递送生物分子至免疫细胞的制作方法
相关申请的引用
本申请根据美国法典第35篇第119条(e)款要求2014年10月31日提交的美国临时申请号62/073,548的优先权的权益,所述申请以引用的方式整体并入本文。
联邦政府资助研究的声明
本发明是在由美国国立卫生研究院(nationalinstitutesofhealth)授予的授权号gm101420、ai112521、ai111595和ai069259下由政府资助完成。政府享有本发明的某些权利。
发明领域
本发明涉及递送材料至细胞。
序列表的引用
本申请以引用的方式并有存在于名为“38172-508001wo_sequence_listing.txt”的文件中的核苷酸和/或氨基酸序列,所述文件大小是2.31千字节并且在2015年10月30日以与ms-windows具有操作系统相容性的ibm-pc机器格式创建,所述文件包含在2015年10月30日作为本申请的一部分提交的文本文件中。
发明背景
将大分子(例如多糖、蛋白质或核酸)递送到细胞质可以暂时地或永久地改变细胞功能用于研究或治疗目的。然而,用于细胞内递送到原代免疫细胞(特别是静息淋巴细胞)的现有技术具有局限性。电穿孔导致相当大的细胞毒性。病毒载体不能感染静息淋巴细胞。细胞膜穿透(或转导)肽不能有效地转染原代淋巴细胞。抗体-药物复合物和缀合物需要针对每种细胞类型和不同设计的特异性抗体以携带不同的有效载荷。此外,它们生产成本高且潜在地具有免疫原性。已经显示适体-sirna嵌合rna在体内引起靶向基因敲低且没有任何毒性或免疫激活,但是仅用于递送小rna,并且它们需要鉴定每个目标细胞的特异性靶向适体。基于纳米颗粒和脂质体的技术的进步已经导致了细胞内递送至药物和抗原到吞噬抗原呈递细胞(诸如树突细胞和单核细胞/巨噬细胞)改进,但对淋巴细胞无效。这些方法大多数导致了有效载荷的内体摄取,并且只有非常小比例的有效载荷(估计为约1-2%)从内体逃逸到细胞溶质,在细胞溶质中需要用于生物活性的迁移。这些技术中的许多还导致不可生物降解的包装或递送材料在细胞中的积累,这可能影响细胞功能。因此,需要能够将多种大分子有效且无毒地递送到免疫细胞的替代技术。
发明概述
本发明提供了与将化合物或组合物递送到免疫细胞相关的先前问题的解决方案。在本发明之前,引入化合物(例如蛋白质、核酸、碳水化合物)是困难且无效率的,并且/或者需要存在不需要的介质(诸如毒性化合物或病毒载体)。根据本发明,免疫细胞功能的工程化方法包括通过暂时破裂免疫细胞的细胞质周围的膜来细胞内递送化合物。例如,用于优先递送化合物到免疫细胞的细胞溶质的无病毒载体方法包括使包含靶免疫细胞的细胞悬浮液穿过微流体装置并使所述悬浮液与待递送的化合物或有效载荷接触的步骤。所述装置包括10-60μm的收缩长度和3-8μm(例如3-4μm或4μm)的收缩宽度。
例如,所述装置包括2μm-10μm直径的收缩。在涉及初始t细胞和b细胞的优选实施方案中,所述装置包括具有约10、15、20、25、30或10-30μm的长度,约3、3.5、4或3-4μm的宽度,约15、20、25或15-25μm的深度,和/或约5、6、7、8、9、10、11、12、13、14、15或5-15度角的收缩。
与递送到非免疫细胞的化合物的量或与在不存在细胞挤压(例如仅通过胞吞作用)的情况下递送到免疫细胞的量相比,在穿过所述收缩之后,递送到免疫细胞的化合物的量大至少10%(例如,20%、50%、2倍、5倍、10倍或更大)。例如,免疫细胞包括b细胞、t细胞、巨噬细胞或树突细胞。为了将有效载荷优先递送到免疫细胞,示例性装置的特征在于细胞穿过的一个或多个通道,该通道包括30μm的收缩长度和4μm的收缩宽度。
在细胞处理期间使用0至45摄氏度的温度,例如0-25℃。例如,初始t细胞、b细胞和/或单核细胞的处理在4-8℃的温度下(例如在冰上)进行。在另一个实例中,在20-25℃的温度下(例如在典型的环境室温下)使用所述装置处理树突细胞、激活的t细胞和/或激活的b细胞。
有效载荷含有试图递送到免疫细胞的细胞质的任何分子或化合物。例如,所述化合物包括抗原,例如疾病相关抗原诸如肿瘤抗原、病毒抗原、细菌抗原或真菌抗原。抗原可以被纯化或者在其它组分的混合物中,例如,抗原存在于细胞裂解物中,诸如肿瘤细胞裂解物或来自受试者(例如患有传染病的受试者)的活检感染的组织或受疾病影响的组织的裂解物。在一些实例中,抗原包括整个的全长的(或未加工的)蛋白质抗原,例如超过7、8、9或10个氨基酸长度的蛋白质或肽。其它货物分子包括核酸诸如sirna、mrna、mirna、编码或非编码寡核苷酸以及小分子(例如小分子探针)。核酸(诸如dna),例如表达载体(诸如质粒)也以这种方式递送,而不需要病毒载体。
在一些实例中,与激活状态相比,免疫细胞处于静息状态。例如,细胞的特征在于表达以下标志物:cd25、klrg1、cd80、cd86、pd-1、pdl-1、ctla-4、cd28、cd3、mhc-i、mhc-ii、cd62l、ccr7、cx3cr1和cxcr5,其中每一种都可以被操控(通过使用所述方法将分子引入免疫细胞来增加或减少)。细胞悬浮液包括经加工的细胞,例如重悬的血沉棕黄色层细胞(buffycoatcells)(分级的白细胞)或全血。初始免疫细胞(例如t细胞)的特征在于相对低水平的cd25、cd80、cd86、pd-1和ctla-4的表达以及相对高水平的ccr7(与激活的细胞相比)。
用于优先递送化合物到免疫细胞(与非免疫细胞相比)的装置包括至少一个微流体通道(例如以注射器的形式)或多个通道(例如以微芯片或微流体装置的形式)。例如,所述通道包括30μm的收缩长度和4μm的收缩宽度。
