d)mahadevan,i.;rasmussen,m.tetrahedron,1993,49,7337来合成。另外,以下化合物被合成,并且先前已经在美国专利第9,000,167号中被报告:4-(三乙基甲硅烷基)二苯并呋喃(3);4,6-双(三乙基甲硅烷基)二苯并呋喃(4);3-(三乙基甲硅烷基)联苯-2-醇(5);(3’-三乙基甲硅烷基)联苯-2-醇(6);3,3’-双(三乙基甲硅烷基)联苯-2-醇(7);邻三乙基甲硅烷基二苯醚
标准nmr光谱学实验用以下进行:varianmercury(1h,300mhz)光谱仪、varianinova400mhz光谱仪、装配有autox探针的varian500mhz光谱仪、或装配有triax探针的varian600mhz光谱仪。化学位移通过使用残留溶剂峰作为内标依据距me4si的低磁场ppm来报告。光谱使用第7版mestrenova来分析和处理。ir光谱在perkinelmerspectrumbxii光谱仪上使用沉积在nacl板上的薄膜来获得,并且以吸收频率(cm–1)被报告。uhplc-lcms分析在装配有agilenteclipseplusc18rrhd1.8μm柱的agilent1290超高效液相色谱/质谱仪上获得。gc-fid分析在装配有hp-5(5%-苯基)-甲基聚硅氧烷毛细管柱的agilent6890n气相色谱仪(agilent)上获得。gc-ms分析在装配有hp-5(5%-苯基)-甲基聚硅氧烷毛细管柱的agilent6850气相色谱仪(agilent)上获得。高分辨率质谱(ei和fab)通过加州理工学院(californiainstituteoftechnology)的质谱设备获取。epr光谱在brukerems光谱仪上记录。
实施例2:碱性活化剂的评估
贯穿本说明书,n-甲基吲哚被示出充当与本发明的化学有关的反应性的优良的示例。各种碱的影响在以下的标称条件下被评估,其中表1中提供了结果:
来自表1的结果揭示,用于c-h甲硅烷基化反应的良好的催化剂通过体积大的碱性阴离子和钾阳离子的组合来分类:kot-bu证明是理想的催化剂,并且在纯的条件(neatcondition)下或在thf和meot-bu(条目18、20和22)中操作,但khmds(条目21)和kotms(条目24)也是有效的。在liot-bu和naot-bu(条目1和条目2)下的完全没有反应性以及当18-冠-6被添加到kot-bu(条目23)时反应性的急剧降低支持钾阳离子的重要的、虽然未知的作用。在化学计量的反应中,转化率大致与碱度相关联(即,ot-bu>oet>ome;条目5–7)。在这些条件下,在催化剂不存在下或当使用kh、koh、koac以及cs2co3(条目9–12)时,没有产物被观察到。注意,koh在这些反应中是不起反应的先前的发现现在已经被确证,但通过改变反应条件,现在可能的是,在此催化剂下实现这些转化(参见对于koh的实施例9)。有机碱dabco和用于硅的常见基于氟的活化剂-tbaf、csf、以及kf–也被研究,并且未能使起始材料转化(条目13–16)。成功的甲硅烷基化反应的顶部空间gctcd分析指示h2的形成。
令人关注地,其他潜在的螯合剂不抑制并且在许多情况下改进了体系的收率和选择性二者。这种效应不被很好地理解。在不受这个或任何其他理论的正确性的束缚的情况下,可以提出这些配体螯合了钾阳离子。基于联吡啶的配体骨架以及tmeda(未示出)被证实在促进甲硅烷基化反应中的高的选择性和效率中是最有效的。这通过与不能螯合钾的1,7-菲咯啉的反应来支持,该反应给出较低的产物收率。
本发明的体系和方法的活性对不同的碱载量是显著地耐受的。在n-甲基吲哚模型体系中,例如,减少碱载量仅略微地降低了效率。显著地,甚至下降至1mol%的kotbu是有效的并且得到了65%收率和相应的89%c2选择性的主要c2产物。这种载量甚至低于或等于技术现状的基于过渡金属的芳族c-h甲硅烷基化体系所需要的载量。
实施例3:对照实验和痕量金属分析
实施例3.1:采用可商购的kot-bu、再升华的kot-bu、以及新鲜制备的kot-bu的对照反应。三个反应平行地进行(thf,45℃,1-甲基吲哚,20mol%kot-bu,0.2mmol规模):a)kot-bu(aldrich,升华级别,99.99%,痕量金属基础)按原样被使用;b)kot-bu(aldrich,升华级别,99.99%痕量金属基础)在通过在真空下加热该材料而再升华之后来使用;以及c)通过钾金属与无水t-buoh的反应,随后是t-buoh的蒸发和固体的升华而制备的kot-bu被使用。在这些反应中在转化率和选择性方面没有明显的差异被观察到。
实施例3.2:采用购自不同的供应商的不同级别的kot-bu的对照反应。四个反应平行地进行(thf,45℃,1-苄基吲哚,20mol%kot-bu,0.2mmol规模):a)kot-bu(aldrich,升华级别,99.99%,痕量金属基础),b)kot-bu(strem,98%),c)kot-bu(tci,>97%),以及d)kot-bu(alfa-aesar,97%)。反应通过uhplc-lcms来监测。产物的转化率在90小时之后完成大于90%,并且在这四个反应中在转化率和选择性方面没有明显的差异被观察到。
实施例3.3:分析500mg样品,各自具有来自四个不同供应商(strem,aldrich,tci,alfa-aesar)的kot-bu、1-苄基吲哚、et3sih、thf,以及标准反应混合物(0.5mmol规模混合物,遵循一般程序用103.5mg的1-bn-吲哚、来自aldrich的11.2mg的kot-bu、在0.5ml的thf中的173.5mg的et3sih制备的并且在手套箱中搅拌持续72h)。将每个样品添加到50mldigitube消化管(digestiontube)(scpscience),随后添加3.0ml的等离子体纯硝酸(plasmapurenitricacid)(scpscience),并且加热到75℃持续36小时。在消化之后,使用milliq水将每个样品稀释到50ml并且在agilent7900icp-ms光谱仪上进行样品分析。lod指示,分析物浓度低于仪器的最低检测限。值以ppb(微克每升)计。
表3.
实施例4:对于kot-bu-催化的c-h甲硅烷基化的自由基性质的研究
进行若干实验以获得对于反应机理的洞察。作为第一项研究,反应在自由基捕获剂(radicaltrap)tempo和加尔万氧基(galvinoxyl)的存在下进行。在以其他方式有益于n-甲基吲哚的甲硅烷基化的条件下,两种添加剂都阻碍c-h甲硅烷基化。
在第二组实验中,三个对照实验试图探查tempo的作用。在23℃下,在1当量的tempo下,观察到痕量的三乙基甲硅烷基保护的产物ii,推测由甲硅烷基自由基和tempo自身的自由基组合引起。当温度升高到65℃时,产物ii变成混合物的主要组分,这支持在甲硅烷基化反应中涉及甲硅烷基自由基物质。相比之下,在kot-bu不存在下,没有观察到此被保护的化合物ii,这指示催化剂对于产生甲硅烷基自由基是重要的。
为了评估极性机理(即,甲硅烷基阴离子的形成)的可能的贡献,用苯并噻吩3h作为基质,在作为添加剂的环己烯氧化物(已知环氧化物(包括环己烯氧化物)通过甲硅烷基阴离子经历亲核开环)的存在下对kot-bu–催化的反应进行实验。然而,在该测试中使用的标准环境条件下,环氧化物在反应之后被定量回收,并且以中等收率获得期望的甲硅烷基化产物4h,这提供对抗离散的甲硅烷基阴离子的形成的证据。
实施例5:一般程序
在氮气填充的手套箱中,使2打兰闪烁管(scintillationvial)或4ml螺口管(screwcapvial)装载有对应的基质(0.1mmol-0.5mmol,1当量)、碱(例如kot-bu或koh,0.1当量-5当量)和磁性搅拌棒,随后注射添加溶剂(1ml)和硅烷(1当量-5当量,在使用之前,通过短的活性氧化铝垫过滤)。将反应管密封,并且将混合物在指示的温度下搅拌持续指示的时间。将该管从手套箱中移出,将反应混合物用二乙醚(2ml)稀释,并且在减压下浓缩。区域选择性(c2甲硅烷基化产物与c3甲硅烷基化产物:c2:c3)通过粗制混合物的1hnmr分析或gc分析来确定。将残余物通过硅胶快速色谱法来纯化,以给出期望的产物。
除非另有指示,否则在制备实验中仅具有超过2%的总收率的产物被分离并表征。在萘基烷基醚的情况下,使用不同的后处理程序(workupprocedure)。在冷却之后,反应用二氯甲烷(5ml)来稀释并且小心地用2ml的1n含水的hcl来猝灭。添加十三烷,并且将混合物转移至分液漏斗。分离出有机相,并且用二氯甲烷(3ml)萃取水层。合并的有机层经无水mgso4干燥并且过滤。对于所有的反应而言,产物通过与可信样品比较使用gc/ms和gc/fid和nmr来确定。在萘基烷基醚还原中观察到的痕量的可溶性副产物包括萘、1,2,3,4-四氢萘和5,6,7,8-四氢-2-萘酚。
在大多数情况下,产物在通过经由独立的光谱分析或与可信样品比较、或二者的nmr和/或gc-ms的表征之前被分离并且纯化。在其中产物不被分离和纯化的那些情况下,表征基于gc-ms和/或gc-fid分析来进行。
实施例6:选定的反应
实施例6.1:4-(三乙基甲硅烷基)二苯并呋喃的反应
反应根据一般程序通过在100℃下,使4-et3si-二苯并呋喃(3,141mg,0.5mmol,1当量)、kot-bu(112mg,1mmol,2当量)和et3sih(401微升,2.5mmol,5当量)在2ml的甲苯中加热持续20小时来进行。在酸性水溶液后处理之后,粗制反应混合物通过使用己烷和己烷-醚(10:1)的二氧化硅色谱法来纯化,以分离2-苯基苯酚(2,30mg,0.177mmol,35%)、2-三乙基甲硅烷基-6-苯基苯酚(5,37mg,0.134mmol,26%)、2-(3-三乙基甲硅烷基苯基)苯酚(6,17mg,0.063mmol,12%)。未消耗的3以及产物1、4和7的量使用对应的混合级分的色谱法后(post-chromatography)gc-fid分析来获得。
实施例6.2:作为中间体的甲硅烷基化的二苯并呋喃朝向c-o键裂解的研究:用kot-bu的裂解尝试
在100℃下,在j.young管中在氮气下将起始材料3(14.1mg,0.05mmol,1当量)与kot-bu(分别为5.6mg或11.2mg,1当量或2当量)在0.8ml的氘代甲苯中一起加热持续20小时。通过1hnmr监测反应进程显示出在这两种情况下无3的转化。同样地,在160℃下,使起始材料3(28.2mg,0.1mmol,1当量)或4(39.6mg0.1mmol,1当量)与kot-bu(36.6mg)在0.3ml的均三甲苯中加热持续20小时。粗制反应混合物的通过gc-fid或1hnmr的后续分析揭示出在3的情况下至1的3%转化率和从4至3的5%转化率。
实施例6.3:4-(甲氧基)二苯并呋喃在升高的温度下的反应
反应根据一般程序通过在100℃下将4-meo-二苯并呋喃(8,89mg,0.5mmol,1当量)、kot-bu(112mg,1mmol,2当量)和et3sih(401微升,2.5mmol,5当量)在2ml的甲苯中加热持续20小时来进行。在水溶液后处理之后,粗制反应混合物通过使用己烷和己烷-醚的硅胶色谱法来纯化以回收未消耗的起始材料8(3mg,0.015mmol,3%)并且分离二苯并呋喃(1,8.4mg,0.05mmol,10%;因为1的级分包含少量的起始材料8,所以定量通过1h-nmr使用ch2br2作为内标来进行)、1,1’-联苯-2-醇(2,4.3mg,0.025mmol,5%)、4-et3si-二苯并呋喃(3,11mg,0.039mmol,8%)、2-甲氧基-6-苯基-苯酚(9,mg,0.025mmol,5%)、2-(3’-甲氧基苯基)苯酚(10,47mg,0.235mmol,47%)。注意:仅表征了具有超过2%的收率的化合物。9和10的1h和13cnmr光谱归属(spectralassignment)与文献报告一致。
实施例6.4:芳烃的三乙基甲硅烷基化
实施例6.4.1.在升高的温度下
在许多例子中,当在升高的温度下使用甲苯或均三甲苯作为在还原裂解反应中使用的溶剂时,溶剂衍生的甲硅烷基化产物的形成在升高的温度下在目的在于c-o键、c-n键或c-s键裂解的实验期间被观察到。因为不可能通过柱色谱法或蒸馏使得到的产物从它们的相应的母溶剂分离,所以就此而言,难以评估它们的收率,但是它们被试验性地估计为基于et3sih的5%-10%的范围内。在甲苯的情况下,产物的确定通过使所获得的nmr光谱与文献数据(rychnovsky等人,j.org.chem.2003,68,10135.)比较而确证。因此,结论是,主要产物是苄基三乙基硅烷(17),这也与异构产物的碎裂模式的gc-ms分析一致。同样地,似乎均三甲苯的甲硅烷基化主要在苄(或α)位中进行。hrms[c15h26si]计算为234.1804,测量为234.1804)。
实施例6.4.2.直接c(sp3)–h甲硅烷基化反应
苄基三乙基硅烷18a:反应根据一般程序通过在65℃下将kot-bu(11.2mg,0.1mmol,20mol%)、甲苯(46mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和dme(0.5ml)加热持续108h来进行。c(sp3):c(sp2)=18:1。期望的产物18a的gc收率是53%。在真空(60毫托,23℃)下对起始材料和挥发物的蒸发之后,分析纯产物(25.0mg,24%收率)作为无色油来获得。注意:化合物18a是易挥发的,并且在真空下容易除去。rf=0.8(100%己烷);1hnmr(500mhz,cdcl3)δ7.22(m,2h),7.09–7.05(m,1h),7.05–7.02(m,2h),2.12(s,2h),0.96–0.91(t,9h),0.53(q,j=7.9hz,6h)。
三乙基((4’-甲基-[1,1’-联苯]-4-基)甲基)硅烷18b:反应根据一般程序通过在85℃下将kot-bu(11.2mg,0.1mmol,23mol%)、4,4’-二甲基-1,1’-联苯(80.0mg,0.44mmol)、et3sih(240μl,1.5mmol,3.4当量)、以及0.5ml的thf加热持续96h来进行。单甲硅烷基化产物与双甲硅烷基化产物的比率是16:1。期望的产物18b和起始材料4,4’-二甲基-1,1’-联苯(69.7mg的混合物,包含56.6mg的18b,43%收率,基于1hnmr计算的)在通过硅胶快速色谱法(100%己烷)纯化之后获得。小部分的分析纯化合物18b在随后通过硅胶快速色谱法的纯化之后作为无色油获得。rf=0.5(100%己烷);1hnmr(500mhz,cdcl3)δ7.50–7.47(m,2h),7.46–7.42(m,2h),7.25–7.21(m,2h),7.11–7.04(m,2h),2.39(s,3h),2.14(s,2h),0.95(t,j=7.9hz,9h),0.54(q,j=8.0hz,6h);13cnmr(126mhz,cdcl3)δ139.7,138.5,136.7,136.5,129.6,128.6,126.8,126.7,21.4,21.2,7.5,3.1;ir(纯的膜(neatfilm),nacl)3022,2951,2909,2873,1610,1497,1455,1416,1238,1209,1153,1005,845,806,773,729cm–1;hrms(ei+)对于c20h28si[m+·]计算为:296.1960,实测为296.1954。
2-甲基-6-((三乙基甲硅烷基)甲基)吡啶18c:反应根据一般程序通过在65℃下将kot-bu(11.2mg,0.1mmol,20mol%)、2,6-卢剔啶(53.5mg,0.5mmol)、et3sih(240μl,1.5mmol,3当量)、以及0.5ml的thf加热持续120h来进行。期望的产物18c(58.6mg,53%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中5%→10%etoac)纯化之后作为无色油获得。注意:化合物18c是易挥发的,并且在真空下容易除去。rf=0.3(在己烷中5%etoac);1hnmr(500mhz,cdcl3)δ7.36(t,j=7.6hz,1h),6.90–6.73(m,2h),2.47(s,3h),2.32(s,2h),0.98–0.83(m,9h),0.58–0.48(m,6h);13cnmr(126mhz,cdcl3)δ160.8,157.4,135.9,119.0,118.4,25.4,24.5,7.2,3.3;ir(纯的膜,nacl)3060,2951,2874,1587,1575,1450,1414,1372,1269,1238,1145,1078,1016,919,796,748,726cm–1;hrms(ei+)对于c13h24nsi[m+h]+计算为:222.1678,实测为222.1666。
2,5-二甲基噻吩的甲硅烷基化:反应根据一般程序通过在65℃下将2,5-二甲基噻吩(56mg,0.5mmol,1当量)、kot-bu(11.2mg,0.1mmol,0.2当量)和et3sih(3当量)在四氢呋喃中加热持续45小时来进行。粗制产物混合物的gc-ms获得了与单甲硅烷基化衍生物相关联的质量峰。1hnmr数据与2-甲基-5-(三乙基甲硅烷基甲基)噻吩的形成一致。1hnmr(500mhz,thf-d8)δ6.52–6.42(m,1h),6.41–6.29(m,1h),2.35(s,3h),2.23(s,2h),1.00–0.92(m,9h),0.63–0.53(m,6h)。13cnmr(126mhz,thf-d8)δ140.78,136.28,125.96,124.03,15.73,15.45,7.97,4.08。hrms:[c12h22ssi]计算为226.1212,测量为226.1220。
n-甲基-2-甲基吲哚的甲硅烷基化:反应根据一般程序通过在65℃下将1,2-二甲基吲哚(73mg,0.5mmol,1当量)、kot-bu(17mg,0.15mmol,0.3当量)和et3sih(319微升,2.0mmol,4当量)在1ml的四氢呋喃中加热持续65小时来进行。在水溶液后处理之后,粗制反应混合物通过分别使用己烷:et2o:et3n的80:1:4混合物的硅胶色谱法来纯化,以获得作为无色油的74mg(57%)的标题化合物。1hnmr(500mhz,thf-d8)δ7.35–7.29(m,1h),7.19(dd,j=8.1,0.9hz,1h),6.97(ddd,j=8.2,7.1,1.2hz,1h),6.90(ddd,j=8.0,7.1,1.1hz,1h),6.06(d,j=0.8hz,1h),3.64(s,3h),2.25(d,j=0.7hz,2h),0.96(t,j=7.9hz,9h),0.71–0.58(m,6h)。13cnmr(126mhz,thf-d8)δ139.50,138.30,129.69,120.24,119.70,119.47,109.27,98.96,29.75,11.73,7.62,4.16。hrms:[c16h25nsi]计算为259.1756,测量为259.1754。此反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
三乙基(苯氧基(苯基)甲基)硅烷22:反应根据一般程序通过在65℃下将kot-bu(11.2mg,0.1mmol,20mol%)、(苄氧基)苯21(92.0mg,0.5mmol)、et3sih(240μl,1.5mmol,3当量)、以及0.25ml的thf加热持续120h来进行。期望的产物22(68.4mg,46%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.3(100%己烷);1hnmr(500mhz,cdcl3)δ7.46–7.37(m,4h),7.38–7.28(m,4h),7.30–7.20(m,2h),5.80(s,1h),0.92(t,j=7.9hz,9h),0.66–0.55(m,6h);13cnmr(126mhz,cdcl3)δ145.3,128.1,128.1,126.9,126.9,126.4,126.3,6.7,4.9;ir(纯的膜,nacl)3063,3026,2954,2875,1598,1492,1454,1413,1302,1239,1188,1090,1065,1006,974,833,740,700cm–1;hrms(ei+)对于c19h25osi[(m+h)-h2]+计算为:297.1675,实测为297.1668。
芳族胺也经得起甲硅烷基化。在以下的情况下,gc-ms确定出的以下方案在所提供的条件下是可操作的:
在较低的温度下,该反应似乎提供产物的混合物,而无可确定的单个产物。虽然未确认,但是仍可能的是,对环外的胺的邻位甲硅烷基化的表观正常倾向(apparentnormalproclivity)受到与两个甲基基团相关联的位阻效应(stericbulk)的抑制。
实施例6.5:二苯并呋喃在升高的温度下的甲硅烷基化
除非另外指示,否则使用一般程序进行实验。收率在±2%内是可再现的。在此值得注意的是,低水平的碱,尤其是相对于基质的亚化学计量的量的碱,即使在这些高温下也导致甲硅烷基化产物相对于裂解产物的最高收率。
实施例6.6:芳基烷基醚在升高的温度下的甲硅烷基化
芳基烷基醚在升高的温度下的甲硅烷基化在应用于二芳基醚的条件下进行以探查sp2相对于sp3c-o键的裂解选择性。在这些实验的升高的温度下,2-甲氧基萘的反应以中等收率得到2-萘酚作为主要产物(方案1)。粗制反应混合物的gc-ms分析指示出存在痕量的萘连同2-甲基萘和另外的被还原的物质,包括部分芳族还原的产物。还检测到大概衍生自2-萘酚甲硅烷基化的化合物。同样地,2-乙氧基萘在相同条件下的裂解得到以略微更高的收率的2-萘酚,但是具有相同的或类似的副产物。空间上更大的醚被研究以探查c-o键裂解的多用性和可能机理。虽然大的烷基取代基邻近于醚氧,但是2-新戊氧基萘的反应以与使用较不庞大的基质近似相同的收率提供2-萘酚。甚至2-叔丁氧基萘也被裂解以得到预期的萘酚,收率为55%(方案1)。在相同的条件下进行的但是不使用三乙基硅烷的对照实验在2-乙氧基萘和2-叔丁氧基萘的情况下提供了2-萘酚,虽然具有显著减小的收率。因为2-甲氧基-基质和2-新戊氧基-基质在这种无硅烷的裂解中保持完整,所以b消除机理可能是有效的。当尝试在标准条件下还原4-叔丁基苯甲醚和4-甲基苯甲醚时,相应的酚类的收率是高的,可能是因为被取代的苯环由于空间原因的更具挑战性的甲硅烷基化(方案2)。