本发明还包括通过细胞内递送化合物来使免疫细胞功能工程化的方法,所述递送通过暂时破裂免疫细胞的细胞质周围的膜并将抗原递送到细胞溶质中来介导。例如,抗原包括大于2、3、4、5、6、7、8、9或10个氨基酸的长度,并且其中免疫细胞将抗原加工成小于11个氨基酸长度的肽。然后细胞在免疫细胞的表面上展示较短的经加工的肽,即抗原的i类组织相容性抗原限制性加工形式。例如,加工用于i类呈递至cd8+t细胞或细胞毒性t细胞的mhc/hla的肽在8-10个残基的范围内,并且加工用于mhc/hlaii类呈递至cd4+t细胞或辅助细胞的肽在14-20个残基的范围内。短于8个残基(例如2、3、4、5、6或7个残基)的肽可用于呈递至其它t细胞类型或nk细胞。
例如,通过使免疫细胞穿过2μm-10μm直径的收缩来破裂细胞膜。通过细胞挤压入细胞溶质来递送的抗原是全长的未加工的蛋白质或肽,其必须被加工成适合结合用于抗原呈递细胞的抗原呈递的组织相容性抗原的大小/长度。使免疫细胞功能工程化的方法还可以包括使负载抗原的免疫细胞与效应t细胞接触并激活细胞毒性t细胞免疫应答。在一些实例中,挤压的负载抗原的免疫细胞包括b细胞、树突细胞或巨噬细胞。在其它实例中,挤压的负载抗原的免疫细胞包括t细胞。在任一种情况下,与在不存在挤压(例如仅通过胞吞作用或胞饮作用摄取)的情况下与相同抗原或有效载荷接触的免疫细胞相比,挤压的负载抗原的免疫细胞包含至少10%、25%、50%、2倍、5倍、10倍、20倍、25倍、50倍或更多抗原或其它有效载荷组合物。
在此种治疗之后,改变免疫细胞的功能、活性或激活状态。例如,赋予t细胞抗原呈递表型的方法由通过使t细胞穿过如上所述的微流体装置将抗原(例如整个的未加工的蛋白质或其片段)递送到t细胞的细胞溶质来进行。例如,所述装置包括约2μm-10μm直径的收缩,并且在穿过微流体装置之后t细胞包含t细胞表面上的抗原的i类组织相容性抗原限制性加工形式。所述方法还可以包括使第一(挤压的,负载抗原的)t细胞与第二t细胞接触,所述第二t细胞包含i类组织相容性抗原限制性细胞毒性t细胞表型。示例性抗原包括一种或多种肿瘤抗原,例如肿瘤抗原(诸如肿瘤活检裂解物或病毒抗原)的混合物。以这种方式产生负载抗原的t细胞在体外和/或体内使用以引发细胞毒性t细胞应答。因此,本发明包括使用细胞挤压的负载抗原的t细胞来激活抗原特异性的细胞毒性t细胞应答。例如,此类t细胞通过根据递送/负载的抗原杀伤肿瘤细胞和/或杀伤病毒感染的细胞来赋予临床益处。
纯化本文所述的组合物。纯化的化合物为目标化合物的至少60重量%(干重)。优选地,制品为目标化合物的至少75重量%,更优选至少90重量%,并且最优选至少99重量%。通过任何合适的标准方法,例如通过柱色谱、聚丙烯酰胺凝胶电泳或hplc分析,来测量纯度。例如,化合物(例如蛋白质抗原)已经与天然存在的一种或多种化合物分离。在细胞的情况下,纯化的群为选择的细胞类型的至少75%、85%、90%、95%、98%、99%或100%。纯化或富集特定细胞类型的方法是熟知的,包括使用装置(诸如细胞分选机)通过大小或细胞表面标志物表达来分开。
根据以下本发明的优选实施方案的描述和权利要求书,本发明的其它特征和优点将显而易见。本文中引用的所有参考文献均以引用的方式并入本文。
附图简述
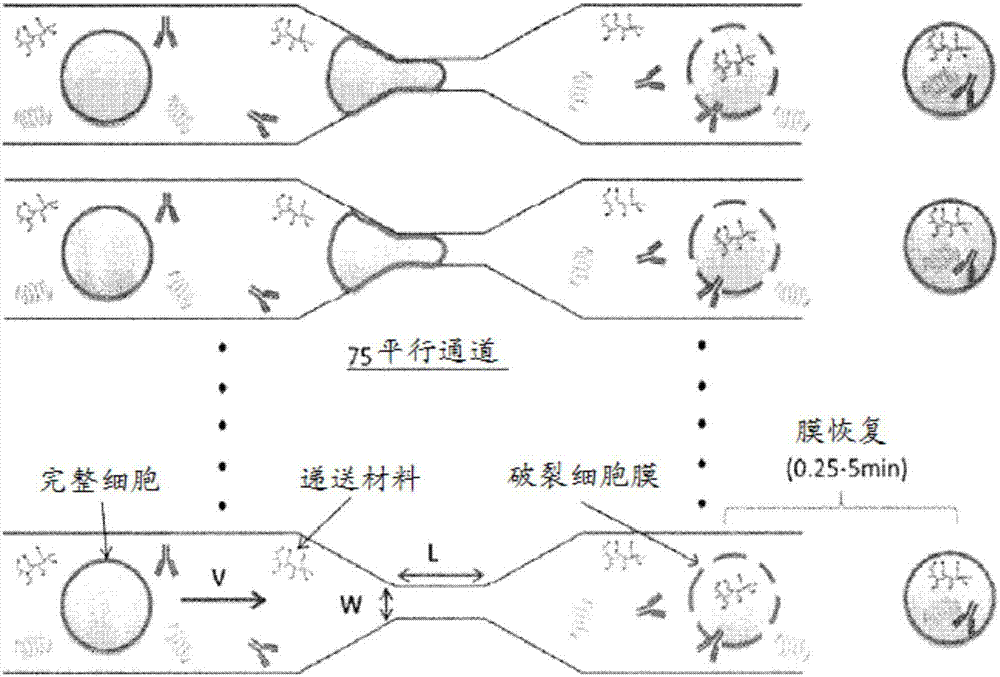
图1a是示出递送系统的一系列图。为了建立所述系统,每个微流体芯片都安装在支架中,这使微流体芯片能与聚碳酸酯流体贮存器相连接。为了操作所述系统,将待递送的大分子与细胞混合,以约30-150μl的体积负载到装置贮存器中,并且然后连接到压力源以诱导流体通过微流体通道。图1b是示出细胞变形和有效载荷递送的图。当细胞流过通道时,它们在收缩处变形,导致膜破裂。然后流体中的大分子通过破裂的膜扩散,并且在细胞膜被修复之后仍然被捕获在细胞中。这些图证明了通过细胞挤压的递送。
图2a和b是示出葡聚糖和抗体递送到鼠免疫细胞的直方图和柱状图。图2a示出了由细胞挤压(cellsqueeze)装置处理以递送apc标记的igg1的t细胞、b细胞和髓样细胞(cd11b+)的代表性直方图。图2b示出了递送效率。所有结果在处理1小时内通过流式细胞术测量。死细胞通过碘化丙啶染色来排除。图2b中的数据(平均值±sd)来自3个独立实验。未处理的细胞未穿过所述装置或暴露于生物分子。将“无装置”样品与生物分子一起孵育,但未被所述装置处理。这种对照意在考虑表面结合、胞吞作用和其它背景效应。
图3a-d是示出葡聚糖、抗体和sirna递送到人免疫细胞的图。在图3a中,测试人t细胞和mddc的瀑布蓝(cascadeblue)标记的3kda葡聚糖、荧光素标记的70kda葡聚糖和apc标记的igg1的递送。展示了30-4(t细胞)和10-7(mddc)装置(左)和跨装置设计的重复(右)的代表性直方图。图3b示出了分别在cd4+t细胞和mddc中sirna介导的cd4和dc-sign蛋白水平的敲低。测试不同的sirna浓度和装置设计以评估敲低对剂量或收缩大小的依赖性。图3c示出了人调节性t细胞也响应于30-4装置的治疗示出了cd4表达的显著敲低。排除死亡细胞用于递送或敲低分析。图3d示出了t细胞的装置性能与amaxa的核转染的比较。对于两种系统,示出了sirna针对cd4的递送后72小时的蛋白质表达。还示出了通过两种方法处理后的细胞活力。
图4a-b是示出通过靶向敲低内源基因和病毒基因来抑制hiv感染的图。在图a中,p24抗原的细胞内染色被用作感染后24小时处理的人cd4+t细胞中hiv感染水平的指标。在这些研究中,vifsirna和/或gagsirna在感染前24小时递送,而cd4sirna在感染前48小时递送。图4n示出了实验条件的重复(最小n=4)中的p24抗原染色的中值荧光强度。数据表示为平均值+1标准误差。
图5是疫苗接种方法的图。
图6是示出3kda和70kda葡聚糖以及鼠原代免疫细胞的抗体的摄取的一系列曲线图。示出用于计算递送效率值的门控。这些数据对应于图2中所呈现的实验。灰色填充的直方图表示未处理的细胞,黑色表示暴露于材料但未被装置处理的细胞,未填充灰色表示在靶生物分子的存在下由装置处理的细胞。
图7是示出对应于图2中所呈现的实验的细胞活力数据的一系列柱状图。***指示当将30-4装置处理的细胞的活力与无装置或未处理的情况进行比较时,p<0.001。用装置处理的b细胞和髓样细胞的活力变化与未处理的或无装置的情况没有显著差异。
图8是示出葡聚糖和抗体递送到骨髓来源的树突细胞(bmdc)的一系列图。通过在含有gm-csf的培养基中培养骨髓细胞8天,从c57bl6小鼠中生成bmdc。使用两种装置设计10-6和30-6递送瀑布蓝标记的3kda葡聚糖、荧光素标记的70kda葡聚糖和apc标记的igg1。
图9是示出抗体与葡聚糖递送的相关性的图。将使用30-4装置(灰点;中心,右上)使葡聚糖(3kda和70kda)和抗体递送至t细胞与用材料孵育(即,无装置(黑点;左下))相比。
图10是示出人cd4+t细胞活力的柱状图。与未处理的对照相比,穿过装置的细胞具有降低的活力,但是比经历核转染的细胞具有更好的活力。使用单因素方差分析和接着的boneferroni检验来计算统计显著性。*指示p<0.05,并且***指示p<0.001。其它比较组没有示出显著不同的活力(即10-4与未处理的或30-4相比,且30-4与核转染相比)。
图11是示出测试不同的装置设计对人mddc的递送(上)和活力(下)的结果的一系列柱状图。使用6种不同的装置设计以及使用amaxa核转染递送瀑布蓝标记的3kda葡聚糖、荧光素标记的70kda葡聚糖和apc标记的igg1同种型对照抗体。处理后立即测量活力和递送结果。
图12是示出通过将alexa488或alexa647标记的sirna和3kda瀑布蓝标记的葡聚糖由10-4i装置同时递送至人cd4t细胞并且由30-5x5i装置递送鼠b细胞的一系列图。数据指示两种材料的递送密切相关。此结果与所提出的扩散递送机制一致,即递送功效主要取决于材料大小而不是化学结构。
图13是示出cd45ra表达的图。针对cd45ra的sirna通过10-4装置递送到人t细胞。在治疗后72小时通过流式细胞术测量敲低。
图14是示出cd4mrna敲低的柱状图(如递送后48小时通过pcr所测量的)。
图15是示出如通过流式细胞术测量的治疗后2周cd4+人t细胞中cd4的表达水平的一系列柱状图。还测量cd3水平作为对照基因。
图16是示出模型货物葡聚糖递送到人类单核细胞的一系列图。单核细胞来源于人血液。在两个不同的操作压力下使用四种不同的装置设计递送瀑布蓝标记的3kda葡聚糖和荧光素标记的70kda葡聚糖。0psi情况对应于仅暴露于葡聚糖但未由装置处理的对照。通过碘化丙啶染色测量活力。
图17是示出葡聚糖递送到人b细胞的一系列图。b细胞来源于人血液。在两个不同的操作压力下使用五种不同的装置设计递送瀑布蓝标记的3kda葡聚糖和荧光素标记的2mda葡聚糖。0psi情况对应于仅暴露于葡聚糖但未由装置处理的对照。通过碘化丙啶染色测量活力。
图18是示出处理后72小时测定的dc-sign的蛋白质水平的柱状图。通过6种不同的装置设计测量蛋白质敲低,并与核转染进行比较。注意,即使在对照sirna递送的情况下,核转染似乎也导致dc-sign的约50%非特异性敲低。这可能指示由于电穿孔处理的脱靶效应,即细胞暴露于电场导致对细胞的损伤,具体地说是对靶蛋白的损伤,这导致在不存在针对蛋白质的sirna的情况下测量的表达水平的降低。这些结果指示,相比与非特异性(脱靶)效应相关的电穿孔/核转染方法,膜变形方法更具有特异性。
图19是示出关于不可渗透的alexa-488标记的10kda葡聚糖染料递送到全血中的细胞的数据的一系列图。将荧光标记的染料与全血混合,并将全血+标记的染料的混合物穿过装置,然后在rbc裂解后通过facs测量到血细胞的递送。结果证明,染料被成功地递送到细胞中,并且已被表征为“细胞不可渗透”的货物化合物使用细胞挤压被有效地递送到免疫细胞。令人惊奇的是递送过程在全血中起作用。全血在没有纯化(例如从红细胞分级或分离外周血单核细胞)的情况下非常难以操控,但本文公开的装置能够很好地将化合物递送到全血中的免疫细胞中。对于非限制性实例,参见图33和34。
图20是微流体膜破裂系统的图。
图21a-b是显示在optimem缓冲液中在120psi下用10-4芯片递送后一天的mrna表达的点图。
图22a和图22b是示出以不同芯片角度在不同压力下的10kdaalexa488标记的葡聚糖的递送的图和柱状图。括号中的数字是收缩角度。芯片角度范围为0-180度,例如11-105度。示意图指示芯片角度。深度参数范围为2μm至1mm,例如约20pm,并且在美国专利公布号20140287509(以引用的方式并入本文)中进一步描述。示例性参数包括对于初始t细胞和b细胞的0-30μm长度/3-4μm宽度/20μm深度/11度角。
图23是示出转录因子递送到nk细胞和pdc的曲线图。在oct4重组蛋白递送到脾脏小鼠nk细胞后4小时测量oct4mrna表达。这些数据指示转录因子是有活性的,并且能够诱导内源性oct4的表达。oct4转录因子是ips生成所需的四个因子(oct4、klf4、cmyc、sox2)之一,并对其自身进行正反馈控制。活性oct4蛋白的递送产生增加的oct4mrna表达。活性转录因子的递送是基于蛋白质的方法的重编程过程中的关键步骤。这些方法具有优于基于病毒和dna的系统的许多优点,因为它们可以使整合的风险最小化。
图24a-b是示出未观察到响应于挤压的检测到的正常dc功能的抑制的曲线图。图24a:通过单独抗原胞吞作用的挤压处理后在lps中培养的脾脏dc(lug/ml)和未处理细胞在上调cd80和cd86表达能力方面没有可检测的差异。未受激的胞吞作用条件和维持在4℃的未处理细胞被用作对照。图24b:将来自cd45同类系小鼠的脾脏dc与lps(1um/ml)注射到c57bl6小鼠的足垫中,并且在注射后18小时在引流淋巴结中回收。检测到淋巴结归巢能力没有显著差异。