总体地,对于烷基c-o键切断的选择性与其中发生芳基c-o还原的ni和硼烷催化的c-o裂解反应中观察到的选择性形成对比。还值得注意的是,在这些条件下仅观察到痕量的萘环氢化产物,这与文献中报告的基于硅烷的离子氢化的结果形成对比。
有指导性的是,使甲氧基取代的二芳基醚8和11的裂解(方案2)与上文呈现的结果相比较。虽然芳基烷基醚对于还原烷基氧键比还原芳基氧键表现出强的偏好,但是方案2中的两种甲氧基基质都表明区域选择性的逆转,几乎专门地提供芳基氧键破裂产物。虽然不意图受该理论的正确性的束缚,但是这种效果可以归因于在经历破裂的c-o键邻位的给体氧原子的存在。支持这种推断是主要地产生10的二苯并呋喃衍生物8的还原开环的高选择性。同样地,在木质素模型11的裂解中观察到苯酚和苯甲醚的优选形成,其中对于酚类12和13具有相似的选择性。人们可以推测,这种效果可以通过在正被破坏的c-o键的亲电子活化期间的正电荷积聚的氧原子共振稳定化来合理地说明。为了测试这种假设,使化合物3经历该反应条件并且分离开环的酚类5和6以及脱甲硅烷基化的产物1和2(方案2,插图c)。在共振稳定化不存在时,裂解的选择性被逆转为有利于异构体5。还值得注意的是,如1和2的形成表明的,甲硅烷基化反应因此在典型的反应条件下是可逆的。在已例证具挑战性的4-o-5木质素模型8和11的可能性之后,这种方法用含有六个car-o键的低聚醚14来测试(方案2,插图d)。值得注意地,在165℃下在均三甲苯中实现了14的定量转化并且得到了苯酚、苯、间苯二酚和其他未确定的产物,每芳基氧键具有仅0.5当量的硅烷。
在方案2中,化合物1至7指的是实施例6.5中描述的对应的化合物。
实施例6.7:芳基烷基醚和硫酯在环境温度或接近环境温度下的甲硅烷基化
实施例6.7.1:三乙基(2-甲氧基苯基)硅烷
反应根据一般程序通过在65℃下将苯甲醚(54mg,0.5mmol,1当量)、kot-bu(11mg,0.1mmol,0.2当量)和et3sih(239微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续65小时来进行。在水溶液后处理之后,粗制反应混合物通过使用己烷(等度的(isochratic))的硅胶色谱法来纯化以获得59mg(54%)作为无色的油的标题化合物。1hnmr(500mhz,thf-d8)δ7.40–7.17(m,2h),7.01–6.81(m,2h),3.77(s,3h),1.02–0.85(m,9h),0.87–0.74(m,6h)。13cnmr(126mhz,thf-d8)δ164.58,135.52,130.42,123.92,120.08,109.23,54.09,6.93,3.22。
反应根据一般程序通过在85℃下将kot-bu(11.2mg,0.1mmol,20mol%)、苯甲醚(54.0mg,0.5mmol,1当量)、和et3sih(243μl,1.5mmol,3当量)而没有任何添加的溶剂加热持续72h来进行。邻:(间+对)>20:1。期望的产物17a的gc收率是65%。在真空(60毫托,23℃)下对起始材料和挥发物的蒸发之后,分析纯产物(47.7mg,43%收率)作为无色油来获得。注意:化合物17a是易挥发的,并且可以在真空下被除去。rf=0.3(在己烷中10%et2o)。1hnmr(500mhz,cdcl3)δ7.41–7.30(m,2h),6.97(m,1h),6.87–6.81(m,1h),3.80(s,3h),1.05–0.90(m,9h),0.91–0.77(m,6h)。
实施例6.7.2:三乙基(3-甲氧基萘-2-基)硅烷
反应根据一般程序通过在65℃下将2-甲氧基萘(79mg,0.5mmol,1当量)、kot-bu(19.6mg,0.18mmol,0.35当量)和et3sih(319微升,2.0mmol,4当量)在1ml的四氢呋喃中加热持续48小时来进行。在水溶液后处理之后,粗制反应混合物通过用己烷(等度的)洗脱的硅胶色谱法来纯化以获得79mg(58%)作为无色的油的标题化合物。1hnmr(500mhz,thf-d8)δ7.84(s,1h),7.78–7.73(d,1h),7.73–7.68(d,1h),7.38(ddd,j=8.2,6.8,1.3hz,1h),7.27(ddd,j=8.1,6.8,1.2hz,1h),7.15(s,1h),3.90(s,3h),1.01–0.90(m,9h),0.68–0.53(m,6h)。13cnmr(126mhz,thf-d8)δ163.03,137.88,136.83,130.10,128.58,128.09,127.29,127.21,124.03,104.57,55.25,8.02,7.48。
hrms:[c17h24osi]计算为272.1608,测量为272.1596。2-甲氧基萘和其反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
令人关注地,以1-甲氧基萘起始的反应不产生甲硅烷基化产物:
反应根据一般程序在65℃下将1-甲氧基萘(79mg,0.5mmol,1当量)、kot-bu(11.2mg,0.1mmol,0.1当量)和et3sih(240微升,1.5mmol,3当量)在1ml的四氢呋喃中加热65小时来进行。反应用二乙醚(1ml)来稀释,用水(0.5ml)猝灭并且有机相通过gc-ms、gc-fid和1hnmr分析来分析。通过gc-ms和gc-fid(十三烷标准物)的分析展现了芳基c-o裂解产物萘和烷基c-o键裂解产物萘酚分别以百分之13和百分之8的收率的形成,显著地完全排除了任何甲硅烷基化物质。
实施例6.7.3二苯基醚的甲硅烷基化
反应根据一般程序通过在65℃下将苯基醚(85mg,0.5mmol,1当量)、kot-bu(11mg,0.10mmol,0.2当量)和et2sih2(194微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续65小时来进行。在水溶液后处理之后,粗制反应混合物通过使用己烷:三乙胺的80:2混合物的硅胶色谱法来纯化以获得68mg(20%)作为无色的油状固体的标题化合物。1hnmr(500mhz,thf-d8)δ7.64–7.57(m,2h),7.55(dd,j=7.3,1.8hz,1h),7.41(ddd,j=8.3,7.2,1.8hz,1h),7.15(dd,j=8.3,1.0hz,1h),7.14–7.09(m,2h),4.34(si-h)(p-like,j=1.2hz,1h),1.06–0.95(m,12h),0.92–0.82(m,8h)。13cnmr(126mhz,thf-d8)δ166.04,161.43,139.74,137.00,135.55,135.05,132.12,130.19,128.79,123.56,123.37,118.41,9.06,7.93,6.70,4.83。hrms:[c20h27osi2]计算为339.1601,测量为339.1607。
反应混合物的第二级分产生34mg(39%)的环化衍生物。1hnmr(500mhz,thf-d8)δ7.57–7.50(m,2h),7.40(ddd,j=8.3,7.2,1.8hz,2h),7.15(dd,j=8.6,0.7hz,2h),7.11(td,j=7.2,1.0hz,2h),0.99–0.95(m,4h),0.92–0.86(m,6h)。13cnmr(126mhz,thf-d8)δ161.54,134.96,132.07,123.41,118.80,117.39,7.95,6.72。hrms:[c16h19osi]计算为255.1205,测量为255.1206。这些反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
获得含有以低收率(约7%)的产物的第三级分,其光谱特性表现为与上文所示的单甲硅烷基化产物的结构一致。
在第二组实验中,当二苯醚(oxydibenzene)被用作溶剂时,反应更清洁地产生单甲硅烷基化的衍生物,三乙基(2-苯氧基苯基)硅烷17b:
反应根据一般程序通过在85℃下将kot-bu(11.2mg,0.1mmol,20mol%)、二苯醚(85.0mg,0.5mmol)、和et3sih(240μl,1.5mmol,3当量)在没有溶剂下加热持续120h来进行。期望的产物17b(84.5mg,55%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.4(100%己烷);1hnmr(500mhz,cdcl3)δ7.52–7.46(m,1h),7.38–7.25(m,3h),7.10(t,j=7.4hz,2h),7.00(d,j=7.9hz,2h),6.81(d,j=8.1hz,1h),0.97(t,j=7.9hz,9h),0.85(q,j=7.9hz,6h)。
实施例6.7.4:1,4-二甲氧基苯的甲硅烷基化:
反应根据一般程序通过在65℃下将kot-bu(11.2mg,0.1mmol,20mol%)、1,4-二甲氧基苯(69.1mg,0.5mmol)、和et3sih(240μl,1.5mmol,3当量)在0.5ml的thf中加热持续72h来进行。期望的产物17c(53.1mg,42%收率)和双甲硅烷基化的副产物si-17c(16.1mg,8%收率)在通过硅胶快速色谱法(100%己烷)纯化之后获得。
(2,5-二甲氧基苯基)三乙基硅烷17c:无色油,rf=0.5(100%己烷);1hnmr(500mhz,cdcl3)δ6.93(d,j=3.1hz,1h),6.85(dd,j=8.8,3.1hz,1h),6.76(d,j=8.8hz,1h),3.80(s,3h),3.74(s,3h),0.99–0.91(m,9h),0.85–0.74(m,6h);13cnmr(126mhz,cdcl3)δ158.8,153.3,126.7,122.2,122.3,114.1,55.7,55.5,7.6,3.7;ir(纯的膜,nacl)2952,2873,1580,1478,1463,1398,1272,1220,1177,1050,1026,872,800,769,732cm–1;hrms(ei+)对于c14h24o2si[m+·]计算为:252.1546,实测为252.1540。
(2,5-二甲氧基-1,4-亚苯基)二(三乙基硅烷)si-17c:白色固体,rf=0.8(100%己烷);1hnmr(500mhz,cdcl3)δ6.81(s,2h),3.75(s,6h),0.95(td,j=7.9,0.9hz,9h),0.85–0.77(m,6h);13cnmr(126mhz,cdcl3)δ158.5,127.1,116.9,55.6,7.7,3.8;ir(纯的膜,nacl)2948,2870,1459,1418,1345,1262,1203,1107,1045,999,868,727,700cm–1;hrms(ei+)对于c20h38si2o2[m+·]计算为:366.2410,实测为366.2415。
三乙基(2-甲氧基-5-甲基苯基)硅烷20:反应根据一般程序通过在85℃下将kot-bu(11.2mg,0.1mmol,20mol%)、1-甲氧基-4-甲基苯19(61.0mg,0.5mmol)、和et3sih(240μl,1.5mmol,3当量)加热持续120h来进行。期望的产物20(38.5mg,32%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.4(100%己烷);1hnmr(500mhz,cdcl3)δ7.17–7.08(m,2h),6.74(dt,j=8.7,1.3hz,1h),3.76(s,3h),2.30(s,3h),0.97–0.92(m,9h),0.85–0.79(m,6h);13cnmr(126mhz,cdcl3)δ162.7,136.7,130.9,129.2,125.0,109.5,55.2,20.8,7.8,3.7;ir(纯的膜,nacl)2951,2873,1595,1480,1464,1385,1238,1175,1147,1081,1034,1004,876,806,708cm–1;hrms(ei+)对于c14h24osi[m+·]计算为:236.1596,实测为236.1598。
实施例6.7.5:三乙基((苯基硫代)甲基)硅烷
反应根据一般程序通过在65℃下将苯甲硫醚(62mg,0.5mmol,1当量)、kot-bu(11mg,0.1mmol,0.2当量)和et3sih(239微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续65小时来进行。在水溶液后处理之后,粗制反应混合物通过使用己烷(等度的)的硅胶色谱法来纯化以获得81mg(68%)作为无色的油的标题化合物。1hnmr(500mhz,thf-d8)δ7.31–7.26(m,2h),7.25–7.19(m,2h),7.11–7.01(m,1h),1.03(t,j=7.9hz,9h),0.78–0.60(m,6h)。13cnmr(126mhz,thf-d8)δ140.73,128.31,125.69,124.19,13.01,6.62,3.06。hrms:[c13h21ssi]计算为237.1140,测量为237.1133。苯硫基甲烷及其反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
实施例6.8:用c-n和c-s杂芳基化合物在升高的温度下的实验
实验还用c-n和c-s杂芳基化合物来进行。在包含c-n键的化合物的情况下,反应性表现为相似于对于c-o键所看到的反应性,并且合理地预期的是,用于后者的宽范围方法将导致前者中的相似反应性的结果:
在包含c-s化合物的化合物的情况下,该方法似乎通常导致分子的完全脱硫,至少在这些实验的攻击性条件下,这反映这些类型的基质的较高的反应性(仅与实施例6.9.34至实施例6.9.38比较)。反应性的这种差异可以反映c-o键、c-n键和c-s键之间的键能的差异(比较苯酚(111)、苯胺(104)和苯硫酚(85,全部以kcal/mol计)中的c-x键离解能)。特别受关注的是均匀受阻的二苯并噻吩类在相对温和的条件下的脱硫。在这些转化中没有检测到单一的c-s产物:
(产物未确定,除了通过cc-ms没有观察到ar-s;通过nmr没有观察到rs-h)
实施例6.9:用杂芳基化合物在环境温度或接近环境温度下的实验
在环境温度或接近环境温度(65℃或低于65℃)下进行一系列实验以测试更具反应性的杂芳基化合物中的几种的区域选择性。下文示出了测试条件和结果。所有反应的收率通过分离(硅胶色谱法,或球管至球管的蒸馏(bul-to-bulbdistillation))或通过使用定量用的内标的gc-fid或nmr分析。注意,c-3甲硅烷基化杂芳烃在某些情况下被发现倾向于在硅胶上原脱甲硅烷基化(protodesilylation)。在这些情况下,使用球管至球管的蒸馏或可选择地用加入洗脱液中的约3%三乙胺的硅胶色谱法、或这两种方法的组合。产物如通过1h、13cnmr和异核单量子相干(hsqc)光谱或gc-ms、或二者的组合所指示的来确定,如果可能的话使用与可信样品的比较。
实施例6.9.1:1-甲基-2-(三乙基甲硅烷基)-1h-吲哚2a
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基吲哚1a(65.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)加热持续96h来进行。c2:c3>20:1。期望的产物2a(95.6mg,78%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中2%→3%ch2cl2)纯化之后作为无色油获得。rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.70(dt,j=7.9,1.1hz,1h),7.40(dq,j=8.3,1.0hz,1h),7.30(ddd,j=8.3,7.0,1.3hz,1h),7.16(ddd,j=7.9,6.9,1.0hz,1h),6.81(d,j=1.1hz,1h),3.90(s,3h),1.13–1.05(m,9h),1.03–0.95(m,6h);13cnmr(125mhz,cdcl3)δ140.4,138.3,128.7,122.0,120.7,119.1,113.1,109.1,33.1,7.7,4.2。ir(纯的膜,nacl)2953,2909,2874,1492,1464,1415,1372,1356,1299,1233,1166,1101,1069,1007,973,797cm–1;hrms(esi+)对于c15h24nsi[m+h]+计算为:246.1673,实测为246.1674。此反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
此材料还以如下规模被制备。将装配有搅拌棒并且用橡胶隔片塞好的500ml烘箱干燥的舒伦克瓶排空,并且用氩气重新填充一次。将kot-bu(18.8克,167.9mmols,20mol%)在工作台上称重,并且在强的氩气流下添加到该烧瓶。然后,将装载的烧瓶排空,并且用氩气重新填充。将先前被脱气的1-甲基吲哚(95%纯度,aksci,未蒸馏的、黄色的油;95.1ml,762.4mmol,1.0当量)和et3sih(182.6ml,1142mmol,1.5当量)通过注射器穿过隔片添加。然后,将混合物冷却到-78℃(干冰/丙酮),并且排空/用氩气回填持续三个循环。将冷却浴移除,并且允许烧瓶在氩气的正压下加温至室温。然后将烧瓶转移到设定在45℃下的加热套,并且搅拌持续72小时。将具有产生的深红紫色溶液的烧瓶从加热移出,并且允许冷却至室温,用无水et2o(50ml)稀释,并且过滤,以除去固体残余物。在溶剂在真空中被除去之后,添加搅拌棒,并且将透明的深琥珀色溶液在高真空(100毫托)下搅拌持续若干小时,以除去剩余的挥发物。然后,使混合物经历在真空下的蒸馏。期望的产物2a作为浅黄色油(141.88g,76%收率)被获得。
实施例6.9.2:1-甲基-3-(三乙基甲硅烷基)-1h-吲哚
反应根据一般程序通过在23℃下将n-甲基吲哚(66mg,0.5mmol,1当量)、kot-bu(56mg,0.5mmol,1当量)和et3sih(88微升,0.55mmol,1.1当量)在1ml的四氢呋喃中加热持续312小时来进行。在水溶液后处理之后,粗制反应混合物通过用95:5己烷:net3(等度的)洗脱的硅胶色谱法来纯化以获得作为无色的油的103mg(84%)的标题化合物。1hnmr(500mhz,thf-d8)δ7.63(dt,j=7.9,1.0hz,1h),7.32(dt,j=8.2,0.9hz,1h),7.15(s,1h),7.12(ddd,j=8.2,7.0,1.1hz,1h),7.01(ddd,j=8.0,7.0,1.1hz,1h),3.78(s,3h),1.06–0.95(m,9h),0.95–0.83(m,6h)。13cnmr(126mhz,thf-d8)δ138.63,135.94,133.37,121.44,120.88,118.79,108.96,104.39,31.61,7.04,4.11。此反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
实施例6.9.3:1-甲基-3-(三乙基甲硅烷基)-1h-吲哚2b
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苄基吲哚1b(103.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续40h来进行。c2:c3>20:1。期望的产物2b(132.2mg,82%收率)在通过硅胶快速色谱法(在己烷中10%ch2cl2)纯化之后作为无色油获得。rf=0.3(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δδ7.81–7.77(m,1h),7.38–7.29(m,3h),7.26–7.19(m,3h),7.02(ddd,j=6.9,2.2,1.0hz,2h),6.97(s,1h),5.59(s,2h),1.08–1.04(m,9h),0.94–0.89(m,6h);13cnmr(125mhz,cdcl3)δ140.2,138.5,138.3,129.1,128.7,127.3,125.9,122.3,120.7,119.5,114.1,110.2,50.2,7.5,4.0。ir(纯的膜,nacl)3060,3029,2954,2909,2875,1606,1495,1466,1452,1416,1377,1353,1333,1300,1238,1196,1164,1115,1096,1014,798,734cm–1;hrms(esi+)对于c21h28nsi[m+h]+计算为:322.1986,实测为322.1985。
反应根据一般程序通过在23℃下将1-苄基吲哚(62mg,0.5mmol,1当量)、kot-bu(11mg,0.1mmol,0.2当量)和et3sih(239微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续48小时来进行。在水溶液后处理之后,粗制反应混合物通过使用己烷(等度的)的硅胶色谱法来纯化以获得50mg(31%)作为无色的油状固体的标题化合物。
1hnmr(500mhz,thf-d8)δ7.56(ddd,j=7.7,1.3,0.7hz,1h),7.25–7.07(m,4h),7.02(ddd,j=8.2,6.9,1.3hz,1h),6.98(ddd,j=7.9,6.9,1.1hz,1h),6.92–6.86(m,2h),6.80(d,j=0.9hz,1h),5.52(s,2h),1.06–0.88(m,9h),0.85–0.69(m,6h)。
实施例6.9.4:1-乙基-2-(三乙基甲硅烷基)-1h-吲哚2c:
反应根据一般程序通过在60℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-乙基吲哚1c(72.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)加热持续84h来进行。c2:c3>20:1。期望的产物2c(92.4mg,71%收率)在通过硅胶快速色谱法(在己烷中5%ch2cl2)纯化之后作为无色油获得。rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.67(dt,j=7.9,0.9hz,1h),7.40(dt,j=8.2,0.9hz,1h),7.25(ddd,j=8.2,7.0,1.2hz,1h),7.13(ddd,j=7.9,7.0,1.0hz,1h),6.75(d,j=1.0hz,1h),4.31(q,j=7.2hz,2h),1.46(t,j=7.2hz,3h),1.08–1.04(m,9h),0.99–0.92(m,6h);13cnmr(125mhz,cdcl3)δ139.0,137.4,129.1,121.7,120.7,119.0,113.0,109.4,41.5,15.5,7.5,4.0。ir(纯的膜,nacl)2953,2909,2874,1491,1466,1416,1378,1347,1335,1299,1218,1165,1090,1069,1012,956,900,820,787,773,750,733cm–1;hrms(mm:esi-apci+)对于c16h26nsi[m+h]+计算为:260.1829,实测为260.1829。
实施例6.9.5:1-苯基-2-(三乙基甲硅烷基)-1h-吲哚2d:
反应根据一般程序通过在60℃下将kot-bu(7.4mg,0.07mmol,20mol%)、n-苯基吲哚1d(63.2mg,0.33mmol,1当量)和et3sih(160μl,1.0mmol,3当量)加热持续84h来进行。c2:c3>20:1。期望的产物2d(45.6mg,45%收率)在通过硅胶快速色谱法(在己烷中3%ch2cl2)纯化之后作为白色固体获得。rf=0.5(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.74–7.67(m,1h),7.58–7.47(m,3h),7.44–7.36(m,2h),7.21–7.12(m,2h),7.12–7.05(m,1h),6.93(d,j=0.9hz,1h),0.92(t,j=7.9hz,9h),0.68–0.55(m,6h);13cnmr(125mhz,cdcl3)δ141.6,140.8,139.1,129.2,128.8,128.7,128.3,122.4,120.5,119.8,114.9,110.5,7.5,4.0。ir(纯的膜,nacl)3058,2952,2909,2873,1597,1498,1465,1428,1362,1297,1237,1214,1122,1071,1012,976,922,820,793,736cm–1;hrms(mm:esi-apci+)对于c20h26nsi[m+h]+计算为:308.1829,实测为308.1824。
实施例6.9.6:1-(甲氧基甲基)-2-(三乙基甲硅烷基)-1h-吲哚2e:
反应根据一般程序通过在60℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲氧基甲基吲哚1e(80.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)加热持续84h来进行。c2:c3>10:1。期望的产物2e(75.1mg,55%收率)在通过硅胶快速色谱法(在己烷中3%etoac)纯化之后作为无色油获得。rf=0.3(在己烷中5%etoac);1hnmr(500mhz,cdcl3)δ7.67(dt,j=7.8,1.0hz,1h),7.53(dq,j=8.3,0.9hz,1h),7.28(ddd,j=8.3,7.0,1.2hz,1h),7.17(ddd,j=7.9,7.0,1.0hz,1h),6.86(d,j=0.9hz,1h),5.55(s,2h),3.30(s,3h),1.10–1.01(m,9h),1.01–0.92(m,6h);13cnmr(125mhz,cdcl3)δ140.7,138.3,129.2,122.6,120.8,120.0,115.6,109.8,76.8,55.6,7.5,4.1。ir(纯的膜,nacl)2952,2908,2874,1495,1466,1416,1393,1344,1311,1299,1224,1166,1126,1104,1091,1045,1004,961,913,797,762,735cm–1;hrms(mm:esi-apci+)对于c16h26nosi[m+h]+计算为:276.1778,实测为276.1769。
实施例6.9.7:2-(三乙基甲硅烷基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1h-吲哚2f:
反应根据一般程序通过在60℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-(2-三甲基甲硅烷基-乙氧基甲基)-1h-吲哚1f(123.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)加热持续84h来进行。c2:c3>20:1。期望的产物2f(121.4mg,67%收率)在通过硅胶快速色谱法(在己烷中15%ch2cl2)纯化之后作为无色油获得。rf=0.2(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.62(dt,j=7.8,1.0hz,1h),7.50(dq,j=8.3,0.9hz,1h),7.24(ddd,j=8.3,7.0,1.2hz,1h),7.12(ddd,j=7.9,7.0,0.9hz,1h),6.80(d,j=0.9hz,1h),5.54(s,2h),3.54–3.48(m,2h),1.04–0.98(m,9h),0.96–0.90(m,8h),-0.02(s,9h);13cnmr(125mhz,cdcl3)δ140.5,138.1,129.1,122.4,120.7,119.9,115.3,109.8,75.2,65.6,18.1,7.6,4.0,-1.3。ir(纯的膜,nacl)2952,2875,1495,1466,1443,1417,1378,1343,1312,1299,1249,1167,1081,1003,972,939,894,859,836,796,760,749,734cm–1;hrms(mm:esi-apci+)对于c20h36nosi2[m+h]+计算为:362.2330,实测为362.2340。
实施例6.9.8:4-甲基-n-甲基吲哚与et3sih的反应:
反应根据一般程序进行。对于条件a:反应用kot-bu(11.2mg,0.1mmol,20mol%)、4-甲基-n-甲基吲哚1g(72.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf在25℃下进行持续120h。c2:c3>20:1。期望的单甲硅烷基化产物2g(61.8mg,48%收率)和双甲硅烷基化产物16(9.7mg,5%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中2%→3%ch2cl2)纯化之后获得。对于条件b:反应用kot-bu(11.2mg,0.1mmol,20mol%)、4-甲基-n-甲基吲哚1g(72.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)在45℃下进行持续84h。c2:c3>20:1。仅形成单甲硅烷基化产物2g(89.7mg,69%收率),并且在通过硅胶快速色谱法(在己烷中3%ch2cl2)纯化之后获得。
1,4-二甲基-2-(三乙基甲硅烷基)-1h-吲哚2g:无色油;rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.23–7.14(m,2h),6.91(dt,j=6.7,1.0hz,1h),6.75(d,j=0.9hz,1h),3.85(s,3h),2.60(s,3h),1.07–1.00(m,9h),0.98–0.92(m,6h);13cnmr(125mhz,cdcl3)δ140.2,137.6,130.2,128.6,122.2,119.4,111.5,106.8,33.2,18.8,7.7,4.3。ir(纯的膜,nacl)2953,2910,2874,1586,1502,1454,1415,1366,1323,1280,1238,1160,1140,1077,1004,953,765,752,735cm–1;hrms(mm:esi-apci+)对于c16h26nsi[m+h]+计算为:260.1829,实测为260.1823。
1-甲基-2-(三乙基甲硅烷基)-4-((三乙基甲硅烷基)甲基)-1h-吲哚16:无色油;rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.28(dd,j=8.2,7.1hz,1h),6.98(d,j=8.3hz,1h),6.97–6.94(m,2h),3.31(s,3h),2.50(s,2h),1.01(t,j=7.8hz,9h),0.95(t,j=7.9hz,9h),0.83(q,j=7.8hz,6h),0.58(q,j=7.9hz,6h);13cnmr(125mhz,c6d6)δ141.1,136.0,133.3,122.8,118.9,113.0,105.8,32.9,19.2,7.7,4.5,4.1。ir(纯的膜,nacl)2952,2909,2874,1579,1498,1454,1443,1414,1359,1322,1285,1237,1151,1070,1008,980,774,734cm–1;hrms(ei+)对于c22h39nsi2[m·+]计算为:373.2621,实测为373.2624。
实施例6.9.9:1,5-二甲基-2-(三乙基甲硅烷基)-1h-吲哚2h:
反应根据一般程序通过在25℃下将kot-bu(11.2mg,0.1mmol,20mol%)、5-甲基-n-甲基吲哚1h(72.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续96h来进行。c2:c3>20:1。期望的产物2h(88.7mg,68%收率)在通过硅胶快速色谱法(在己烷中10%ch2cl2)纯化之后作为无色油获得。rf=0.3(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.39(s,1h),7.25–7.19(m,1h),7.05(dd,j=8.4,1.6hz,1h),6.63(d,j=0.8hz,1h),3.81(s,3h),2.45(s,3h),1.03–0.97(m,9h),0.93–0.86(m,6h)。13cnmr(125mhz,cdcl3)δ138.8,138.3,128.9,128.3,123.6,120.2,112.4,108.8,33.1,21.5,7.7,4.1。ir(纯的膜,nacl)2952,2909,2873,1505,1456,1358,1321,1236,1181,1104,1069,1003,833,788,736cm–1;hrms(esi+)对于c16h26nsi[m+h]+计算为:260.1826,实测为260.1827。
实施例6.9.10:5-甲基-n-甲基吲哚与et3sih的反应:
反应根据一般程序进行。对于条件a:反应用kot-bu(11.2mg,0.1mmol,20mol%)、6-甲基-n-甲基吲哚1i(72.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf在25℃下进行持续120h。c2:c3>20:1。期望的单甲硅烷基化产物2i(69.5mg,54%收率)和双甲硅烷基化产物si-2i(5.2mg,3%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中2%→3%ch2cl2)纯化之后获得。对于条件b:反应用kot-bu(11.2mg,0.1mmol,20mol%)、6-甲基-n-甲基吲哚1i(72.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)在45℃下进行持续84h。c2:c3>20:1。仅形成单甲硅烷基化产物2i(108.1mg,83%收率),并且在通过硅胶快速色谱法(在己烷中3%ch2cl2)纯化之后获得。
1,6-二甲基-2-(三乙基甲硅烷基)-1h-吲哚2i:无色油;rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.55(d,j=8.0hz,1h),7.18(s,1h),6.98(ddd,j=8.0,1.4,0.7hz,1h),6.73(d,j=0.9hz,1h),3.85(s,3h),2.57(s,3h),1.08–1.03(m,9h),0.98–0.92(m,6h);13cnmr(125mhz,cdcl3)δ140.9,137.6,131.8,126.7,121.0,120.3,113.0,109.1,33.0,22.0,7.6,4.2。ir(纯的膜,nacl)2953,2910,2874,1617,1480,1451,1413,1376,1360,1333,1296,1233,1065,1003,941,808,781,736cm–1;hrms(esi+)对于c16h26nsi[m+h]+计算为:260.1826,实测为260.1823。
1-甲基-2-(三乙基甲硅烷基)-6-((三乙基甲硅烷基)甲基)-1h-吲哚si-2i:无色油;rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.64(dd,j=7.9,0.8hz,1h),6.99–6.93(m,2h),6.81(d,j=0.9hz,1h),3.41(s,3h),2.31(s,2h),1.02–0.93(m,18h),0.79(q,j=7.7hz,6h),0.58(q,j=7.9hz,6h);13cnmr(125mhz,c6d6)δ141.9,136.3,134.6,126.7,121.2,120.9,114.0,108.3,32.7,22.4,7.8,7.7,4.5,3.7。ir(纯的膜,nacl)2952,2909,2874,1615,1568,1479,1463,1414,1361,1336,1319,1299,1234,1195,1157,1090,1065,1009,948,842,817,787,771,736cm–1;hrms(ei+)对于c22h39nsi2[m·+]计算为:373.2621,实测为373.2609。
实施例6.9.11:1,7-二甲基-2-(三乙基甲硅烷基)-1h-吲哚2j:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、7-甲基-n-甲基吲哚1j(72.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)加热持续84h来进行。c2:c3>20:1。期望的产物2j(78.9mg,61%收率)在通过硅胶快速色谱法(在己烷中3%ch2cl2)纯化之后作为无色油获得。rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.43(d,j=7.7hz,1h),6.94–6.87(m,2h),6.66(s,1h),4.11(s,3h),2.80(s,3h),1.03–0.97(m,9h),0.92–0.85(m,6h);13cnmr(125mhz,cdcl3)δ139.2,139.1,129.7,125.0,121.0,119.4,119.0,113.6,36.8,20.6,7.7,4.2。ir(纯的膜,nacl)2953,2909,2873,1503,1459,1415,1396,1377,1358,1340,1315,1304,1238,1156,1113,1086,1063,1004,861,798,742cm–1;hrms(esi+)对于c16h26nsi[m+h]+计算为:260.1826,实测为260.1828。
实施例6.9.12:n-甲基-5-甲氧基吲哚1k与et3sih的反应:
反应根据一般程序进行。对于条件a:反应用kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基-5-甲氧基吲哚1k(80.7mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf在25℃下进行持续120h。c2:c3>20:1。c2-甲硅烷基化产物2k(58.7mg,43%收率)、c6-甲硅烷基化产物15(12.5mg,9%收率)和双甲硅烷基化产物si-2k(42.9mg,22%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中5%→10%→25%ch2cl2)纯化之后获得。对于条件b:反应用kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基-5-甲氧基吲哚1k(80.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf在25℃下进行持续72h。c2:c3>20:1。期望的产物2k(87.6mg,64%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中5%→10%→25%ch2cl2)纯化之后获得,并且观察到少量(<5%)的副产物。
5-甲氧基-1-甲基-2-(三乙基甲硅烷基)-1h-吲哚2k:白色固体;rf=0.2(在己烷中33%ch2cl2);1hnmr(500mhz,cdcl3)δ7.21(s,1h),7.07(d,j=2.4hz,1h),6.89(dd,j=8.8,2.5hz,1h),6.63(d,j=0.8hz,1h),3.85(s,3h),3.81(s,3h),1.03–0.96(m,9h),0.93–0.86(m,6h);13cnmr(125mhz,cdcl3)δ154.0,139.0,135.9,128.8,112.6,112.3,109.8,102.0,56.1,33.2,7.7,4.1。ir(纯的膜,nacl)2950,2909,2872,1503,1450,1413,1334,1237,1208,1173,1147,1102,1072,1027,997,843,801,735,716cm–1;hrms(esi+)对于c16h26nosi[m+h]+计算为:276.1778,实测为276.1776。
5-甲氧基-1-甲基-2,6-二(三乙基甲硅烷基)-1h-吲哚si-2k:白色固体,rf=0.6(在己烷中33%ch2cl2);1hnmr(500mhz,cdcl3)δ7.30(s,1h),7.01(s,1h),6.64(d,j=0.8hz,1h),3.85(s,3h),3.83(s,3h),1.06–0.97(m,18h),0.95–0.86(m,12h);13cnmr(125mhz,cdcl3)δ159.1,138.9,136.1,130.1,120.8,116.3,112.2,99.7,55.5,33.2,7.9,7.7,4.3,4.1。ir(纯的膜,nacl)2952,2874,2908,1608,1556,1475,1454,1407,1363,1337,1236,1205,1172,1144,1123,1072,1004,971,837cm–1;hrms(esi+)对于c22h40nosi2[m+h]+计算为:390.2643,实测为390.2632。
5-甲氧基-1-甲基-6-(三乙基甲硅烷基)-1h-吲哚15:无色油;rf=0.4(在己烷中33%ch2cl2);1hnmr(500mhz,cdcl3)δ7.27(s,1h),7.01(s,1h),7.00(d,j=3.0hz,1h),6.38(dd,j=3.0,0.8hz,1h),3.82(s,3h),3.78(s,3h),1.00–0.94(m,9h),0.91–0.83(m,6h);13cnmr(125mhz,cdcl3)δ159.2,132.5,130.1,129.3,120.2,116.5,100.4,100.3,55.5,33.0,7.9,4.1。ir(纯的膜,nacl)2950,2908,2873,1612,1554,1505,1471,1414,1310,1268,1231,1190,1148,1123,1059,1017,984,831cm–1;hrms(esi+)对于c16h26nosi[m+h]+计算为:276.1778,实测为276.1765。
实施例6.9.13:5-(苄氧基)-1-甲基-2-(三乙基甲硅烷基)-1h-吲哚2l:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基-5-苄氧基吲哚1l(118.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续64h来进行。c2:c3>20:1。期望的产物2l(119.4mg,68%收率)在通过硅胶快速色谱法(在己烷中25%ch2cl2)纯化之后作为黄色固体获得。rf=0.4(在己烷中5%etoac)。1hnmr(500mhz,cdcl3)δ7.48(d,j=7.0hz,2h),7.41–7.36(m,2h),7.35–7.29(m,1h),7.22(d,j=8.9hz,1h),7.14(d,j=2.4hz,1h),6.97(dd,j=8.8,2.4hz,1h),6.62(d,j=0.8hz,1h),5.11(s,2h),3.81(s,3h),1.04–0.96(m,9h),0.96–0.84(m,6h);13cnmr(125mhz,cdcl3)δ153.3,139.1,138.1,136.2,129.0,128.6,127.8,127.6,113.4,112.5,109.8,104.0,71.3,33.2,7.6,4.2。ir(纯的膜,nacl)2951,2908,2872,1492,1452,1422,1336,1288,1237,1192,1150,1102,1075,1018,840,812,751,735cm–1;hrms(mm:esi-apci+)对于c22h30nosi[m+h]+计算为:352.2091,实测为352.2093。