图25a-d是示出与其它抗原递送方法相比,使用膜变形装置系统和方法导致更有效的抗原呈递的图。图25a:过继转移的cd8+ot-it细胞响应于皮下dc疫苗接种的体内增殖。相对于允许胞吞抗原处理的bmdc,装置处理的bmdc(0.1mg/m1的ova)证明了显著的t细胞增殖增加(p<0.0001)。图25b:已经与用表达ova的黑色素瘤细胞系(b16f10)的细胞裂解物处理的bmdc共培养的cd8+ot-it细胞的体外增殖。%指示在通过10-6细胞挤压装置处理之前添加到bmdc细胞悬浮液中的细胞裂解材料的分数。在与apc接触之前,用羧基荧光素琥珀酰亚胺酯(cfse)标记cd8t细胞。如果它们增殖,cfse染料分布在子细胞中,并且导致每个细胞的荧光强度较低。未增殖的t细胞保持高的cfse强度。图25c:测量分泌抗原特异性ifnγ的cd8t细胞的分数。在这些实验中,小鼠在不存在过继转移的ot-it细胞的情况下针对ova疫苗接种。通过在疫苗接种后7天分离疫苗接种的小鼠脾脏并用siinfekl肽(ova表位)体外再刺激它们来测量内源性抗原特异性应答。抗原特异性cd8t细胞响应于再刺激而分泌ifnγ。图25d;已经与用两种不同装置设计(0.1mg/m1ova)处理的b细胞共培养的cd8+ot-it细胞的体外增殖。在这些实验中cpg被用作佐剂。
图26是示出体内cfse增殖测定结果的图:此图测量抗原特异性cd8t细胞的增殖。当cd8t细胞在小鼠中被激活并增殖时,它们稀释cfse染料并具有较低的荧光强度。在这种情况下,通过装置或阳性对照处理的供体t细胞在受体小鼠中产生了更大的cd8t细胞的激活和增殖。胞吞作用对照示出了最小效应。
图27是示出小鼠中抗原特异性ot-it细胞响应于用抗原处理的野生型t细胞疫苗接种的增殖的一系列直方图。通过cfse染色测量t细胞增殖应答,所述染色剂随着细胞增殖而稀释。较低的强度指示较大的应答,较高的强度峰值指示无/少应答。每列代表使用来源于同一只小鼠的淋巴结和脾脏的实验的重复。每列实验涉及3只小鼠(总计9只小鼠)。
图28a-b是示出dq-ova+3kda-葡聚糖+t细胞两者门控的直方图。
图29是示出使用小鼠t细胞作为抗原呈递细胞的柱状图。通过细胞挤压处理以负载未加工的卵白蛋白抗原的t细胞与ot-1(siinfekl特异性t细胞系)一起培养,并且评估激活标志物cd25和cd69。
图30是制备和表征挤压介导的作为抗原呈递细胞的b细胞的产生的示例性程序的示意图。
图31是示出细胞挤压的b细胞诱导有效的cd8+t细胞增殖的一系列直方图。
图32是示出细胞挤压的树突细胞(递送ova的)分泌γ干扰素的一系列直方图。
图33示出了10kda材料直接递送到未修饰的全血中的人b细胞和t细胞的数据和动画。
图34是示出全血中的活力和递送效率的图。
详述
使用无载体的微流体递送平台(细胞挤压)将大分子以最小的细胞毒性直接递送到原代免疫细胞(例如小鼠、人)的细胞溶质中。这种方法的基础原理是通过靶细胞的快速机械变形或挤压暂时破裂膜,这允许大分子在流体介质中的扩散而摄取,并且随后进行细胞膜修复(参见例如美国专利公布号20140287509,其以引用的方式并入本文。使用微流体设计库,将测试化合物(诸如葡聚糖聚合物、抗体和小干扰rna(sirna))的摄取递送到原代人和鼠t细胞、b细胞、单核细胞/巨噬细胞和树突细胞+中。结果证明所述平台递送多种不同大小和类型的大分子的效用。材料向具有大范围的细胞直径(约8-30μm)、不同形态、各向异性和膜柔韧性的不同类别的免疫细胞的有效递送需要鉴别不同细胞类型的具体条件。对于细胞挤压,收缩宽度是关键参数,并且其它参数(诸如几何元素、速度、缓冲液和温度)也可能影响货物的递送。用于将货物递送到初始t细胞或b细胞的示例性收缩宽度在3-4μm的范围内;用于递送到激活的t细胞或b细胞的示例性收缩宽度在4-6μm的范围内;并且用于递送到树突细胞的示例性收缩宽度在6-8μm的范围内。
sirna的递送导致稳健的基因敲低。此外,递送抗病毒sirna到cd4+t细胞抑制hiv复制,证明了基于微流体的递送的功能效用。类似地,递送抗原蛋白到树突细胞和b细胞在体外和体内导致抗原特异性cd8+t细胞的更有效的抗原呈递和更大的激活/增殖。通过提供具有最小的活力损失的稳健的细胞内递送的平台,细胞挤压代表了探索和控制免疫细胞功能用于研究和临床应用的灵活且有用的工具。
生物分子(诸如蛋白质和sirna)细胞内递送到原代免疫细胞(特别是静息淋巴细胞)中是困难的。本文所述的无载体微流体平台通过细胞的快速机械变形引起暂时的膜破裂,这导致大分子细胞内递送到免疫细胞(诸如t细胞、b细胞、单核细胞/巨噬细胞和树突细胞)。测试16个微流体装置设计的库的递送葡聚糖聚合物、sirna和抗体到人和鼠免疫细胞的能力。通过测量sirna介导的cd45、dc-sign和cd4蛋白的敲低来验证递送材料的活性。既不需要病毒载体又不需要电场的微流体递送导致与电穿孔相当或更好的递送且更少的细胞毒性。通过在用定向针对病毒vif和gag基因的sirna处理的原代人cd4t细胞中抑制hiv病毒复制来示出所述技术在疾病应用中的效用。因此,无载体微流体递送提供了克服大分子细胞溶质递送到过去难以工程化的细胞(例如,原代免疫细胞)的障碍的一种方法。因此,所述方法和装置可用于免疫细胞功能的工程化。
在某些方面,本公开内容涉及将化合物优先递送到免疫细胞的细胞溶质的方法,所述方法包括使包含免疫细胞的细胞悬浮液穿过微流体装置并使所述悬浮液与化合物接触,其中所述装置包括约2μm、3μm、4μm、5μm、6μm、7μm、8μm、9μm、10μm或2μm-10μm直径的收缩,并且其中递送到免疫细胞的化合物的量比递送到非免疫细胞的量大至少10%。在某些方面,本公开内容涉及将化合物递送到免疫细胞的细胞溶质的方法,所述方法包括使包含免疫细胞的细胞悬浮液穿过微流体装置并使所述悬浮液与化合物接触,其中所述装置包括约2μm、3μm、4μm、5μm、6μm、7μm、8μm、9μm、10μm或2μm-10μm直径的收缩。
术语‘细胞挤压’是指包括使细胞悬浮液穿过包括收缩的微流体装置的方法。在一些实施方案中,细胞悬浮液在穿过微流体装置之前、同时或之后与化合物接触。在一些实施方案中,与在不穿过所述装置的情况下与化合物接触的免疫细胞相比,免疫细胞在穿过所述装置后包含至少10%、25%、50%、2倍、5倍、10倍、20倍、25倍、50倍或更多的化合物。
工程化免疫细胞功能
通过暂时破裂或使膜完整性变形将材料递送到免疫细胞的细胞内空间中,可以查询和表征内部机制,并且它们的功能被操控或改变以用于多种应用。
工程化细胞功能和/或了解细胞内部工作的有效方法是将材料(例如生物活性分子)引入细胞并直接操控细胞内过程。本文所述的方法包括与现有方法或先前方法相比的优点,其主要集中在操控细胞所在的介质的内容物和/或通过结合表面受体进行信号传导。细胞内递送到免疫细胞是使用现有技术或先前技术的重大挑战。本文所述的细胞挤压平台将不同的材料递送到免疫细胞中,并已经证明了在体内和体外影响细胞功能的能力。这些方法可用于编程(或重编程)用于临床使用(例如过继转移疗法)的免疫细胞功能。此外,所述方法可用于测试和阐明免疫学机制以鉴定药物靶标和/或诊断。
本发明的方面涉及令人惊奇的发现,当化合物保持在全血中时,它们可以被递送到免疫细胞(诸如人b细胞和t细胞)。全血在没有纯化(例如从红细胞分级或分离外周血单核细胞)的情况下难以操控。然而,本文公开的装置和方法将化合物递送到全血中的免疫细胞内。本发明能够在不需要在使不均匀混合物(免疫细胞、红细胞、血浆/血清)穿过细胞挤压装置之前从全血中分离免疫细胞的情况下,将化合物递送到免疫细胞。这种值得注意的结果具有重要的技术益处,和在一些情况下使患者能够床边治疗以及在无法使用细胞分级(例如战场)的情况下加工细胞的能力。例如,受试者可以将全血除去,通过本发明的装置进行加工,然后(例如在连续过程中)再灌注。由于不需要免疫细胞的分离或富集,因此需要更少的细胞操控,并且不需要使用介质(诸如人造介质)来加工细胞。另外,可以高效率地进行处理全血中的免疫细胞,同时保持高水平的活力。参见,例如图33和34。
在可以与先前实施方案组合的一些实施方案中,细胞悬浮液包含哺乳动物细胞。在一些实施方案中,细胞悬浮液包含混合的细胞群。在一些实施方案中,细胞悬浮液为全血。在一些实施方案中,细胞悬浮液包含血沉棕黄色层细胞。在一些实施方案中,细胞悬浮液为淋巴液。在一些实施方案中,细胞悬浮液包含外周血单核细胞。在一些实施方案中,细胞悬浮液包含纯化的细胞群。在一些方面,所述细胞为原代细胞或细胞系细胞。在一些实施方案中,所述细胞为血细胞。在一些实施方案中,所述血细胞为免疫细胞。在一些实施方案中,所述免疫细胞为淋巴细胞。在一些实施方案中,免疫细胞为t细胞、b细胞、自然杀伤(nk)细胞、树突细胞(dc)、nkt细胞、肥大细胞、单核细胞、巨噬细胞、嗜碱性粒细胞、嗜酸性粒细胞或嗜中性粒细胞。在一些实施方案中,所述免疫细胞为适应性免疫细胞,诸如t细胞和b细胞。在一些实施方案中,所述免疫细胞为先天免疫细胞。示例性先天免疫细胞包括先天淋巴样细胞(ilc1、ilc2、ilc3)、嗜碱性粒细胞、嗜酸性粒细胞、肥大细胞、nk细胞、嗜中性粒细胞和单核细胞。在一些实施方案中,所述免疫细胞为记忆细胞。在一些实施方案中,所述免疫细胞为原代人t细胞。在一些实施方案中,所述细胞是小鼠、狗、猫、马、大鼠、山羊、猴或兔细胞。在一些实施方案中,所述细胞为人细胞。在一些实施方案中,所述细胞悬浮液包含非哺乳动物细胞。在一些实施方案中,所述细胞为鸡、青蛙、昆虫或线虫细胞。
在一些实例中,与激活状态相比免疫细胞处于静息状态,例如,与静息状态下相同表型的细胞相比,激活状态的细胞通常包含较大的直径。例如,细胞的特征在于表达以下标志物:cd25、klrg1、cd80、cd86、pd-1、pdl-1、ctla-4、cd28、cd3、mhc-i、mhc-ii、cd62l、ccr7、cx3cr1和cxcr5,其中每一种都可以被调控(通过使用所述方法将分子引入免疫细胞来增加或减少)。在一些实施方案中,通过将化合物递送到免疫细胞中,免疫细胞上的一种或多种标志物的表达增加。在一些实施方案中,通过将化合物递送到免疫细胞中,免疫细胞上的一种或多种标志物的表达减少。在一些实施方案中,通过将化合物递送到免疫细胞中,免疫细胞上的一种或多种标志物的表达增加,并且一种或多种标志物的表达减少。在一些实施方案中,所述免疫细胞为初始免疫细胞。初始免疫细胞(例如t细胞)的特征在于与激活的免疫细胞的表达水平相比相对低水平的cd25、cd80、cd86、pd-1和ctla-4的表达以及相对高水平的ccr7(与激活的细胞相比)。
本主题的方面涉及主要组织相容性复合物(mhc)。mhc的主要功能是结合来源于病原体的肽片段,并将它们展示在细胞表面上以由适当的t细胞识别。在人类中,mhc也被称为人白细胞抗原(hla)。对应于mhci类(hla-a、hla-b和hla-c)的hla呈递来自细胞内的肽。例如,如果细胞被病毒感染,则hla系统将病毒的片段带到细胞的表面,使得细胞可以被免疫系统破坏。这些肽由蛋白酶体中分解的消化蛋白产生。一般来讲,并且关于mhc-1,这些特定的肽是长度约8-10个氨基酸的小聚合物。由mhci类呈递的外来抗原引发破坏细胞的杀伤t细胞(也被称为cd8阳性细胞或细胞毒性t细胞)。对应于mhcii类(hla-dp、hla-dm、hla-doa、hla-dob、hla-dq和hla-dr)的hla将抗原从细胞外呈递到t淋巴细胞。这些特定抗原刺激辅助t细胞的增殖,这进而刺激产生抗体的b细胞以产生针对此特异性抗原的抗体。自身抗原被调节性t细胞抑制。