实施例6.9.14:5-(甲氧基甲基)-n-甲基吲哚1m与et3sih的反应:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、5-(甲氧基甲基)-n-甲基吲哚1m(87.5mg,0.5mmol,1当量)和et3sih(243μl,1.5mmol,3当量)加热持续84h来进行。c2:c3>20:1。期望的产物2m(69.3mg,48%收率)、副产物1h(2.5mg,2%收率)和2h(11.3mg,9%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中25%→50%ch2cl2)纯化之后获得。
5-(甲氧基甲基)-1-甲基-2-(三乙基甲硅烷基)-1h-吲哚2m:无色油,rf=0.4(在己烷中50%ch2cl2);1hnmr(500mhz,cdcl3)δ7.59(d,j=0.8hz,1h),7.33(d,j=8.4hz,1h),7.25(d,j=8.4hz,1h),6.73(d,j=0.8hz,1h),4.59(s,2h),3.85(s,3h),3.38(s,3h),1.06–0.99(m,9h),0.96–0.90(m,6h);13cnmr(125mhz,cdcl3)δ140.0,138.9,128.8,128.5,122.6,120.5,113.0,109.1,75.6,57.6,33.2,7.6,4.1。ir(纯的膜,nacl)2952,2873,2817,1504,1455,1415,1357,1324,1297,1236,1188,1153,1137,1094,1069,1004,971,878,840,798,783,726cm–1;hrms(esi+)对于c17h28nosi[m+h]+计算为:290.1935,实测为290.1948。
实施例6.9.15:1-甲基-5-苯基-2-(三乙基甲硅烷基)-1h-吲哚2n:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、5-苯基-n-甲基吲哚1n(103.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续108h来进行。c2:c3>20:1。期望的产物2n(77.8mg,48%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中5%→10%ch2cl2)纯化之后作为白色固体获得。rf=0.3(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.90(s,1h),7.72(d,j=7.6hz,2h),7.55(d,j=8.5hz,1h),7.53–7.47(m,2h),7.44(d,j=8.5hz,1h),7.37(t,j=7.4hz,1h),6.85(s,1h),3.91(s,3h),1.09(t,j=7.8hz,9h),1.03–0.95(m,6h);13cnmr(125mhz,cdcl3)δ142.9,140.0,139.3,132.8,129.2,128.7,127.5,126.3,122.0,119.2,113.5,109.4,33.2,7.6,4.2。ir(纯的膜,nacl)2950,2908,2873,1600,1485,1455,1361,1325,1301,1214,1162,1074,1004,1086,887,820,807,787,759,733cm–1;hrms(mm:esi-apci+)对于c21h28nsi[m+h]+计算为:322.1986,实测为322.1984。
实施例6.9.16:n-甲基吲哚1a与et2sih2的反应:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基吲哚1a(65.5mg,0.5mmol,1当量)、et2sih2(194μl,1.5mmol,3当量)和0.5ml的thf加热持续72h来进行。c2:c3>20:1。甲硅烷基化产物2o(73.4mg,68%收率)和少量的双吲哚基硅烷si-2o在通过硅胶快速色谱法(梯度洗脱,在己烷中1%→2%→5%ch2cl2)纯化之后获得。
2-(二乙基甲硅烷基)-1-甲基-1h-吲哚2o:无色油;rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.66(dt,j=7.9,1.0hz,1h),7.37(dt,j=8.3,1.1hz,1h),7.28–7.25(m,1h),7.16–7.09(m,1h),6.79(d,j=0.9hz,1h),4.50–4.43(m,1h),3.88(s,3h),1.14–1.06(m,6h),1.00–0.93(m,4h);13cnmr(125mhz,cdcl3)δ140.2,136.6,128.6,122.2,120.8,119.3,112.8,109.3,32.8,8.4,3.7。ir(纯的膜,nacl)2954,2908,2872,2110,1492,1464,1412,1371,1357,1327,1301,1233,1166,1101,1071,1009,974,987,815,785cm–1;hrms(mm:esi-apci+)对于c13h20nsi[m+h]+计算为:218.1360,实测为218.1354。
二乙基双(1-甲基-1h-吲哚-2-基)硅烷si-2o:无色油;rf=0.2(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.68(dt,j=7.9,1.0hz,2h),7.31(dt,j=8.3,1.0hz,2h),7.25(ddd,j=8.2,6.9,1.2hz,2h),7.13(ddd,j=7.9,6.9,1.1hz,2h),6.92(d,j=0.9hz,2h),3.57(s,6h),1.31(q,j=8.4hz,4h),1.07(t,j=7.9hz,6h);13cnmr(125mhz,cdcl3)δ140.7,136.5,128.7,122.5,120.9,119.4,113.8,109.4,32.7,7.5,4.5。ir(纯的膜,nacl)2955,2874,1492,1463,1414,1355,1327,1299,1233,1166,1101,1072,1008,799,751cm–1;hrms(mm:esi-apci+)对于c22h27n2si[m+h]+计算为:347.1938,实测为347.1934。
实施例6.9.17:1-苄基-2-(二乙基甲硅烷基)-1h-吲哚2p:
反应根据一般程序通过在60℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苄基吲哚1b(103.5mg,0.5mmol,1当量)和et2sih2(194μl,1.5mmol,3当量)加热持续72h来进行。c2:c3>20:1。期望的产物2p(114.1mg,78%收率)在通过硅胶快速色谱法(在己烷中5%ch2cl2)纯化之后作为无色油获得。rf=0.5(在己烷中25%ch2cl2);1hnmr(500mhz,cdcl3)δ7.75(dt,j=7.7,1.0hz,1h),7.36–7.26(m,4h),7.26–7.15(m,2h),7.07–7.01(m,2h),6.94(d,j=0.9hz,1h),5.56(s,2h),4.44(p,j=3.3hz,1h),1.12–1.03(m,6h),0.94–0.79(m,4h)。13cnmr(125mhz,cdcl3)δ140.1,138.5,136.7,129.0,128.7,127.4,126.1,122.5,120.8,119.6,113.7,110.1,49.8,8.3,3.6。ir(纯的膜,nacl)2954,2873,2114,1605,1494,1466,1450,1413,1353,1334,1301,1233,1198,1164,1116,1095,972,815cm–1;hrms(mm:esi-apci+)对于c19h24nsi[m+h]+计算为:294.1673,实测为294.1668。
实施例6.9.18:2-(二乙基甲硅烷基)-1-苯基-1h-吲哚2q:
反应根据一般程序通过在55℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苯基吲哚1d(96.5mg,0.5mmol,1当量)、et2sih2(194μl,1.5mmol,3当量)和0.5ml的meot-bu加热持续96h来进行。c2:c3>20:1。期望的产物2q(76.9mg,55%收率)在通过硅胶快速色谱法(在己烷中10%ch2cl2)纯化之后作为黄色油获得。rf=0.6(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.76–7.74(m,1h),7.60–7.55(m,2h),7.53–7.47(m,3h),7.30–7.17(m,3h),7.03(d,j=0.9hz,1h),4.30(p,j=3.3hz,1h),1.02–0.98(m,6h),0.79–0.63(m,4h);13cnmr(125mhz,cdcl3)δ141.1,140.3,137.1,129.4,128.8,128.1,128.0,122.8,120.7,120.1,115.1,110.5,8.2,3.4。ir(纯的膜,nacl)3058,2953,2872,2117,1597,1498,1466,1433,1415,1363,1300,1215,1202,1146,1121,1072,1013,978,921,902,823,759,748,737cm–1;hrms(mm:esi-apci+)对于c18h22nsi[m+h]+计算为:280.1516,实测为280.1515。
实施例6.9.19:2-(二乙基甲硅烷基)-1-(甲氧基甲基)-1h-吲哚2r:
反应根据一般程序通过在60℃下加热kot-bu(11.2mg,0.1mmol,20mol%)、n-甲氧基甲基吲哚1e(80.5mg,0.5mmol,1当量)和et2sih2(193μl,1.5mmol,3当量)持续96h来进行。c2:c3>20:1。期望的产物2r(81.0mg,66%收率)在通过硅胶快速色谱法(在己烷中3%etoac)纯化之后作为无色油获得。rf=0.3(在己烷中5%etoac);1hnmr(500mhz,cdcl3)δ7.67(dt,j=7.9,1.0hz,1h),7.54(ddd,j=8.3,2.0,0.9hz,1h),7.29(ddd,j=8.3,7.0,1.2hz,1h),7.18(ddd,j=7.9,7.0,1.0hz,1h),6.88(d,j=0.9hz,1h),5.60(s,2h),4.49(p,j=3.3hz,1h),3.29(s,3h),1.14–1.08(m,6h),1.03–0.94(m,4h);13cnmr(125mhz,cdcl3)δ140.4,136.6,129.2,122.8,120.9,120.2,115.1,109.9,76.6,55.6,8.3,3.8。ir(纯的膜,nacl)2954,2874,2819,2115,1496,1467,1443,1413,1393,1360,1344,1314,1300,1282,1226,1190,1166,1127,1102,1091,1047,1009,974,914,896,818,749,736cm–1;hrms(mm:esi-apci+)对于c14h22nosi[m+h]+计算为:248.1465,实测为248.1459。
实施例6.9.20:2-(二乙基甲硅烷基)-1-((2-(三甲基甲硅烷基)乙氧基)甲基)-1h-吲哚2s:
反应根据一般程序通过在60℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-(2-三甲基甲硅烷基-乙氧基甲基)-1h-吲哚1f(123.5mg,0.5mmol,1当量)和et2sih2(194μl,1.5mmol,3当量)加热持续84h来进行。c2:c3>20:1。期望的产物2s(106.7mg,64%收率)在通过硅胶快速色谱法(在己烷中14%ch2cl2)纯化之后作为无色油获得。rf=0.2(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.65(dt,j=7.9,1.0hz,1h),7.53(dt,j=8.3,0.9hz,1h),7.27(ddd,j=8.3,7.0,1.2hz,1h),7.15(ddd,j=7.9,7.0,0.9hz,1h),6.84(d,j=0.8hz,1h),5.61(s,2h),4.48(p,j=3.3hz,1h),3.55–3.48(m,2h),1.14–1.04(m,6h),1.03–0.88(m,6h),-0.02(s,9h);13cnmr(125mhz,cdcl3)δ140.2,136.5,129.1,122.7,120.8,120.1,114.7,110.1,75.0,65.6,18.0,8.4,3.7,-1.3。ir(纯的膜,nacl)2953,2874,2116,1496,1466,1443,1413,1379,1343,1318,1300,1249,1219,1165,1081,1010,974,922,895,859,835,748,735cm–1;hrms(mm:esi-apci+)对于c18h32nosi2[m+h]+计算为:334.2017,实测为334.2028。
实施例6.9.21:2-(二乙基甲硅烷基)-1,3-二甲基-1h-吲哚2t:
反应根据一般程序通过在65℃下将kot-bu(11.2mg,0.1mmol,20mol%)、1,3-二甲基-1h-吲哚1t(72.6mg,0.5mmol,1当量)、et2sih2(193μl,1.5mmol,3当量)、以及0.5ml的thf加热持续120h来进行。期望的产物2t(84.2mg,65%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.6(100%己烷);1hnmr(500mhz,c6d6)δ7.67(d,j=7.9hz,1h),7.30(dd,j=8.3,6.9hz,1h),7.22(t,j=7.4hz,1h),7.10(d,j=8.2hz,1h),4.59(p,j=3.7hz,1h),3.31(s,3h),2.46(s,3h),0.98(t,j=7.8hz,6h),0.77(qd,j=7.9,3.9hz,4h);13cnmr(125mhz,c6d6)δ140.6,131.5,129.8,122.7,122.3,119.4,119.0,109.4,32.4,10.9,8.8,4.7。ir(纯的膜,nacl)2952,2871,2125,1509,1460,1351,1317,1237,1167,1138,1011,975,839,803,737cm–1;hrms(ei+)对于c14h21nsi[m·+]+计算为:231.1443,实测为231.1446。
实施例6.9.22:2-(乙基二甲基甲硅烷基)-1-甲基-1h-吲哚2u:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基吲哚1a(66.8mg,0.5mmol,1当量)、etme2sih(197μl,1.5mmol,3当量)和0.5ml的meot-bu加热持续120h来进行。c2:c3>20:1。期望的产物2u(58.5mg,54%收率)在通过硅胶快速色谱法(在己烷中3%ch2cl2)纯化之后作为无色油获得。rf=0.4(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.67(dt,j=7.8,1.0hz,1h),7.37(dd,j=8.3,0.9hz,1h),7.28(ddd,j=8.2,6.9,1.1hz,1h),7.14(ddd,j=7.9,6.9,1.0hz,1h),6.77(d,j=0.9hz,1h),3.89(s,3h),1.11–1.02(m,3h),0.95–0.90(m,2h),0.43(s,6h);13cnmr(125mhz,cdcl3)δ140.3,140.2,128.5,122.1,120.7,119.2,112.0,109.1,33.1,7.8,7.6,-2.6。ir(纯的膜,nacl)2954,2908,2873,1492,1464,1418,1356,1326,1300,1249,1233,1166,1131,1101,1071,1007,958,897,821cm–1;hrms(mm:esi-apci+)对于c13h19nsi[m+h]+计算为:217.1280,测量为217.1287。
此产物也通过在23℃下将n-甲基吲哚1a(62mg,0.5mmol,1当量)、kot-bu(11mg,0.1mmol,0.2当量)和etme2sih(198微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续48小时来制备。在水溶液后处理之后,粗制反应混合物通过使用己烷(等度的)的硅胶色谱法来纯化以获得80mg(74%)作为无色的油的标题化合物。1hnmr(500mhz,thf-d8)δ7.48(d,j=7.9hz,1h),7.31(dd,j=8.4,1.0hz,1h),7.10(ddd,j=8.2,6.9,1.2hz,1h),6.95(ddd,j=7.9,6.9,0.9hz,1h),6.64(d,j=0.9hz,1h),3.84(s,3h),1.05–0.95(m,3h),0.89(d,j=7.9hz,2h),0.38(s,6h)。13cnmr(126mhz,thf-d8)δ140.45,138.94,128.58,121.45,120.10,118.51,113.53,111.90,108.67,32.17,7.37,6.77,-3.67。
实施例6.9.23:1-苄基-2-(乙基二甲基甲硅烷基)-1h-吲哚2v:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苄基吲哚1b(102.5mg,0.5mmol,1当量)、etme2sih(197μl,1.5mmol,3当量)和0.5ml的thf加热持续96h来进行。c2:c3>20:1。期望的产物2v(87.9mg,60%收率)在通过硅胶快速色谱法(在己烷中10%ch2cl2)纯化之后作为无色油获得。rf=0.3(在己烷中10%ch2cl2);1hnmr(500mhz,cdcl3)δ7.75–7.69(m,1h),7.34–7.23(m,3h),7.23–7.11(m,3h),6.96(ddd,j=6.8,2.2,1.2hz,2h),6.88(s,1h),5.54(s,2h),1.00(t,j=7.9hz,3h),0.79(q,j=7.8hz,2h),0.32(s,6h);13cnmr(125mhz,cdcl3)δ140.5,140.1,138.4,128.9,128.7,127.3,125.9,122.4,120.8,119.6,112.9,110.1,50.1,7.8,7.5,-2.6。ir(纯的膜,nacl)3060,3028,2954,2910,2873,1605,1495,1466,1450,1377,1353,1334,1300,1249,1196,1164,1115,1096,1014,958,823,780,725cm–1;hrms(mm:esi-apci+)对于c19h23nsi[m+h]+计算为:293.1600,实测为293.1590。
在第二实验中,在25℃下,将1-苄基吲哚(104mg,0.5mmol,1当量),kot-bu(17mg,0.15mmol,0.3当量)和etme2sih(198微升,1.5mmol,3当量)在1ml的四氢呋喃中搅拌持续65小时。在水溶液后处理之后,粗制反应混合物通过相应地使用己烷:二乙醚:三乙胺的80:1:4混合物的硅胶色谱法来纯化以获得作为无色油的107mg(73%)的标题化合物。1hnmr(500mhz,thf-d8)δ7.55(ddd,j=7.7,1.4,0.8hz,1h),7.22–7.16(m,2h),7.16–7.09(m,2h),7.02(ddd,j=8.2,6.9,1.4hz,1h),6.97(ddd,j=8.0,6.9,1.2hz,1h),6.86(ddd,j=7.2,1.3,0.7hz,2h),6.78(d,j=0.9hz,1h),5.51(d,j=1.1hz,2h),0.95–0.90(m,3h),0.24(s,6h)。13cnmr(126mhz,thf-d8)δ141.31,140.50,139.94,130.09,129.39,127.90,126.71,122.96,121.45,120.10,113.93,110.81,50.62,8.50,7.93,-2.40。
实施例6.9.24:1-苄基-2-(二甲基(苯基)甲硅烷基)-1h-吲哚2w:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苄基吲哚1b(103.5mg,0.5mmol,1当量)、phme2sih(230μl,1.5mmol,3当量)和0.5ml的thf加热持续96h来进行。c2:c3>20:1。起始材料1b和产物2w的混合物(174.5mg的混合物,包含133.9mg的2w,78%收率,基于1hnmr计算的)在通过硅胶快速色谱法(在己烷中2%etoac)纯化之后获得。分析纯化合物2w在随后通过制备型hplc(在己烷中3%etoac)的纯化之后作为白色固体获得。rf=0.4(在己烷中5%etoac);1hnmr(500mhz,cdcl3)δ7.71–7.66(m,1h),7.51–7.48(m,2h),7.40–7.35(m,1h),7.34–7.29(m,2h),7.21–7.16(m,3h),7.14–7.08(m,3h),6.90(d,j=0.7hz,1h),6.78–6.75(m,2h),5.25(s,2h),0.50(s,6h);13cnmr(125mhz,cdcl3)δ140.4,139.4,138.3,137.5,134.2,129.6,128.9,128.6,128.1,127.2,125.9,122.6,121.0,119.6,114.1,110.2,50.0,-1.7。ir(纯的膜,nacl)3064,3027,2956,1605,1587,1494,1466,1450,1427,1353,1335,1301,1250,1197,1164,1116,1106,1096,1014,905,822cm–1;hrms(mm:esi-apci+)对于c23h24nsi[m+h]+计算为:342.1673,实测为342.1676。