在某些方面,本公开涉及用于将化合物或组合物递送到细胞中的方法。在一些实施方案中,所述化合物为单一化合物。在一些实施方案中,所述化合物为化合物的混合物。在一些实施方案中,所述化合物包括核酸。在一些实施方案中,所述化合物为核酸。示例性核酸包括但不限于重组核酸、dna、重组dna、cdna、基因组dna、rna、sirna、mrna、sarna、mirna、lncrna、trna和shrna。在一些实施方案中,所述核酸与细胞中的核酸同源。在一些实施方案中,所述核酸与细胞中的核酸异源。在一些实施方案中,所述化合物为质粒。在一些实施方案中,所述核酸为治疗性核酸。在一些实施方案中,所述核酸编码治疗性多肽。
在一些实施方案中,所述核酸编码报告标志物或选择性标志物。示例性报告标志物包括但不限于绿色荧光蛋白(gfp)、红色荧光蛋白(rfp)、水母素、β-半乳糖苷酶、尿卟啉原(uroporphyrinogen/urogen)iii甲基转移酶(umt)和荧光素酶。示例性选择性标志物包括但不限于杀稻瘟菌素、g418/遗传霉素、潮霉素b、嘌呤霉素、博莱霉素、腺嘌呤磷酸核糖转移酶和胸苷激酶。在一些实施方案中,所述化合物是编码mhc复合物的核酸。在一些实施方案中,所述化合物是编码mhci类复合物或mhcii类复合物的核酸。在一些实施方案中,所述核酸编码嵌合抗原受体,诸如嵌合t细胞受体。在一些实施方案中,所述核酸编码重组t细胞受体。例如,编码嵌合抗原受体的核酸以无病毒的方式(即通过细胞挤压)引入到t细胞中,以维持car-t的表达。例如,在不使用病毒颗粒的情况下完成dna的引入。然而,核酸构建体可以包括病毒基因组元件,其可以帮助整合或作为染色体外核酸维持。
在一些实施方案中,所述化合物包括蛋白质或多肽。在一些实施方案中,所述化合物为蛋白质或多肽。在一些实施方案中,所述蛋白质或多肽为治疗性蛋白质、抗体、融合蛋白、抗原、合成蛋白质、报告标志物或选择性标志物。在一些实施方案中,所述蛋白质为基因编辑蛋白或核酸酶诸如锌指核酸酶(zfn)、转录激活因子样效应物核酸酶(talen)、大核酸酶或cre重组酶。在一些实施方案中,所述融合蛋白可以包括但不限于嵌合蛋白药物(诸如抗体药物缀合物)或重组融合蛋白(诸如用gst或链霉亲和素示踪的蛋白质)。在一些实施方案中,所述化合物为转录因子。示例性转录因子包括但不限于oct5、sox2、c-myc、klf-4、t-bet、gata3、foxp3和rorγt。在一些实施方案中,所述核酸为转座子。转座子或转座元件是将其自身插入到基因组内的另一位置的dna片段。
在一些实施方案中,所述化合物包括抗原。在一些实施方案中,所述化合物为抗原。抗原是刺激特异性免疫应答(诸如细胞或抗体介导的免疫应答)的物质。抗原与由免疫细胞表达的受体(诸如t细胞受体(tcr))结合,所述受体对特定抗原或抗原呈递分子(诸如mhc/hla异二聚体)具有特异性。抗原-受体结合随后触发导致下游免疫效应途径(诸如细胞激活、细胞因子产生、细胞迁移、细胞毒性因子分泌和抗体产生)的细胞内信号传导途径。在一些实施方案中,所述化合物包括疾病相关的抗原。在一些实施方案中,抗原来源于外来来源,诸如细菌、真菌、病毒或过敏原。在一些实施方案中,抗原来源于内部来源,诸如肿瘤细胞或自身蛋白(即自身抗原)。在一些实施方案中,所述肿瘤抗原在肿瘤裂解物中。自身抗原是生物体自身细胞上存在的抗原。自身抗原通常不会刺激免疫应答,但在自身免疫疾病诸如i型糖尿病或类风湿关节炎、多发性硬化症(和其它脱髓鞘病症)的情况下可能会刺激免疫应答。在一些实施方案中,所述抗原为新抗原。新抗原是正常人类基因组中不存在但是由于导致新型蛋白质序列形成的肿瘤特异性dna修饰而在致癌细胞内产生的抗原。示例性病毒抗原包括hiv抗原、埃博拉(ebola)抗原、hpv抗原和ebv抗原,这些抗原作为混合物被纯化或递送或者作为杀灭的或减毒的病毒或病毒片段被递送。在一些实施方案中,hpv抗原来源于hpv16的致癌基因e6和e7。在一些实施方案中,所述化合物包括来自感染未知病原体的组织的细胞裂解物。在一些实施方案中,所述抗原为非蛋白质抗原,诸如脂质、糖脂或多糖。
在一些实施方案中,所述蛋白质或多肽为报告标志物或选择性标志物。示例性报告标志物包括但不限于绿色荧光蛋白(gfp)、红色荧光蛋白(rfp)、水母素、β-半乳糖苷酶、尿卟啉原(uroporphyrinogen/urogen)iii甲基转移酶(umt)和荧光素酶。示例性选择性标志物包括但不限于杀稻瘟菌素、g418/遗传霉素、潮霉素b、嘌呤霉素、博莱霉素、腺嘌呤磷酸核糖转移酶和胸苷激酶。
在一些实施方案中,所述化合物包括小分子。在一些实施方案中,所述化合物为小分子。示例性的小分子包括但不限于荧光标志物、染料、药剂、代谢物或放射性核素。在一些实施方案中,所述药剂是治疗性药物和/或细胞毒性剂。在一些实施方案中,所述化合物包括纳米颗粒。纳米颗粒的实例包括金纳米颗粒、量子点、碳纳米管、纳米壳、树枝状大分子和脂质体。在一些实施方案中,所述纳米颗粒含有治疗性分子或与治疗性分子连接(共价或非共价)。在一些实施方案中,所述纳米颗粒含有核酸,诸如mrna或cdna。在一些实施方案中,所述纳米颗粒含有标记,诸如荧光标记或放射性标记。
本发明的方面涉及改进的细胞内抗体的递送。细胞内抗体的非限制性实例描述于1999年12月21日授予的美国专利号6,004,940;2001年12月11日授予的美国专利号6,329,173;2010年6月10日公布的美国专利公布号2010/0143371;和2006年2月16日公布的美国专利公布号2006/0034834中,所述专利各自的内容以引用的方式并入本文。影响细胞内抗体的有用性的限制因素是在目标细胞(包括全血中的细胞)中表达它们。本发明克服了这个限制因素,并使分离的抗体或编码抗体的构建体能够被递送到免疫细胞的细胞溶质中。
本发明不仅包括完整单克隆抗体的递送,还包括免疫活性抗体片段的递送,例如fab或(fab)2片段;工程化的单链fv分子;或嵌合分子,例如含有一种抗体(例如鼠来源)和另一种抗体(例如人源)的其余部分的结合特异性的抗体。在另一个实例中,所述嵌合分子是来源于单克隆抗体的单链可变片段(scfv)融合到cd3-ζ跨膜结构域和内结构域的融合物。此类分子响应于其靶标的scfv的识别而导致ζ信号的传输。免疫球蛋白重链和轻链的可变部分通过柔性接头融合以形成scfv。此scfv之前是信号肽,以将新生蛋白质定向到内质网和随后的表面表达。柔性间隔区允许scfv在不同方向上取向以使抗原能够结合。跨膜结构域是典型的疏水性α螺旋,通常来源于信号传导内结构域的原始分子,所述信号传导内结构域突出到细胞中并传输所需的信号。使用本文所述的微流体挤压方法将此类嵌合抗原受体递送到t细胞。
在一些实施方案中,所述化合物包括嵌合抗原受体(car)。在一些实施方案中,所述化合物为嵌合抗原受体(car)。在一些实施方案中,car是细胞外识别结构域(例如,抗原结合结构域)、跨膜结构域和一个或多个细胞内信号传导结构域的融合物。在抗原接合时,car的细胞内信号传导部分可以在免疫细胞中引发激活相关的应答,诸如细胞因子或细胞溶解分子的释放。在一些实施方案中,car为嵌合的t细胞抗原受体。在一些实施方案中,car含有对肿瘤抗原具有特异性的抗原结合结构域。在一些实施方案中,所述car抗原结合结构域是单链抗体可变片段(scfv)。在一些实施方案中,所述化合物增强t细胞功能。在一些实施方案中,增强t细胞功能的所述化合物为免疫检查点途径抑制剂。示例性免疫检查点途径抑制剂包括但不限于程序性死亡-1途径抑制剂、程序性死亡配体-1途径抑制剂和抗细胞毒性t淋巴细胞抗原4途径抑制剂。例如,免疫检查点途径抑制剂可以靶向shp2,即涉及pd-1和ctla-4信号传导的酪氨酸磷酸酶。
在一些实施方案中,所述化合物包括荧光示踪的分子。在一些实施方案中,所述化合物为荧光标记的分子,诸如用荧光染料(诸如太平洋蓝、alexa288、cy5或瀑布蓝)示踪的分子。在一些实施方案中,所述化合物为放射性核素、葡聚糖颗粒、磁珠或不可渗透的染料。在一些实施方案中,所述化合物为用pacblue标记的3kda葡聚糖颗粒。在一些实施方案中,所述化合物为用alexa488标记的10kda葡聚糖颗粒。在一些实施方案中,所述化合物为小分子荧光团示踪的蛋白质。在一些实施方案中,所述化合物为用alexa647示踪的小分子。在一些实施方案中,所述化合物包括病毒或病毒样颗粒。在一些实施方案中,所述病毒为治疗性病毒。在一些实施方案中,所述病毒为溶瘤病毒。在一些实施方案中,所述病毒或病毒样颗粒含有编码治疗性分子(诸如治疗性多肽)的核酸。
在一些实施方案中,所述化合物包括耐受原因子。在一些实施方案中,所述化合物包括佐剂。在一些实施方案中,所述化合物包括分化因子。待递送到t细胞的细胞溶质以促进t细胞的分化和/或激活/成熟的示例性分化因子包括t盒转录因子t-bet和脱中胚蛋白(eomesodermin/eomes)、nfκb和/或叉头框p3(foxp3)。
用于细胞内递送的示例性化合物和组合物包括:
·核酸,特别是:在化学上、生物学上或以另外方式被修饰的dna(质粒或其它寡聚物)和rna(例如sirna、mrna、trna、sarna、lncrna、mirna、指导rna)。蛋白质:例如抗体、抑制剂、酶(例如激酶)、转录因子、核糖体、抗原、细胞裂解物;
·肽:长的(100-10,000个氨基酸)和短的(1-100个氨基酸)纳米材料:例如基于脂质的纳米颗粒、聚合物纳米颗粒、碳纳米管、量子点、金属纳米颗粒(包括金);
·病毒:病毒(或病毒样)颗粒的细胞质递送产生成功的细胞的基因递送,否则所述细胞对感染有抵抗力。使用无复制能力的病毒表示操控细胞功能的另外的手段;
·其它材料:聚合物、染料、trisnta、小分子药物、佐剂、探针;
·上述任何组合的混合物。
化合物/组合物递送的示例性细胞类型信息(所有适应性免疫细胞和先天免疫细胞)包括:
·所有哺乳动物物种,例如人、小鼠、狗、猫、马、猴
·b细胞(例如初始b细胞、浆母细胞);
·t细胞(例如th1、th17、th2、treg、cd8、cd4、trm、tem、tcm);
·树突细胞(例如pdc、单核细胞衍生的dc、cdc、cd8+dc、cd11b+dc;
·单核细胞、巨噬细胞;
·嗜中性粒细胞、nk细胞、先天淋巴样细胞(ilc1、ilc2、ilc3)、嗜碱性粒细胞、粒细胞和肥大细胞。
·前体细胞(造血干细胞、clp、间充质干细胞)
工程化免疫细胞抗原呈递
本公开的某些方面涉及免疫细胞功能的工程化方法,所述方法包括通过暂时破裂免疫细胞的细胞质周围的膜并将抗原递送到细胞溶质中来细胞内递送化合物。在一些实施方案中,所述抗原包括大于7、8、9或10个氨基酸的长度,并且其中所述免疫细胞加工所述抗原并在所述免疫细胞的表面上展示所述抗原的i类组织相容性抗原限制性加工形式。
本公开的某些方面涉及免疫细胞功能的工程化方法,所述方法包括通过使免疫细胞穿过包括收缩的微流体装置来细胞内递送化合物并使所述免疫细胞与所述化合物接触。在一些实施方案中,所述化合物包括抗原,并且所述免疫细胞加工所述抗原并将所述抗原展示在免疫细胞的表面上。在一些实施方案中,所述免疫细胞在免疫细胞的表面上展示所述抗原的i类组织相容性抗原限制性加工形式。在一些实施方案中,所述免疫细胞在免疫细胞的表面上展示所述抗原的ii类组织相容性抗原限制性加工形式。在一些实施方案中,通过使所述免疫细胞穿过2μm-10μm直径的收缩来破裂细胞膜。在一些实施方案中,所述抗原包括全长未加工的蛋白质。在一些实施方案中,所述免疫细胞与效应t细胞(诸如cd8+t细胞)接触并激活细胞毒性t细胞免疫应答。在一些实施方案中,所述免疫细胞与效应t细胞(诸如cd4+t细胞)接触并激活辅助t细胞免疫应答。在一些实施方案中,免疫细胞与效应t细胞接触并激活耐受原性t细胞免疫应答。在一些实施方案中,所述免疫细胞包括b细胞、树突细胞或巨噬细胞。在一些实施方案中,所述免疫细胞包括t细胞。