实施例6.9.25:1-甲基-2-(三丁基甲硅烷基)-1h-吲哚2x:
反应根据一般程序通过在35℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基吲哚1a(65.6mg,0.5mmol,1当量)、n-bu3sih(385μl,1.5mmol,3当量)和0.5ml的thf加热持续65h来进行。c2:c3>20:1。期望的产物2x(123.5mg,75%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为白色固体获得。rf=0.5(100%己烷)。1hnmr(500mhz,cdcl3)δ7.61(dt,j=7.9,1.0hz,1h),7.37–7.30(m,1h),7.22(ddd,j=8.2,6.9,1.1hz,1h),7.08(ddd,j=7.9,6.9,1.0hz,1h),6.69(d,j=0.9hz,1h),3.84(s,3h),1.38–1.27(m,12h),0.94–0.86(m,15h);13cnmr(125mhz,cdcl3)δ140.2,139.0,128.6,121.7,120.5,118.9,112.7,108.9,32.9,26.6,26.1,13.6,12.7;ir(纯的膜,nacl)2955,2922,2871,2855,1492,1464,1411,1375,1356,1325,1298,1232,1196,1166,1102,1070,897,885,799,788,749,732cm–1;hrms(ei+)对于c21h35nsi[m+·]计算为:329.2539,实测为329.2523。
实施例6.9.26:1-甲基-2-(三乙基甲硅烷基)-1h-吡咯并[3,2-b]吡啶4a:
反应根据一般程序通过在45℃下将kot-bu(4.5mg,0.04mmol,20mol%)、n-甲基-4-氮杂吲哚3a(26.4mg,0.2mmol,1当量)、et3sih(98μl,0.6mmol,3当量)和0.2ml的thf加热持续96h来进行。c2:c3=6:1。c2-和c3-甲硅烷基化产物的混合物(16.2mg,33%收率)在通过硅胶快速色谱法(在己烷中50%etoac)纯化之后获得。分析纯c2-甲硅烷基化产物4a在随后通过制备型tlc(在己烷中50%etoac)的纯化之后作为无色油获得。rf=0.1(在己烷中33%etoac);1hnmr(500mhz,cdcl3)δ8.44(dd,j=4.6,1.4hz,1h),7.60(dt,j=8.3,1.2hz,1h),7.09(dd,j=8.3,4.6hz,1h),6.90(d,j=0.9hz,1h),3.83(s,3h),1.03–0.97(m,9h),0.96–0.89(m,6h);13cnmr(125mhz,cdcl3)δ147.0,143.0,142.7,133.0,116.4,116.1,113.8,33.1,7.6,4.0。ir(纯的膜,nacl)2953,2909,2874,1596,1557,1455,1434,1413,1355,1317,1288,1237,1134,1064,1004,800cm–1;hrms(esi+)对于c14h23n2si[m+h]+计算为:247.1625,实测为247.1621。
实施例6.9.27:1-甲基-2-(三乙基甲硅烷基)-1h-吡咯并[3,2-c]吡啶4b:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基-5-氮杂吲哚3b(66.0mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续120h来进行。c2:c3>20:1。期望的产物4b(37.9mg,31%收率)在通过硅胶快速色谱法(100%etoac)纯化之后作为黄色油获得。rf=0.2(100%etoac);1hnmr(500mhz,cdcl3)δ8.87(d,j=1.1hz,1h),8.28(d,j=5.9hz,1h),7.24–7.18(m,1h),6.80(d,j=0.9hz,1h),3.82(s,3h),1.02–0.96(m,9h),0.94–0.87(m,6h);13cnmr(125mhz,cdcl3)δ143.7,143.6,140.8,140.4,125.7,112.9,104.5,32.9,7.6,4.0。ir(纯的膜,nacl)2953,2909,2874,1597,1563,1485,1463,1435,1415,1368,1334,1310,1291,1219,1184,1123,1069,1004,900,809cm–1;hrms(esi+)对于c14h23n2si[m+h]+计算为:247.1625,实测为247.1626。
实施例6.9.28:1-甲基-2-(三乙基甲硅烷基)-1h-吡咯并[2,3-c]吡啶4c:
反应根据一般程序通过在45℃下将kot-bu(5.8mg,0.52mmol,20mol%)、n-甲基-6-氮杂吲哚3c(35.0mg,0.26mmol,1当量)、et3sih(126μl,0.78mmol,3当量)和0.3ml的thf加热持续94h来进行。c2:c3>20:1。期望的产物4c(32.9mg,50%收率)在通过硅胶快速色谱法(梯度洗脱,在ch2cl2中2.5%→5%meoh)纯化之后作为黄色油获得。rf=0.3(在ch2cl2中5%meoh);1hnmr(500mhz,cdcl3)δ8.76(s,1h),8.20(d,j=5.5hz,1h),7.47(dd,j=5.5,1.1hz,1h),6.68(d,j=0.8hz,1h),3.93(s,3h),1.03–0.97(m,9h),0.95–0.89(m,6h);13cnmr(125mhz,cdcl3)δ143.5,138.1,137.2,133.0,132.6,114.7,112.0,33.3,7.5,3.9。ir(纯的膜,nacl)2952,2909,2874,1594,1559,1496,1475,1457,1415,1358,1333,1315,1286,1241,1167,1120,1070,1004,817,808cm–1;hrms(esi+)对于c14h23n2si[m+h]+计算为:247.1625,实测为247.1620。
实施例6.9.29:1-甲基-2-(三乙基甲硅烷基)-1h-吡咯并[2,3-b]吡啶4d:
反应根据一般程序通过在35℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-甲基-7-氮杂吲哚3d(66mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续63h来进行。c2:c3>20:1。期望的产物4d(87.1mg,71%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中0%→10%etoac)纯化之后作为无色油获得。rf=0.3(在己烷中10%etoac);1hnmr(500mhz,cdcl3)δ8.33(dd,j=4.7,1.6hz,1h),7.87(dd,j=7.8,1.6hz,1h),7.02(dd,j=7.8,4.7hz,1h),6.67(s,1h),3.95(s,3h),1.04–0.97(m,9h),0.96–0.88(m,6h);13cnmr(125mhz,cdcl3)δ151.0,143.2,139.2,128.3,120.7,115.3,111.0,31.4,7.6,3.9。ir(纯的膜,nacl)3052,2953,2910,2874,1590,1570,1489,1444,1403,1302,1286,1226,1162,1134,1107,1066,1004,906,804,772,739cm–1;hrms(fab+)对于c14h23n2si[m+h]+计算为:247.1631,实测为247.1637。这些反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
实施例6.9.30:1-甲基-2-(三乙基甲硅烷基)-1h-吡咯并[2,3-b]吡啶4e:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苄基-7-氮杂吲哚3e(104.0mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续144h来进行。c2:c3>20:1。期望的产物4e(89.4mg,56%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中2.5→5%etoac)纯化之后作为无色油获得。rf=0.3(在己烷中5%etoac);1hnmr(500mhz,cdcl3)δ8.34(dd,j=4.7,1.6hz,1h),7.94(dd,j=7.8,1.6hz,1h),7.25–7.16(m,3h),7.07(dd,j=7.8,4.6hz,1h),6.87–6.85(m,2h),6.79(s,1h),5.69(s,2h),0.91–0.83(m,9h),0.74–0.69(m,6h);13cnmr(125mhz,cdcl3)δ151.2,143.7,139.04,138.96,128.6,128.4,127.0,125.9,120.5,115.7,112.2,47.8,7.4,3.7。ir(纯的膜,nacl)2954,2874,1589,1570,1495,1452,1439,1422,1378,1357,1309,1239,1157,1103,1004,909,803,777cm–1;hrms(mm:esi-apci+)对于c20h27n2si[m+h]+计算为:323.1938,实测为323.1947。
实施例6.9.31:1-苄基-2-(二乙基甲硅烷基)-1h-吡咯并[2,3-b]吡啶4f:
反应根据一般程序通过在60℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苄基-7-氮杂吲哚3e(104.5mg,0.5mmol,1当量)和et2sih2(194μl,1.5mmol,3当量)加热持续84h来进行。c2:c3>20:1。期望的产物4f(96.2mg,65%收率)在通过硅胶快速色谱法(在己烷中3%etoac)纯化之后作为黄色油获得。rf=0.4(在己烷中10%etoac);1hnmr(500mhz,cdcl3)δ8.37(dd,j=4.7,1.6hz,1h),7.95(dd,j=7.8,1.6hz,1h),7.30–7.16(m,3h),7.09(dd,j=7.8,4.6hz,1h),7.01–6.99(m,2h),6.80(s,1h),5.71(s,2h),4.32(p,j=3.3hz,1h),0.95(t,j=7.9hz,6h),0.78–0.63(m,4h);13cnmr(125mhz,cdcl3)δ150.9,143.8,138.9,137.4,128.6,128.5,127.2,126.6,120.5,115.8,111.7,47.6,8.1,3.4.ir(纯的膜,nacl)2955,2873,2120,1590,1568,1495,1453,1439,1422,1358,1300,1235,1156,1100,1009,973,910,808cm–1;hrms(mm:esi-apci+)对于c18h23n2si[m+h]+计算为:295.1625,实测为295.1636。
实施例6.9.32:1-苄基-2-(二甲基(苯基)甲硅烷基)-1h-吡咯并[2,3-b]吡啶4g:
反应根据一般程序通过在60℃下将kot-bu(11.2mg,0.1mmol,20mol%)、n-苄基-7-氮杂吲哚3e(103.9mg,0.5mmol,1当量)和phme2sih(230μl,1.5mmol,3当量)加热持续96h来进行。c2:c3>20:1。期望的产物4g(118.0mg,69%收率)在通过硅胶快速色谱法(在己烷中3%etoac)纯化之后作为黄色油获得。rf=0.4(在己烷中10%etoac);1hnmr(500mhz,cdcl3)δ8.35(dd,j=4.7,1.6hz,1h),7.97(dd,j=7.8,1.6hz,1h),7.49–7.45(m,2h),7.41–7.38(m,1h),7.37–7.32(m,2h),7.20–7.13(m,3h),7.08(dd,j=7.8,4.6hz,1h),6.84(s,1h),6.77–6.68(m,2h),5.46(s,2h),0.42(s,6h);13cnmr(125mhz,cdcl3)δ151.3,144.0,140.0,138.8,136.9,134.2,129.7,128.8,128.5,128.1,127.0,126.1,120.4,115.9,112.2,47.6,-2.0.ir(纯的膜,nacl)3050,3027,2956,1589,1569,1495,1439,1427,1359,1309,1250,1156,1107,1029,987,910,822cm–1;hrms(mm:esi-apci+)对于c22h23n2si[m+h]+计算为:343.1625,实测为343.1635。
实施例6.9.33:苯并呋喃-2-基三乙基硅烷
反应根据一般程序通过在60℃下将苯并呋喃(59mg,0.5mmol,1当量)、kot-bu(19.6mg,0.18mmol,0.35当量)和et3sih(239微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续45小时来进行。在水溶液后处理之后,粗制反应混合物通过用己烷(等度的)洗脱的硅胶色谱法来纯化以获得44mg(38%)作为无色的油的标题化合物。
1hnmr(500mhz,丙酮-d6)δ7.64(ddd,j=7.7,1.3,0.7hz,1h),7.53(dd,j=8.2,0.9hz,1h),7.30(ddd,j=8.3,7.2,1.3hz,1h),7.22(ddd,j=7.7,7.2,1.0hz,1h),7.16(d,j=1.0hz,1h),1.09–0.98(m,9h),0.92–0.84(m,6h)。此反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
实施例6.9.34:苯并[b]噻吩-2-基三乙基硅烷4h:
(注意:此反应的产物先前被错表征(mischaracterize)为苯并[b]噻吩-3-基三乙基硅烷。光谱数据已经被重新解读以提供此处给出的结构)。反应根据一般程序通过在25℃下将kot-bu(11.2mg,0.1mmol,20mol%)、苯并[b]噻吩3h(67.0mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、以及0.5ml的thf加热持续60h来进行。期望的产物4h(120.3mg,97%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.6(100%己烷)。1hnmr(500mhz,cdcl3)δ7.91(m,1h),7.87–7.81(m,1h),7.49(m,1h),7.41–7.29(m,2h),1.07–1.03(m,9h),0.96–0.85(m,6h)。此反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
此材料还以如下规模被制备。在氮气填充的手套箱中,将kot-bu(1.7g,15mmol,20mol%)、苯并[b]噻吩3h(10.1g,75mmol,1当量)、et3sih(23.3ml,146mmol,2当量)、和75ml的thf添加到装配有磁力搅拌棒并且用聚丙烯盖密封的250ml介质罐(mediajar)。将反应混合物在25℃下搅拌持续60h。然后将罐从手套箱中移出,小心地打开(警告:气体释放!),并且用无水et2o(30ml)稀释。将反应过滤,将溶剂在真空中除去并且将残余的挥发物在高真空(30毫托,23℃)下除去。期望的产物4h(17.3g,93%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。
实施例6.9.35:苯并[b]噻吩-2-基二甲基(苯基)硅烷4i:
(注意:此反应的产物先前被错表征为苯并[b]噻吩-3-基二甲基(苯基)硅烷。光谱数据已经被重新解读以提供此处给出的结构)。反应根据一般程序通过在25℃下将kot-bu(11.2mg,0.1mmol,20mol%)、苯并[b]噻吩3h(67.0mg,0.5mmol,1当量)、phme2sih(230μl,1.5mmol,3当量)、以及0.5ml的thf加热持续60h来进行。期望的产物4i(116.6mg,87%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.5(100%己烷)。1hnmr(500mhz,cdcl3)δ7.94–7.87(m,1h),7.87–7.79(m,1h),7.68–7.59(m,2h),7.51(d,j=0.8hz,1h),7.46–7.39(m,3h),7.38–7.31(m,2h),0.69(s,6h)。
13cnmr(126mhz,cdcl3)δ144.01,141.12,140.18,137.29,134.13,132.41,129.70,128.09,124.45,124.18,123.69,122.33,-1.42。hrms:[c16h16ssi]计算为268.0743,测量为268.0742。
实施例6.9.36:2-(5-(三乙基甲硅烷基)噻吩-2-基)吡啶4j:
反应根据一般程序进行。条件a:反应用kot-bu(11.2mg,0.1mmol,20mol%)、2-(噻吩-2-基)吡啶3j(80.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、和0.5ml的thf在25℃下进行持续35h。期望的产物4j(129.3mg,94%收率)在通过硅胶快速色谱法(在己烷中5%etoac)纯化之后作为无色油获得。条件b:反应用kot-bu(19.6mg,0.18mmol,3.5mol%)、2-(噻吩-2-基)吡啶3j(0.81g,5mmol,1当量)、et3sih(2.43ml,15mmol,3当量)、和3.0ml的thf在25℃下进行持续96h。期望的产物4j(1.13g,82%收率)在通过硅胶快速色谱法(在己烷中5%etoac)纯化之后作为无色油获得。rf=0.3(在己烷中5%etoac);1hnmr(500mhz,cdcl3)δ8.56(d,j=4.7hz,1h),7.61(dt,j=3.9,1.7hz,3h),7.23(d,j=3.3hz,1h),7.08(q,j=4.8hz,1h),1.01(t,j=7.9hz,9h),0.82(q,j=7.9hz,6h)。13cnmr(125mhz,cdcl3)δ152.8,149.8,149.6,139.7,136.6,135.6,125.7,121.8,119.0,7.4,4.5;ir(纯的膜,nacl)3054,3001,2953,2909,2874,1585,1563,1528,1517,1464,1436,1422,1377,1315,1290,1238,1207,1151,1077,1066,1047,1007,990,962,807,774,737cm–1;hrms(fab+)对于c15h22nssi[m+h]+计算为:276.1242,实测为276.1239。
实施例6.9.37:2-(5-(乙基二甲基甲硅烷基)噻吩-2-基)吡啶4k:
反应根据一般程序通过在35℃下将kot-bu(11.2mg,0.1mmol,20mol%)、2-(噻吩-2-基)吡啶3j(80.5mg,0.5mmol,1当量)、etme2sih(198μl,1.5mmol,3当量)、和0.5ml的thf加热持续48h来进行。期望的产物4k(107.4mg,87%收率)在通过硅胶快速色谱法(在己烷中10%et2o)纯化之后作为无色油获得。rf=0.4(在己烷中10%et2o);1hnmr(500mhz,cdcl3)δ8.58(ddd,j=4.9,1.8,1.1hz,1h),7.72–7.63(m,2h),7.62(d,j=3.5hz,1h),7.24(d,j=3.5hz,1h),7.13(ddd,j=6.7,4.9,2.0hz,1h),1.05–0.96(m,3h),0.78(qd,j=7.8,0.8hz,2h),0.32(s,6h);13cnmr(125mhz,cdcl3)δ152.7,149.7,149.6,141.9,136.6,135.0,125.6,121.7,118.9,8.3,7.2,-2.5;ir(纯的膜,nacl)3054,3001,2953,2909,2874,1585,1563,1528,1517,1464,1436,1422,1315,1290,1248,1207,1151,1077,1066,1047,1007,990,964,836,812,774,752,737,712cm–1;hrms(fab+)对于c13h18nssi[(m+h)+–h2]计算为:248.0929,实测为248.0935。
实施例6.9.38:2-(5-(二甲基(苯基)甲硅烷基)噻吩-2-基)吡啶4l:
反应根据一般程序通过在35℃下将kot-bu(11.2mg,0.1mmol,20mol%)、2-(噻吩-2-基)吡啶3j(80.5mg,0.5mmol,1当量)、phme2sih(230μl,1.5mmol,3当量)、和1.0ml的thf加热持续48h来进行。期望的产物4l(118.1mg,80%收率)在通过硅胶快速色谱法(在己烷中10%et2o)纯化之后作为无色油获得。rf=0.3(在己烷中10%et2o);1hnmr(500mhz,cdcl3)δ8.60–8.54(m,1h),7.72–7.56(m,5h),7.43–7.33(m,3h),7.26(m,1h),7.14(m,1h),0.63(s,6h);13cnmr(125mhz,cdcl3)δ152.4,150.3,149.5,140.6,137.3,136.6,136.0,133.8,129.3,127.8,125.6,121.8,118.9,–1.6;ir(纯的膜,nacl)3067,2955,1586,1563,1527,1463,1423,1316,1290,1249,1207,1151,1112,1077,1005,989,963,807,773,731cm–1;hrms(fab+)对于c17h18nssi[m+h]+计算为:296.0929,实测为296.0938。
实施例6.9.39:三乙基(5-戊基噻吩-2-基)硅烷4m:
反应根据一般程序进行。条件a:反应用kot-bu(11.2mg,0.1mmol,20mol%)、2-戊基噻吩3m(77.0mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、和0.5ml的thf在25℃下进行持续48h。期望的产物4m(130.0mg,96%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。条件b:反应用kot-bu(5.6mg,0.05mmol,1mol%)、2-戊基噻吩3m(770.4mg,5.0mmol,1当量)、et3sih(2.43ml,15mmol,3当量)、和3.0ml的thf在25℃下进行持续96h。期望的产物4m(1.23g,92%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.6(100%己烷);1hnmr(500mhz,cdcl3)δ7.12(dd,j=3.3,1.5hz,1h),6.91(dt,j=3.3,1.0hz,1h),2.90(td,j=7.7,1.2hz,2h),1.81–1.71(m,2h),1.48–1.36(m,4h),1.06(t,j=7.8hz,9h),0.99–0.94(m,3h),0.84(qd,j=7.8,1.0hz,6h);13cnmr(125mhz,cdcl3)δ151.6,134.7,134.1,125.5,31.7,31.6,30.2,22.6,14.1,7.5,4.7;ir(纯的膜,nacl)3054,2955,2934,2874,1750,1528,1456,1438,1413,1378,1339,1235,1213,1058,1011,988,799,736cm–1;hrms(fab+)对于c15h27ssi[(m+h)–h2]+计算为:267.1603,实测为267.1609。
实施例6.9.40:三乙基(5-戊基呋喃-2-基)硅烷4n:
反应根据一般程序通过在25℃下将kot-bu(8.4mg,0.075mmol,1.5mol%)、2-戊基呋喃3n(691mg,5.0mmol,1当量)、et3sih(2.43ml,15mmol,3当量)、和3ml的thf加热持续96h来进行。期望的产物4n(1.15g,91%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.6(100%己烷);1hnmr(500mhz,cdcl3)δ6.53(d,j=3.0hz,1h),5.96(dt,j=3.0,0.9hz,1h),2.67–2.60(m,2h),1.64(dq,j=9.4,7.4hz,2h),1.36–1.28(m,4h),1.05–0.95(m,9h),0.92–0.85(m,3h),0.74(qd,j=7.8,0.8hz,6h);13cnmr(125mhz,cdcl3)δ161.2,156.2,121.5,104.6,31.6,28.3,27.9,22.6,14.1,7.5,3.6;ir(纯的膜,nacl)3108,2954,2933,2874,1807,1721,1588,1493,1459,1414,1378,1340,1237,1186,1173,1118,1084,1011,962,923,782,736,724cm–1;hrms(fab+)对于c15h27osi[(m+h)–h2]+计算为:251.1831,实测为251.1821。
此材料还以关于4h的几克规模合成的规模使用相同的程序来制备。反应用kot-bu(1.6g,14.6mmol,20mol%)、2-戊基呋喃3n(10.1g,73mmol,1当量)、et3sih(23.3ml,146mmol,2当量)、和73ml的thf在25℃下进行持续72h。期望的产物4n(17.4g,95%收率)在过滤、挥发物在高真空(30毫托,23℃)下的除去和通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。
实施例6.9.41:2-戊基呋喃3n与et2sih2的反应:
反应根据一般程序通过在25℃下将kot-bu(11.2mg,0.1mmol,20mol%)、2-戊基呋喃3n(69.1mg,0.5mmol,1当量)、et2sih2(195μl,1.5mmol,3当量)和0.5ml的thf加热持续76h来进行。期望的产物4o(87.4mg,78%收率)和硅链接的产物si-4o(12.4mg,8%收率)在通过硅胶快速色谱法(100%己烷)纯化之后获得。
二乙基(5-戊基呋喃-2-基)硅烷4o:无色油,rf=0.6(100%己烷);1hnmr(500mhz,cdcl3)δ6.63(d,j=3.1hz,1h),6.00(dt,j=3.1,0.9hz,1h),4.21(p,j=3.2hz,1h),2.75–2.64(m,2h),1.73–1.62(m,2h),1.38–1.32(m,4h),1.11–1.04(m,6h),0.95–0.90(m,3h),0.88–0.81(m,4h);13cnmr(125mhz,cdcl3)δ161.8,153.7,122.7,105.0,31.6,28.4,27.9,22.6,14.1,8.1,3.2;ir(纯的膜,nacl)2955,2931,2873,2120,1588,1493,1461,1233,1082,1010,974,925,798,715cm–1;hrms(fab+)对于c13h23osi[(m+h)–h2]+计算为:223.1518,实测为223.1519。
二乙基双(5-戊基呋喃-2-基)硅烷si-4o:无色油,rf=0.7(100%己烷);1hnmr(500mhz,cdcl3)δ6.62(d,j=3.1hz,2h),5.98(dt,j=3.1,0.9hz,2h),2.69–2.61(m,4h),1.70–1.59(m,4h),1.36–1.30(m,8h),1.08–1.01(m,6h),1.01–0.93(m,4h),0.93–0.81(m,6h);13cnmr(125mhz,cdcl3)δ161.5,153.7,122.8,104.8,31.4,28.2,27.7,22.4,13.9,7.2,4.2;ir(纯的膜,nacl)2955,2928,2873,2859,1587,1493,1461,1233,1187,1010,961,925,783,726cm–1;hrms(ei+)对于c22h36osi[m·+]计算为:360.2485,实测为360.2468。
实施例6.9.42:三丁基(5-戊基呋喃-2-基)硅烷4p:
反应根据一般程序通过在25℃下将kot-bu(11.2mg,0.1mmol,20mol%)、2-戊基呋喃3n(69.1mg,0.5mmol,1当量)、n-bu3sih(386μl,1.5mmol,3当量)、和0.5ml的thf加热持续108h来进行。期望的产物4p(137.8mg,82%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.71(100%己烷);1hnmr(500mhz,cdcl3)δ6.50(d,j=3.0hz,1h),5.95(d,j=3.0,1h),2.67–2.60(m,2h),1.69–1.59(m,2h),1.39–1.24(m,16h),0.94–0.83(m,12h),0.79–0.69(m,6h);13cnmr(125mhz,cdcl3)δ161.0,156.8,121.3,104.7,31.6,28.3,28.0,26.7,26.2,22.6,14.1,13.9,12.3;ir(纯的膜,nacl)3107,2956,2923,2871,2857,2099,1677,1588,1493,1464,1410,1376,1341,1296,1271,1217,1187,1175,1082,1050,1010,961,925,885,781,759,732cm–1;hrms(ei+)对于c21h40osi[m·+]计算为:336.2848,实测为336.2859。
实施例6.9.43:2,5-双(三乙基甲硅烷基)噻吩4q:
反应根据一般程序通过在25℃下将kot-bu(11.2mg,0.1mmol,20mol%)、噻吩3q(42.1mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、和0.5ml的thf加热持续72h来进行。期望的产物4q(134.2mg,86%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.6(100%己烷)。1hnmr(500mhz,cdcl3)δ7.40(s,2h),1.02–0.99(m,18h),0.83–0.79(m,12h)。
实施例6.9.44:1-苄基-1h-吡咯3s与et3sih的反应:
反应根据一般程序通过在25℃下将kot-bu(11.2mg,0.1mmol,20mol%)、1-苄基-1h-吡咯3s(78.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续108h来进行。期望的产物4s(100.3mg,74%收率)和双甲硅烷基化产物si-4s(9.6mg,5%)在通过硅胶快速色谱法(100%己烷)纯化之后获得。
1-苄基-2-(三乙基甲硅烷基)-1h-吡咯4s:无色油,rf=0.3(100%己烷);1hnmr(500mhz,cdcl3)δ7.40–7.32(m,2h),7.32–7.25(m,1h),7.04–6.98(m,2h),6.86(dd,j=2.4,1.5hz,1h),6.51(dd,j=3.5,1.5hz,1h),6.30(dd,j=3.4,2.4hz,1h),5.22(s,2h),0.95(t,j=7.8hz,9h),0.73(q,j=7.8hz,6h);13cnmr(125mhz,cdcl3)δ139.2,129.9,128.7,127.5,126.62,126.56,120.9,108.9,53.5,7.6,4.2;ir(纯的膜,nacl)3088,3064,3029,2952,2908,2873,1516,1506,1495,1454,1418,1353,1329,1288,1237,1175,1112,1080,1008,969,760cm–1;hrms(ei+)对于c17h25nsi[m·+]计算为:271.1756,实测为271.1755。
1-苄基-2,5-双(三乙基甲硅烷基)-1h-吡咯si-4s:无色油,rf=0.4(100%己烷);1hnmr(500mhz,cdcl3)δ7.29–7.21(m,2h),7.21–7.15(m,1h),6.72(dq,j=7.1,1.0hz,2h),6.52(s,2h),5.28(s,2h),0.85–0.82(m,18h),0.63–0.52(m,12h);13cnmr(125mhz,cdcl3)δ140.4,135.6,128.2,126.9,125.5,121.2,53.3,7.4,3.9;ir(纯的膜,nacl)3027,2952,2909,2874,1605,1498,1485,1454,1416,1377,1343,1277,1237,1161,1075,1002,912,775,764,731cm–1;hrms(ei+)对于c23h39nsi2[m·+]计算为:385.2621,实测为385.2638。
实施例6.9.45:1-甲基-5-(三乙基甲硅烷基)-1h-吡唑4t:
反应根据一般程序通过在25℃下加热kot-bu(11.2mg,0.1mmol,20mol%)、1-甲基-1h-吡唑3t(41.1mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、以及0.5ml的thf持续120h来进行。期望的产物4t(72.6mg,74%收率)在通过硅胶快速色谱法(1:1et2o:己烷)纯化之后作为无色油获得。rf=0.3(1:1et2o:己烷)。(despotopoulou,c.;等人,porg.lett.2009,11,3326)1hnmr(500mhz,cdcl3)δ7.47(d,j=1.9hz,1h),6.37(d,j=1.8hz,1h),3.95(s,3h),0.96(m,9h),0.83(m,6h)。
实施例6.9.46:二苯并[b,d]噻吩-4-基三乙基硅烷4u:
反应根据一般程序通过在85℃下将kot-bu(11.2mg,0.1mmol,20mol%)、二苯并噻吩3u(92mg,0.5mmol,1.0当量)、et3sih(243μl,1.5mmol,3.0当量)、和3ml的二氧六环加热持续72h来进行。期望的产物4u(55.4mg,38%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.7(100%己烷);1hnmr(500mhz,cdcl3)δ8.17(m,2h),7.86(m,1h),7.58(m,1h),7.45(m,3h),1.10–0.93(m,15h);13cnmr(125mhz,cdcl3)δ145.6,139.3,135.4,134.7,133.7,131.5,126.5,124.2,123.7,122.4,122.2,121.4,7.4,3.2。ir(纯的膜,nacl)3060,2953,2908,2873,1450,1440,1415,1366,1283,1250,1238,1098,1080,1042,1019,1003,972,812,749,733cm–1;hrms(ei+)对于c18h22ssi[m·+]计算为:298.1212,实测为298.1214。此反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
实施例6.9.47:二苯并[b,d]呋喃3v与et3sih的反应:
反应根据一般程序通过在65℃下将kot-bu(11.2mg,0.1mmol,20mol%)、二苯并[b,d]呋喃3v(84.1mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续65h来进行。期望的产物4v(100.2mg,71%收率)和双甲硅烷基化的产物si-4v(6.9mg,4%收率)在通过硅胶快速色谱法(100%己烷)纯化之后获得。
二苯并[b,d]噻吩-4-基三乙基硅烷4v:无色油,rf=0.6(100%己烷)。1hnmr(500mhz,cdcl3)δ8.01–7.94(m,2h),7.61–7.50(m,2h),7.46(td,j=7.7,1.4hz,1h),7.34(td,j=7.6,4.4hz,2h),1.02(m,15h)。
4,6-双(三乙基甲硅烷基)二苯并[b,d]呋喃si-4v:白色固体,rf=0.7(100%己烷)。1hnmr(500mhz,cdcl3)δ7.99(dd,j=7.6,1.4hz,2h),7.54(dd,j=7.1,1.4hz,2h),7.35(t,j=7.4hz,2h),1.12–0.96(m,30h)。
实施例6.9.48:三乙基(6-甲氧基二苯并[b,d]呋喃-4-基)硅烷4w:
反应根据一般程序通过在65℃下将kot-bu(11.2mg,0.1mmol,20mol%)、4-甲氧基二苯并[b,d]呋喃3w(99.0mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、和0.5ml的thf加热持续65h来进行。期望的产物4w(99.9mg,64%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色油获得。rf=0.3(100%己烷);1hnmr(500mhz,cdcl3)δ7.94(dd,j=7.6,1.4hz,1h),7.53(ddd,j=15.4,7.4,1.2hz,2h),7.37–7.30(m,1h),7.24(t,j=7.8hz,1h),6.99(dd,j=8.0,1.0hz,1h),4.09(s,3h),1.08–0.95(m,15h);13cnmr(125mhz,cdcl3)δ161.1,145.7,145.3,133.4,126.1,123.0,122.8,122.3,121.5,120.4,112.9,111.0,56.9,7.4,3.5;ir(纯的膜,nacl)3052,2952,2925,2873,2852,2361,1627,1596,1576,1497,1483,1456,1432,1387,1322,1308,1270,1220,1180,1168,1147,1125,1038,1006,854,836,767,752,729cm–1;hrms(ei+)对于c19h24o2si[m·+]计算为:312.1546,实测为312.1555。
实施例6.9.49:吡啶的甲硅烷基化
反应根据一般程序通过在65℃下将吡啶(40mg,0.5mmol,1当量)、kot-bu(17mg,0.15mmol,0.3当量)和et3sih(240微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续65小时来进行。在水溶液后处理之后,粗制反应混合物通过相应地使用己烷:二乙醚:三乙胺的80:1:4混合物的硅胶色谱法来纯化以获得14mg(15%)作为无色的油状固体的标题化合物。1hnmr(500mhz,thf-d8)δ8.99–8.16(m,2h),7.62–7.07(m,2h),1.01–0.93(m,6h),0.91–0.79(m,4h)。13cnmr(126mhz,thf-d8)δ149.88,129.76,129.29,7.70,3.66。hrms:[c11h20nsi]计算为194.1365,测量为194.1367。
使此实验再现的尝试导致吡啶的变化的收率,通常产生小于约5%的指示的产物。采用其他缺电子杂芳烃例如喹啉、异喹啉、和吖啶在可比较的条件下的实验同样地产生低的收率(<5%)或不反应。
实施例6.9.50:尝试的4-甲氧基吡啶的甲硅烷基化
反应根据一般程序通过在65℃下将4-甲氧基吡啶(55mg,0.5mmol,1当量)、kot-bu(17mg,0.15mmol,0.3当量)和et3sih(240微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续65小时来进行。反应用二乙醚(1ml)来稀释,用水(0.5ml)猝灭并且有机相通过gc-ms、gc-fid和1hnmr分析来分析并且揭示出没有起始材料至甲硅烷基化产物的明显转化。
实施例6.9.51:尝试的2,6-二甲氧基吡啶的甲硅烷基化
反应根据一般程序通过在65℃下将2,6-二甲氧基吡啶(70mg,0.5mmol,1当量)、kot-bu(17mg,0.15mmol,0.3当量)和et3sih(240微升,1.5mmol,3当量)在1ml的四氢呋喃中加热持续65小时来进行。反应用二乙醚(1ml)来稀释,用水(0.5ml)猝灭并且有机相通过gc-ms、gc-fid和1hnmr分析来分析。gc-ms分析揭示出相应于2-甲硅烷基化产物异构体以及几种未确定的产物的形成的主要质量峰。
实施例7扩展的反应方案-基质和官能团耐受的灵敏度
本方法的一般性和扩展性效用已经在本文被描述,但为了完整的目的,此处提供另外的具体的实施例和反应方案。还包括用于制备和表征这些材料的新的方法学。
作为较早实施例的扩展,在氮上具有me、乙基(et)、苄基(bn)、苯基(ph)和容易裂解的甲氧基甲基和2-[(三甲基甲硅烷基)乙氧基]甲基的多种吲哚类被评估,并且全部产生以中等到良好的收率的区域选择性c2甲硅烷基化(图2,化合物2a-2f)。测试在吲哚核的各种位置的取代基的影响示出,me、ome、obn、ch2ome和ph全部是相容的,这给出48%-83%收率的期望的产物2g–2n。若干氢硅烷被检查,并且以良好的收率获得甲硅烷基化产物(2o–2x)。许多含n、含o和含s的杂芳族化合物(图3)(包括含吡啶的骨架(4a–4g和4j–4l))经历具有高区域选择性的反应。在减少的催化剂载量(1mol%–3.5mol%;4j,4m和4n)下和大规模(4h和4n)的反应证明该工艺的稳健性和制备规模效用(preparativescaleutility)。在程序上便利的条件下,反应按比例放大到大于100g而没有催化剂活性的损失(图4a)。通常,反应证明对于电中性且富电子的杂环是选择性的;具有吸电子基团的吲哚类似乎是不反应的。
实施例7.1.关于噻吩、呋喃和吡咯的竞争实验
为了研究含氮、含氧、和含硫芳族杂环通过kot-bu-催化的c-h甲硅烷基化的相对反应性,使用一当量的et3sih和一当量的每种杂芳烃进行两个内部竞争实验(方案1)。使反应运行到et3sih的部分消耗,并且甲硅烷基化的杂芳烃的相对量通过1hnmr分析来确定。结果证明对于5-元杂芳烃,反应性的相对速率的趋势为:噻吩3q>呋喃3r>1-甲基吡咯3x。
该趋势在被取代的噻吩3m和呋喃3n之间的竞争中被证实。用于竞争实验的程序包括:
对于反应(a):在氮气填充的手套箱中,将kot-bu(11.2mg,0.1mmol,20mol%)、噻吩3q(42.1mg,0.5mmol,1当量)、呋喃3r(34.0mg,0.5mmol,1当量)和1-甲基吡咯3x(40.5mg,0.5mmol,1当量)添加到装配有磁力搅拌棒的2打兰闪烁管。然后添加thf(0.3ml)和et3sih(81μl,0.5mmol,1当量–在使用之前,通过短的活性氧化铝垫过滤)。将该管密封,并且在23℃下搅拌持续约8小时。将该管从手套箱中移出,用二乙醚(2ml)稀释,并且在减压下浓缩。粗制反应混合物通过1hnmr的分析揭示si-4q:si-4r:4x的比率是5:1:0。
对于反应(b):在氮气填充的手套箱中,将kot-bu(11.2mg,0.1mmol,20mol%)、2-戊基噻吩3m(77.0mg,0.5mmol,1当量)、和2-戊基呋喃3n(34.0mg,0.5mmol,1当量)添加到装配有磁力搅拌棒的2打兰闪烁管。然后添加thf(0.3ml)和et3sih(81μl,0.5mmol,1当量–在使用之前,通过短的活性氧化铝垫过滤)。将该管密封,并且在23℃下搅拌持续约8小时。将该管从手套箱中移出,用二乙醚(2ml)稀释,并且在减压下浓缩。粗制反应混合物通过1hnmr的分析揭示4m:4n的比率是5:1。
实施例7.2.官能团相容性的评估。为了提供对于甲硅烷基化反应的官能团耐受性的广泛的处理,进行按照glorius的方法的“稳健性筛选(robustnessscreen)”(表5,其在下文)。某些概括可以从这些结果做出。例如,羰基使反应停止(条目16、17)。然而,作为缩醛(例如苯甲醛二甲缩醛)的保护是很好地耐受的(条目18)。芳基-x基团,其中x=br、i、cn、no2,同样地阻碍反应性(条目7、8、19和20)。有趣地,这些官能团在大部分情况下保持完整的。然而,烯烃部分、炔烃部分、ar-f部分、ar-cl部分、ar-cf3部分、叔胺部分、吡啶部分、和膦部分是相容的(条目2-6、9、11、23-26)。没有烯烃和炔烃的明显的氢化硅烷化或还原发生。甚至游离的oh和nh在某种程度上是耐受的,推测由于杂原子的偶然的甲硅烷基化的原位保护,这通过使用bnotes作为添加剂被确证(条目12、13和15)。此外,环氧化物和氮丙啶也是耐受的,并且这些添加剂的亲核性开环没有被观察到(条目21、22)。
a反应用0.5mmol的3h和0.5mmol的添加剂在一般程序下进行。0.5mmol的十三烷作为内标物在反应开始时被添加。产物的收率、3h和添加剂的剩余的量通过gc-fid分析来确定。b对照反应不添加添加剂。c不确定(与溶剂峰重叠,由于低沸点)。d形成三乙基甲硅烷基保护的吗啉,并且通过gcms来确证。e形成bnotes。f缩醛部分地水解成phcho。
表5
实施例8.制备的硅烷的转化
实施例8.1.一锅法的si定向的本位取代(si-directedipso–substitution)/suzuki–miyaura交叉偶联
在0℃下,将bcl3(1.0m,0.48ml,0.48mmol)在ch2cl2中的溶液通过注射器在n2下添加到吲哚硅烷2a(98.2mg,0.4mmol)在ch2cl2(4ml)中的搅拌的溶液。使混合物在室温下搅拌持续3h,之后将溶剂在真空下除去。使残余物在高真空下干燥持续20min之后,添加4-碘代苯甲醚(94.0mg,0.4mmol)、pd(pph3)4(23.2mg,5mol%)、dme(4ml,脱气的)和2mna2co3水溶液(1ml,脱气的),并且使混合物在回流下搅拌持续5h。然后,使反应混合物冷却到室温,并且添加水(20ml)。混合物用et2o(3x30ml)萃取,将合并的有机萃取物用盐水洗涤,经na2so4干燥并且浓缩。期望的2-(4-甲氧基苯基)-1-甲基-1h-吲哚5(71.9mg,76%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中10%→33%ch2cl2)纯化之后作为白色固体获得。rf=0.4(在己烷中10%etoac);1hnmr(500mhz,cdcl3)δ7.63(d,j=7.7hz,1h),7.49–7.39(m,2h),7.36(d,j=8.2hz,1h),7.24(dt,j=8.2,1.2hz,1h),7.14(dt,j=7.9,1.0hz,1h),7.05–6.96(m,2h),6.51(brs,1h),3.88(s,3h),3.73(s,3h)。
实施例8.2.杂芳基硅醇的合成和在denmark–hiyama交叉偶联中的应用
将化合物2o(44.5mg,0.2mmol和[rucl2(对伞花烃)]2(6.3mg,0.01mmol)添加到装配有搅拌棒的5ml烧瓶。将烧瓶用隔片密封并且置于高真空下持续5min,之后与o2气囊连接并且用o2回填,然后通过注射器经由隔片添加乙腈(1ml)和h2o(7.4μl,0.4mmol)。将反应混合物在室温下搅拌持续12h。将溶剂蒸发并且产物6(36.0mg,77%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中10%→20%etoac)纯化之后作为无色油获得。rf=0.2(在己烷中10%etoac);1hnmr(500mhz,cdcl3)δ7.66(dt,j=7.9,1.0hz,1h),7.37(dd,j=8.3,1.0hz,1h),7.28(ddd,j=8.3,6.9,1.2hz,1h),7.13(ddd,j=7.9,6.9,1.0hz,1h),6.80(d,j=0.9hz,1h),3.93(s,3h),2.12(brs,1h),1.12–1.05(m,6h),1.02–0.95(m,4h);13cnmr(125mhz,cdcl3)δ140.4,138.1,128.4,122.6,121.1,119.4,112.7,109.4,33.1,7.1,6.7。ir(纯的膜,nacl)3315,2956,2876,1493,1463,1413,1357,1328,1300,1234,1166,1102,1075,1007,960,897,839,798,751,732cm–1;hrms(mm:esi-apci+)对于c13h20nosi[m+h]+计算为:234.1309,实测为234.1305。
实施例8.3.2-(4-甲氧基苯基)-1-甲基-1h-吲哚5:
在氮气填充的手套箱中,将装配有搅拌棒的2打兰的管装载有naot-bu(26.8mg,0.28mmol)和cui(26.6mg,0.14mmol)、4-碘代苯甲醚(33.0mg,0.14mmol)、pd(dba)2(8.2mg,0.014mmol,10mol%)和0.2ml的甲苯。将混合物用盖密封并且搅拌持续10min。然后,将此混合物通过注射器转移到包含硅醇6(33.1mg,0.14mmol)的另一个2打兰管。将该管用甲苯(2x0.4ml)洗涤,并且将该冲洗物添加到反应混合物。在将反应在30℃下搅拌持续4h之后,起始材料完全被转化(通过tlc监测)。期望的产物5(28.1mg,84%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中10%→50%ch2cl2)纯化之后作为白色固体获得。
实施例8.4.通过si保护基策略的直接c7锂化-硼基化此一般转化(即,在苯并呋喃类、吲哚类和噻吩类中的c2位的保护/脱保护,包括这些甲硅烷基化衍生物的c7锂化-硼基化)被认为是在本发明的范围内。
实施例8.4.1.三乙基(7-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼-2-基)苯并[b]噻吩-2-基硅烷7。
向装载有搅拌棒、用隔片加盖的火焰干燥的(flame-dried)圆底烧瓶并且在稳定的氩气流下在23℃添加苯并[b]噻吩-2-基三乙基硅烷4h(992mg,4.0mmol,1当量)、戊烷(5.0ml)和tmeda(0.703g,0.907ml,1.5当量)。逐滴地添加正丁基锂(在己烷中1.6m,3.78ml,1.5当量),使得内温保持在22℃和25℃之间(将热电偶通过隔片直接插入到溶液中,用于温度的内部监测)。允许产生的深棕色溶液在22℃下搅拌持续20h。然后,使溶液冷却到-78℃(干冰/丙酮),并且将i-probpin(1.52g,1.64ml,8.06mmol,2.0当量)作为在thf(8.06ml)中的1m溶液逐滴地添加,使得温度被保持在-75℃以下(小心的温度控制对于再现性是重要的)。允许产生的溶液在-78℃下搅拌持续1h,之后将冷却浴移除。允许溶液自然地加温至23℃,并且在该温度下搅拌持续另外的一小时。将产生的浑浊的黄色反应混合物小心地用nh4cl(5ml)猝灭。混合物用et2o(3x10ml)萃取,将合并的有机级分用盐水洗涤,经mgso4干燥,过滤,并且将溶剂蒸发以给出粘稠的棕色液体。期望的产物7(926mg,64%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中0%→3%etoac)纯化之后作为无色固体获得。rf=0.2(100%己烷);1hnmr(500mhz,cdcl3)δ7.91(dd,j=8.0,1.3hz,1h),7.80(dd,j=7.0,1.3hz,1h),7.48(s,1h),7.35(dd,j=7.9,7.0hz,1h),1.42(s,12h),1.10–1.00(m,9h),0.89(m,6h);13cnmr(125mhz,cdcl3)δ149.7,140.8,139.8,132.0,131.4,126.4,123.4,84.3,25.1,7.6,4.4。ir(纯的膜,nacl)2955,2937,1375,1367,1359,1134,1059,854,735cm–1;hrms(ei+)对于c20h31bssio2[m·+]计算为:374.1907,实测为374.1907。
实施例8.4.2.2-(苯并[b]噻吩-7-基)-4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷8.
在室温下向装载有磁力搅拌棒和三乙基(7-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼-2-基)苯并[b]噻吩-2-基)硅烷7(300mg,0.80mmol)的管添加ch2cl2(0.3ml)和三氟乙酸(306μl,4.0mmol,5.0当量)。允许反应搅拌持续3小时,之后将混合物用水(0.5ml)猝灭,用et2o(3x5ml)萃取,并且将合并的有机级分用盐水(5ml)洗涤。将溶剂除去以给出作为白色固体的8(203.8mg,98%),而不进一步纯化。rf=0.4(在己烷中3%etoac);1hnmr(500mhz,cdcl3)δ7.92(dd,j=7.9,1.3hz,1h),7.83(dd,j=7.1,1.3hz,1h),7.48(d,j=5.5hz,1h),7.38(dd,j=7.9,7.0hz,1h),7.34(d,j=5.5hz,1h),1.41(s,12h);13cnmr(125mhz,cdcl3)δ145.8,139.4,132.0,127.5,126.7,123.7,123.4,84.4,25.1。ir(纯的膜,nacl)2977,1564,1504,1461,1372,1330,1300,1267,1199,1165,1135,1097,1038,969,851,829,801,714,672cm–1;hrms(ei+)对于c14h17bso2[m·+]计算为:260.1042,实测为260.1039。
实施例8.5.硅杂-杂环通过分子间/分子内双c-h甲硅烷基化的合成:9,9-二乙基-9h-苯并[d]吡咯并[1,2-a][1,3]氮杂噻咯9.
反应根据一般程序通过使kot-bu(11.2mg,0.1mmol,20mol%)、1-苯基-1h-吡咯(72.0mg,0.5mmol,1当量)、et2sih2(97μl,0.75mmol,1.5当量)、和0.5ml的thf在35℃下加热持续72h并且然后在65℃下持续72小时来进行。期望的产物9(48.8mg,43%收率)在通过硅胶快速色谱法(100%己烷)纯化之后作为无色针状物获得。rf=0.6(100%己烷);1hnmr(500mhz,cdcl3)δ7.51(ddd,j=7.1,1.4,0.6hz,1h),7.46–7.33(m,2h),7.31(dt,j=7.9,0.7hz,1h),7.09(td,j=7.2,1.0hz,1h),6.52(dd,j=3.3,1.0hz,1h),6.41(dd,j=3.3,2.6hz,1h),1.05–0.96(m,6h),0.96–0.79(m,4h);13cnmr(125mhz,cdcl3)δ148.0,134.1,130.8,129.4,128.5,123.9,117.5,117.1,113.3,111.6,7.5,4.4;ir(纯的膜,nacl)2958,2921,2873,2849,1658,1598,1462,1471,1451,1377,1332,1260,1086,1017,799,755,717cm–1;hrms(fab+)对于c14h18nsi[m+h]+计算为:228.1208,实测为228.1206。此反应产物的hsqc光谱先前在美国专利第9,000,167号中已经被报告。
实施例8.6.三聚噻吩的c-h甲硅烷基化
反应根据一般程序进行。对于条件a:反应用kot-bu(11.2mg,0.1mmol,20mol%)、2,2’:5’2”-三聚噻吩(124mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、和0.5ml的thf在25℃下进行持续40h。产物10(204.7mg,86%收率)和si-10(23.5mg,13%收率)在通过硅胶快速色谱法(100%己烷)纯化之后获得。对于条件b:反应用kot-bu(11.2mg,0.1mmol,20mol%)、2,2’:5’2”-三聚噻吩(124mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)、和0.5ml的thf在45℃下进行持续65h。产物10(228.6mg,96%收率)在通过硅胶快速色谱法(100%己烷)纯化之后获得;si-10通过1hnmr和gc-ms作为痕量产物被观察到,但未分离。
5,5”-双(三乙基甲硅烷基)-2,2’:5’,2”-三聚噻吩10:黄色油,rf=0.5(100%己烷);1hnmr(500mhz,cdcl3)δ7.25(d,j=3.5hz,2h),7.14(d,j=3.5hz,2h),7.10(s,2h),1.03(m,18h),0.82(m,12h)。13cnmr(125mhz,cdcl3)δ142.4,136.7,136.5,135.7,124.9,124.5,7.2,4.4;ir(纯的膜,nacl)3057,2953,2934,2908,2874,1750,1455,1428,1417,1377,1303,1236,1212,1198,1068,988,1009,911,892,792,736,723cm–1;hrms(ei+)对于c24h36s3si2[m·+]计算为:476.1518,实测为476.1534。
[2,2’:5’,2”-三聚噻吩]-5-基三乙基硅烷si-10:黄色油,rf=0.4(100%己烷);1hnmr(500mhz,cdcl3)δ7.24(d,j=3.4hz,1h),7.21(dd,j=5.1,1.2hz,1h),7.17(dd,j=3.6,1.2hz,1h),7.14(dd,j=3.4,1.6hz,1h),7.09(q,j=3.7hz,2h),7.02(dd,j=5.1,3.6hz,1h),1.07–0.98(m,9h),0.82(qd,j=7.8,0.9hz,6h);13cnmr(125mhz,cdcl3)δ142.3,137.5,136.8,136.6,136.4,135.6,128.0,125.0,124.6,124.5,124.5,123.8,7.5,4.6;ir(纯的膜,nacl)3068,2953,2873,1458,1425,1377,1235,1195,1069,1011,989,913,865,836,793,737cm–1;hrms(fab+)对于c18h23s3si[m+h]+计算为:363.0731,实测为363.0742。
实施例8.7.edot的c-h甲硅烷基化:(2,3-二氢噻吩并[3,4-b][1,4]二氧芑-5-基)三乙基硅烷11。
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、edot(2,3-二氢噻吩并[3,4-b][1,4]二氧芑,71.1mg,0.5mmol)、et3sih(240μl,1.5mmol,3当量)、以及0.5ml的thf加热持续72h来进行。期望的产物11(79.3mg,62%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中0%→5%etoac)纯化之后作为混浊的(cloudy)黄色油获得。rf=0.3(100%己烷);1hnmr(500mhz,cdcl3)δ6.56(s,2h),4.17(s,4h),0.98(td,j=7.8,0.8hz,9h),0.84–0.74(m,6h);13cnmr(125mhz,cdcl3)δ147.5,142.5,108.7,105.0,64.5,64.5,7.4,3.9;ir(纯的膜nacl)2952,2873,1468,1440,1422,1361,1244,1181,1151,1072,1042,1009,899,721cm–1;hrms(ei+)对于c12h21o2ssi[m+h]+计算为:257.1032,实测为257.1064。
实施例8.8.活性药物成分(api)的后期甲硅烷基化
实施例8.8.1.1-甲基-n-苯基-n-((5-(三乙基甲硅烷基)噻吩-2-基)甲基)哌啶-4-胺12:
反应根据一般程序通过在45℃下将kot-bu(2.2mg,0.02mmol,20mol%)、西那利定(28.2mg,0.1mmol,1当量)、et3sih(48μl,0.3mmol,3当量)、和0.1ml的thf加热持续72h来进行。期望的产物12(24.9mg,62%收率)在通过硅胶快速色谱法(己烷:etoac:et3n=100:100:1)纯化之后作为无色油获得。rf=0.2(己烷:etoac:et3n=20:20:1);1hnmr(500mhz,cdcl3)δ7.24–7.17(m,2h),7.05(d,j=3.4hz,1h),6.97(d,j=3.3hz,1h),6.82(dt,j=7.8,1.0hz,2h),6.72(tt,j=7.2,1.0hz,1h),4.62(s,2h),3.70(tt,j=11.6,4.0hz,1h),2.96–2.92(m,2h),2.30(s,3h),2.07(td,j=11.9,2.5hz,2h),1.93–1.85(m,2h),1.85–1.73(m,2h),0.97(t,j=7.9hz,9h),0.76(q,j=7.8hz,6h);13cnmr(125mhz,cdcl3)δ151.0,149.0,135.2,134.7,129.3,125.3,117.3,113.8,55.8,55.6,46.4,46.0,29.6,7.5,4.6。ir(纯的膜,nacl)2951,2873,2780,2734,1597,1574,1503,1459,1377,1352,1278,1237,1207,1131,1068,1008,987,850,802,745cm–1;hrms(mm:esi-apci+)对于c23h37n2ssi[m+h]+计算为:401.2441,实测为401.2460。
实施例8.8.2.5-(2-氯苄基)-2-(三乙基甲硅烷基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶13a:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、噻氯匹定(132.5mg,0.5mmol,1当量)、et3sih(243μl,1.5mmol,3当量)和0.5ml的thf加热持续48h来进行。
期望的产物13a(107.7mg,57%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中5%→10%et2o)纯化之后作为无色油获得。rf=0.4(在己烷中10%et2o);1hnmr(500mhz,cdcl3)δ7.56(dd,j=7.5,1.8hz,1h),7.37(dd,j=7.8,1.5hz,1h),7.25(td,j=7.4,1.5hz,1h),7.20(td,j=7.6,1.9hz,1h),6.86(s,1h),3.84(s,2h),3.67(d,j=1.6hz,2h),2.94(t,j=5.9hz,2h),2.87(t,j=5.4hz,2h),1.02–0.98(m,9h),0.80–0.74(m,6h);13cnmr(125mhz,cdcl3)δ139.2,136.5,135.6,134.4,134.0,133.2,130.8,129.6,128.3,126.8,58.7,53.3,51.0,26.1,7.5,4.6。ir(纯的膜,nacl)2952,2908,2873,2805,2763,1462,1443,1413,1375,1360,1347,1303,1289,1234,1169,1125,1106,1047,1032,1018,991,907,835,752cm–1;hrms(mm:esi-apci+)对于c20h29clnssi[m+h]+计算为:378.1473,实测为378.1480。
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、噻氯匹定(134.5mg,0.5mmol,1当量)、et2sih2(194μl,1.5mmol,3当量)和0.5ml的thf加热持续108h来进行。产物13b(97.9mg,56%收率)和si-13b(27.3mg,18%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中5%→50%et2o)纯化之后获得。
5-(2-氯苄基)-2-(二乙基甲硅烷基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶13b:无色油,rf=0.4(在己烷中10%et2o);1hnmr(500mhz,cdcl3)δ7.56(dd,j=7.6,1.8hz,1h),7.38(dd,j=7.8,1.4hz,1h),7.26(td,j=7.4,1.5hz,1h),7.21(td,j=7.6,1.9hz,1h),6.93(s,1h),4.30(p,j=3.2hz,1h),3.84(s,2h),3.67(t,j=1.7hz,2h),2.96–2.94(m,2h),2.88–2.85(m,2h),1.05(t,j=7.8hz,6h),0.83(qd,,j=7.5,3.3hz,4h);13cnmr(125mhz,cdcl3)δ140.0,136.4,135.9,134.4,134.2,131.3,130.8,129.6,128.3,126.8,58.6,53.2,50.9,26.1,8.1,4.5。ir(纯的膜,nacl)2953,2909,2872,2805,2112,1456,1447,1361,1348,1303,1290,1231,1169,1125,1106,1048,1033,1009,992,907,810,752cm–1;hrms(mm:esi-apci+)对于c18h25clnssi[m+h]+计算为:350.1160,实测为350.1155。
双(5-(2-氯苄基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶-2-基)二乙基硅烷si-13b:无色油,rf=0.3(在己烷中50%et2o);1hnmr(500mhz,cdcl3)δ7.55(dd,j=7.6,1.8hz,2h),7.37(dd,j=7.8,1.5hz,2h),7.25(td,j=7.4,1.5hz,2h),7.20(td,j=7.6,1.9hz,2h),6.92(s,2h),3.83(s,4h),3.65(t,j=3.3hz,4h),2.94(t,j=5.4hz,4h),2.86(t,j=5.6hz,4h),1.09–0.95(m,10h);13cnmr(125mhz,cdcl3)δ140.2,136.4,135.8,134.53,134.45,132.4,130.9,129.6,128.3,126.8,58.7,53.2,50.9,26.1,7.5,6.5。ir(纯的膜,nacl)3059,2953,2913,2868,2806,1471,1453,1446,1361,1289,1125,1105,1033,989,907,839,805,753cm–1;hrms(mm:esi-apci+)对于c32h37cl2n2s2si[m+h]+计算为:611.1539,实测为611.1523。
实施例8.8.3.5-(2-氯苄基)-2-(二甲基(苯基)甲硅烷基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶13c:
反应根据一般程序通过在45℃下将kot-bu(11.2mg,0.1mmol,20mol%)、噻氯匹定(134.5mg,0.5mmol,1当量)、phme2sih(230μl,1.5mmol,3当量)、和0.5ml的thf加热持续108h来进行。产物13c(135.4mg,68%收率)在通过硅胶快速色谱法(在己烷中3%et2o)纯化之后作为无色油获得。rf=0.3(在己烷中10%et2o);1hnmr(500mhz,cdcl3)δ7.58–7.52(m,3h),7.39–7.34(m,4h),7.25–7.18(m,2h),6.87(s,1h),3.82(s,2h),3.64(t,j=1.7hz,2h),2.95–2.92(m,2h),2.88–2.84(m,2h),0.56(s,6h);13cnmr(125mhz,cdcl3)δ140.1,138.2,136.4,135.9,135.2,134.4,134.1,133.9,130.8,129.6,129.4,128.3,128.0,126.8,58.6,53.2,50.9,26.1,-1.1。ir(纯的膜,nacl)3067,2953,2918,2806,2764,1652,1471,1446,1427,1361,1248,1169,1109,1033,990,907,832,810,777,753cm–1;hrms(mm:esi-apci+)对于c22h25nsi[m+h]+计算为:398.1160,实测为398.1152。
实施例8.8.4.5-(吡啶-2-基甲基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶sm-14
向火焰干燥的50ml舒伦克瓶添加4,5,6,7-四氢噻吩并[3,2-c]吡啶hcl盐(1.0g,5.7mmol)、2-(溴甲基)吡啶hbr盐(2.18g,8.6mmol,1.5当量)、bu4nhso4(0.20g,0.6mmol,10mol%)、k2co3(3.94g,28.5mmol,5当量)、和10ml的乙腈。将烧瓶用氩气吹扫,并且将反应在70℃下搅拌持续18h。期望的产物sm-14(346.5mg,26%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中50%→100%et2o)纯化之后作为黄色油获得。rf=0.1(在己烷中50%et2o)。1hnmr(500mhz,cdcl3)δ8.58(ddd,j=4.9,1.8,0.9hz,1h),7.67(td,j=7.6,1.8hz,1h),7.51(dt,j=7.9,1.0hz,1h),7.19(ddd,j=7.5,4.8,1.2hz,1h),7.07(dt,j=5.1,0.7hz,1h),6.70(d,j=5.1hz,1h),3.89(s,2h),3.64(t,j=1.7hz,2h),2.96–2.83(m,4h);13cnmr(126mhz,cdcl3)δ158.79,149.20,136.52,133.78,133.36,125.22,123.13,122.63,122.13,63.82,53.22,50.89,25.50;ir(纯的膜,nacl)3403,3062,2918,2813,1648,1588,1569,1473,1431,1356,1320,1236,1167,1109,1053,1015,993,905,840,809,761cm–1;hrms(ei+)对于c13h13sn2[(m+h)-h2]+计算为:229.0799,实测为229.0806。
实施例8.8.5.5-(吡啶-2-基甲基)-2-(三乙基甲硅烷基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶14:
反应根据一般程序通过在45℃下将kot-bu(4.5mg,0.04mmol,20mol%)、5-(吡啶-2-基甲基)-4,5,6,7-四氢噻吩并[3,2-c]吡啶sm-14(46.1mg,0.2mmol)、et3sih(96μl,0.6mmol,3当量)、以及0.2ml的thf加热持续72h来进行。期望的产物14(49.1mg,71%收率)在通过硅胶快速色谱法(梯度洗脱,在己烷中75%→100%et2o)纯化之后作为无色油获得。rf=0.5(在己烷中75%et2o);1hnmr(500mhz,cdcl3)δ8.56(ddd,j=4.9,1.8,0.9hz,1h),7.66(td,j=7.7,1.8hz,1h),7.50(dt,j=7.8,1.0hz,1h),7.17(ddd,j=7.5,4.9,1.2hz,1h),6.83(s,1h),3.87(s,2h),3.64(t,j=1.6hz,2h),2.94(tt,j=5.3,1.5hz,2h),2.86(dd,j=5.9,5.0hz,2h),0.97(t,j=7.9hz,9h),0.74(qd,j=7.7,0.8hz,6h);13cnmr(126mhz,cdcl3)δ158.9,149.1,138.9,136.5,135.3,133.8,133.0,123.1,122.1,63.9,53.2,50.9,25.8,7.4,4.4;ir(纯的膜,nacl)3048,2951,2873,2806,1588,1569,1448,1430,1361,1289,1235,1169,1114,1031,1005,992,908,835,757,735,718cm–1;hrms(ei+)对于c19h29n2ssi[m+h]+计算为:345.1821,实测为345.1835。
实施例9.通过氢氧化钾(koh)催化的甲硅烷基化的选定的实施例
与早期发现相反的是,现在已经发现,koh对于杂芳族物质与氢硅烷在某些条件下的直接甲硅烷基化可以是有效的催化剂。现在看来,通过修改反应条件,此催化剂体系可以与以下情况中的每种基质一起使用:其中叔丁醇钾(或其他强碱)先前被示出是有效的,如在两个均于2013年10月2日提交的美国专利申请序列号14/043,929和国际申请号pct/us2013/062963中所描述的以及如在本说明书中所描述的。然而,koh的使用提供重要的实际益处,例如较低的成本和毒性,并且有利的反应建立和纯化。另外,注意,条件的略微的变化可以可再现地改变取代的程度(参见,例如图7,其中呋喃和联噻吩的操作温度的变化允许选择性的单甲硅烷基取代(>10:1单:双,在45℃下;1.2当量硅烷)和双甲硅烷基取代(>10:1双:单,在65℃下;3当量硅烷)。
如在上文实施例2,表1中所示出的,就这一点而言,koh被发现在扫描测试的条件下是完全无反应性的,并且因此被认为在此化学中是完全无活性的。在表1中描述的条件下进行的反应的失败已经被重复和确证:
然而,通过调整反应条件,反应已经被发现在良好的转化率下进行。随着仅温度的稍微增加的反应性的此显著的变化是完全未预料到的。
还参见表6和图5a/b和图6。
此催化剂体系可以在如对于丁醇盐/氢化物体系所描述的相同范围的基质上操作是通过现在发现的以下范围的基质(图7)的可操作性来支持。
如本领域技术人员将明白的,本发明的许多修改和变化形式根据这些教导是可能的,并且据此设想到了所有的此类修改和变化形式。例如,除本文描述的实施方案之外,本发明设想并且要求保护源自本文中引用的本发明的特征的组合的那些发明以及补充本发明的特征的引用的现有技术参考文献的那些。相似地,将明白,任何所描述的材料、特征或制品都可以与任何其他的材料、特征或制品组合使用,并且此类组合被认为是在本发明的范围内。
以下参考文献的某些或全部可以在理解本发明的某些要素或其背景原理方面是有用的。
1.zhang,f.,wu,d.,xu,y.和feng,x.thiophene-basedconjugatedoligomersfororganicsolarcells.j.mater.chem.21,17590–17600(2011)。
2.showell,g.a.和mills,j.s.chemistrychallengesinleadoptimization:siliconisosteresindrugdiscovery.drugdiscov.today8,551–556(2003)。
3.franz,a.k.和wilson,s.o.organosiliconmoleculeswithmedicinalapplications.j.med.chem.56,388-405(2013)。
4.ball,l.t.,lloyd-jones,g.c.和russell,c.a.gold-catalyzeddirectarylation.science337,1644-1648(2012)。
5.denmark,s.e.和baird,j.d.palladium-catalyzedcross-couplingreactionsofsilanolates:aparadigmshiftinsilicon-basedcross-couplingreactions.chem.eur.j.12,4954-4963(2006)。
6.langkopf,e.和schinzer,d.usesofsilicon-containingcompoundsinthesynthesisofnaturalproducts.chem.rev.95,1375-1408(1995)。
7.whisler,m.c.,macneil,s.,snieckus,v.和beak,p.beyondthermodynamicacidity:aperspectiveonthecomplex-inducedproximityeffect(cipe)indeprotonationreactions.angew.chem.int.ed.43,2206-2225(2004)。
8.cheng,c.和hartwig,j.f.rhodium-catalyzedintermolecularc–hsilylationofareneswithhighstericregiocontrol.science343,853–857(2014)。
9.lu,b.和falck,j.r.efficientiridium-catalyzedc–hfunctionalization/silylationofheteroarenes.angew.chem.int.ed.47,7508-7510(2008)。
10.tamao,k.,uchida,m.,izumizawa,t.,furukawa,k.和yamaguchi,s.silolederivativesasefficientelectrontransportingmaterials.j.am.chem.soc.118,11974-11975(1996)。
11.ting,r.,adam,m.j.,ruth,t.j.和perrin,d.m.arylfluoroboratesandalkylfluorosilicatesaspotentialpetimagingagents:high-yieldingaqueousbiomolecular18f-labeling.j.am.chem.soc.127,13094-13095(2005)。
12.du,w.,kaskar,b.,blumbergs,p.,subramanian,p.-k.和curran,d.p.semisynthesisofdb-67andothersilatecansfromcamptothecinbythiol-promotedadditionofsilylradicals.bioorg.med.chem.11,451-458(2003)。
13.furukawa,s.,kobayashi,j.和kawashima,t.developmentofasila-friedel–craftsreactionanditsapplicationtothesynthesisofdibenzosilolederivatives.j.am.chem.soc.131,14192-14193(2009)。
14.curless,l.d.,clark,e.r.,dunsford,j.j.和ingleson,m.j.e–h(e5r3siorh)bondactivationbyb(c6f5)3andheteroarenes;competitivedehydrosilylation,hydrosilylationandhydrogenation.chem.commun.50,5270-5272(2014)。
15.klare,h.f.t.等人cooperativecatalyticactivationofsi–hbondsbyapolarru–sbond:regioselectivelow-temperaturec–hsilylationofindolesunderneutralconditionsbyafriedel-craftsmechanism.j.am.chem.soc.133,3312-3315(2011)。
16.seregin,i.v.和gevorgyan,v.directtransitionmetal-catalyzedfunctionalizationofheteroaromaticcompounds.chem.soc.rev.36,1173–1193(2007)。
17.fedorov,a.,toutov,a.a.,swisher,n.a.和grubbs,r.h.lewis-basesilaneactivation:fromreductivecleavageofaryletherstoselectiveortho-silylation.chem.sci.4,1640-1645(2013)。
18.weickgenannt,a.和oestreich,m.potassiumtert-butoxide-catalyzeddehydrogenativesi–ocoupling:reactivitypatternandmechanismofanunderappreciatedalcoholprotection.chem.asianj.4,406-410(2009)。
19.song,j.j.等人organometallicmethodsforthesynthesisandfunctionalizationofazaindoles.chem.soc.rev.36,1120–1132(2007)。
20.li,c.-j.和trost,b.m.greenchemistryforchemicalsynthesis.proc.natlacad.sci.usa105,13197–13202(2008)。
21.collins,k.d.和glorius,f.arobustnessscreenfortherapidassessmentofchemicalreactions.naturechem.5,597-601(2013)。
22.seiple,i.b.等人directc2harylationofelectron-deficientheterocycleswitharylboronicacids.j.am.chem.soc.132,13194-13196(2010)。
23.zhao,z.和snieckus,v.directedorthometalation-basedmethodology.halo-,nitroso-,andboro-inducedipso-desilylation.linktoaninsitusuzukireaction.org.lett.7,2523-2526(2005)。
24.lee,m.,ko,s.和chang,s.highlyselectiveandpracticalhydrolyticoxidationoforganosilanestosilanolscatalyzedbyarutheniumcomplex.j.am.chem.soc.122,12011-12012(2000)。
25.hansen,m.m.等人lithiatedbenzothiophenesandbenzofuransrequire2-silylprotectiontoavoidanionmigration.synlett8,1351–1354(2004)。
26.wang,y.和watson,m.d.transition-metal-freesynthesisofalternatingthiopheneperfluoroarenecopolymers.j.am.chem.soc.128,2536-2537(2006)。
27.kuznetsov,a.,onishi,y.,inamoto,y.和gevorgyan,y.fusedheteroaromaticdihydrosiloles:synthesisanddouble-foldmodification.org.lett.15,2498–2501(2013)。
28.oyamada,j.,nishiura,m.和hou,z.scandium-catalyzedsilylationofaromaticc–hbonds.angew.chem.int.ed.50,10720-10723(2011)。
29.kakiuchi,f.,tsuchiya,k.,matsumoto,m.,mizushima,e.和chatani,n.ru3(co)12-catalyzedsilylationofbenzylicc–hbondsinarylpyridinesandarylpyrazoleswithhydrosilanesviac-hbondcleavage.j.am.chem.soc.126,12792-12793(2004)。
30.sakakura,t.,tokunaga,y.,sodeyama,t.和tanaka,m.catalyticc–hactivation.silylationofareneswithhydrosilaneordisilanebyrhcl(co)(pme3)2underirradiation.chem.lett.16,2375-2378(1987)。
出于所有目的,在本文件中引用的或描述的每个专利、专利申请和出版物据此通过引用各自以其整体并入本文。
- 还没有人留言评论。精彩留言会获得点赞!
- 高氯酸铵内嵌的碳/金属氧化物复合催化剂及其制备方法与制造工艺
- 一种复合氧化物负载杂多酸催化合成尿囊素的方法与制造工艺
- 一种无定形铁锌复合氧化物光芬顿催化剂的制备方法与制造工艺
- 具有杂原子的过渡金属化合物、包含该化合物的催化剂组合物和使用该催化剂组合物的聚合物的制备方法与制造工艺
- 配体化合物、过渡金属化合物和包含该化合物的催化剂组合物的制造方法与工艺
- 一种碳/金属氧化物纳米纤维复合催化剂的制备方法与制造工艺
- 改性无机耐热氧化物及其制备方法及加氢脱硫催化剂和降低加氢脱硫催化剂失活速度的方法与制造工艺
- 一种用于莫来石陶瓷晶须制备的氧化物催化剂的制造方法与工艺
- 一种中空金属蜂窝式氧化催化器的制造方法
- 一种处理汽车尾气的三元金属氧化物催化剂制备方法
- 一种含有多取代基的杂环含硼芳香化合物及其合成方法及用途与流程
- 一种以芳杂环并茚芴单元为核的双极性小分子发光材料及其制备方法与应用与流程
- 芳杂环类衍生物及其在药物中的应用的制作方法与工艺
- 芳杂环类衍生物及其在药物中的应用的制作方法与工艺
- 一种含五元杂环的不对称芳香二硫醚类化合物在制备治疗SARS药物中的应用的制作方法与工艺
- 芳族杂环通过地球丰富的无过渡金属的催化剂的甲硅烷基化的制造方法与工艺
- 芳香族杂环衍生物、使用其的有机电致发光元件、照明装置及显示装置的制造方法
- 4-(芳杂环取代)氨基-1H-3-吡唑甲酰胺类FLT3抑制剂及其用途的制造方法与工艺
- 芳香族杂环衍生物、有机电致发光元件用材料以及有机电致发光元件的制造方法与工艺
- 一种芳杂环衍生物及其制备方法以及一种有机电致发光器件与制造工艺