本公开的某些方面涉及赋予t细胞抗原呈递表型的方法,所述方法包括通过使t细胞穿过微流体装置将整个的未加工的抗原递送到t细胞的细胞溶质,其中所述装置包括2μm-10μm直径的收缩,并且其中所述t细胞在穿过微流体装置之后包含免疫细胞表面上的抗原的i类组织相容性抗原限制性加工形式。本公开的某些方面涉及赋予t细胞抗原呈递表型的方法,所述方法包括通过使t细胞穿过微流体装置将整个的未加工的抗原递送到t细胞的细胞溶质,其中所述装置包括2μm-10μm直径的收缩,并且其中所述t细胞在穿过微流体装置之后包含免疫细胞表面上的抗原的ii类组织相容性抗原限制性加工形式。在一些实施方案中,所述抗原包括肿瘤抗原或病毒抗原。在一些实施方案中,所述t细胞还与第二t细胞接触,所述第二t细胞包含i类组织相容性抗原限制性细胞毒性t细胞表型。在一些实施方案中,所述t细胞还与第二t细胞接触,所述第二t细胞包含ii类组织相容性抗原限制性辅助t细胞表型。
本公开的某些方面涉及使用细胞挤压的负载抗原的t细胞来激活对抗原具有特异性的细胞毒性t细胞应答。本公开的某些方面涉及使用细胞挤压的负载抗原的t细胞来激活对抗原具有特异性的辅助t细胞应答。本公开的某些方面涉及使用细胞挤压的负载抗原的t细胞来诱导对抗原具有特异性的耐受原性t细胞应答。
工程化免疫细胞归巢
本公开的某些方面涉及赋予免疫细胞的归巢表型的方法,所述方法包括通过使免疫细胞穿过微流体装置将化合物递送到t细胞的细胞溶质,其中所述装置包括2μm-10μm直径的收缩,并且其中所述化合物赋予所述免疫细胞归巢表型的表达。例如,所递送的化合物可以增加趋化因子受体的表达,所述趋化因子受体将归巢定向到特定位点并下调相互影响的趋化因子受体的表达。
在一些实施方案中,所述化合物包括核酸。在一些实施方案中,所述化合物为核酸。示例性核酸包括但不限于重组核酸、dna、重组dna、cdna、基因组dna、rna、sirna、mrna、sarna、mirna、lncrna、trna和shrna。
在一些实施方案中,所述化合物包括蛋白质或多肽。在一些实施方案中,所述化合物为蛋白质或多肽。在一些实施方案中,所述蛋白质为基因编辑蛋白或核酸酶诸如锌指核酸酶(zfn)、转录激活因子样效应物核酸酶(talen)、大核酸酶或cre重组酶。在一些实施方案中,所述化合物为转录因子。示例性转录因子包括但不限于oct5、sox2、c-myc、klf-4、t-bet、gata3、foxp3和rorγt。在一些实施方案中,所述转录因子诱导mhc复合物的细胞表达。
在一些实施方案中,所述化合物包括嵌合抗原受体(car)。在一些实施方案中,所述化合物为嵌合抗原受体(car)。在一些实施方案中,car是细胞外识别结构域(例如,抗原结合结构域)、跨膜结构域和一个或多个细胞内信号传导结构域的融合物。在抗原接合时,car的细胞内信号传导部分可以在免疫细胞中引发激活相关的应答,例如归巢到特定组织或生理位置。在一些实施方案中,car为嵌合的t细胞抗原受体。用于耐受性的工程化免疫细胞
本公开的某些方面涉及赋予免疫细胞的耐受原表型的方法,所述方法包括通过使免疫细胞穿过微流体装置将化合物递送到t细胞的细胞溶质,其中所述装置包括2μm-10μm直径的收缩,并且其中所述化合物诱导免疫细胞分化成具有耐受原表型的细胞。在一些实施方案中,所述化合物包括核酸。在一些实施方案中,所述化合物为核酸。示例性核酸包括但不限于重组核酸、dna、重组dna、cdna、基因组dna、rna、sirna、mrna、sarna、mirna、lncrna、trna和shrna。
本公开的某些方面涉及通过将根据所述方法修饰的免疫细胞引入患者来治疗患者的方法。在一些实施方案中,所述免疫细胞用于免疫抑制疗法。在一些实施方案中,所述细胞从患者分离,根据本文所述的方法修饰,并被引回到所述患者体内。在一些实施方案中,还向所述患者施用免疫检查点抑制剂。
工程化神风(kamikaze)免疫细胞
本公开的某些方面涉及生成神风免疫细胞的方法,所述方法包括通过使免疫细胞穿过微流体装置将自扩增rna递送到t细胞的细胞溶质,其中所述装置包括2μm-10μm直径的收缩,并且其中所述自扩增rna编码所编码蛋白质的连续产生。在一些实施方案中,所述化合物包括核酸。在一些实施方案中,所述化合物为核酸。在一些实施方案中,所述核酸是自扩增rna(sarna)。
筛选用于疫苗开发的抗原
在一些实施方案中,根据本文所述的方法修饰的免疫细胞用来筛选用于疫苗开发的抗原。例如,使用本文所述的挤压方法将肿瘤细胞裂解物递送到抗原呈递细胞、t细胞或b细胞。将apc与来源于患者的t细胞或t细胞克隆/系一起孵育以确定疫苗候选抗原的身份。在另一种方法中,由负载肿瘤裂解物的apc加工和呈递的抗原通过质谱来鉴定。随后将以这种方式鉴定的抗原用于疫苗接种。
免疫细胞迁移
本公开的某些方面涉及确定患者体内的t细胞迁移的方法,所述方法包括根据本文所述的方法将标记递送至t细胞并将标记的t细胞施用于患者体内,其中所述患者体内的t细胞迁移可以通过检测标记的t细胞来确定。在一些实施方案中,所述标记为荧光标记或放射性标记。在一些实施方案中,所述t细胞迁移是迁移到肿瘤。例如,t细胞可以从患者处分离,穿过微流体装置以将同位素递送到t细胞中,并且被注射回患者体内。然后可以使用成像方法(诸如pet扫描)来检测所述标记并追踪贯穿身体的t细胞的迁移。
疫苗的抗原呈递
因为所述系统能够将蛋白质优先递送到免疫细胞,因此工程化此类细胞对于针对目标靶标的患者(人、小鼠、非人类灵长类动物等)的疫苗接种来说是有用的。通过将特异性抗原蛋白(或抗原蛋白的混合物、蛋白质+佐剂的混合物或对应于蛋白质片段的肽)直接递送至靶细胞(例如dc、t细胞或b细胞)的细胞溶质,诱导抗原的mhci类呈递,其随后驱动cd8t细胞介导的针对靶疾病(例如,癌症或病原微生物感染诸如病毒感染)的免疫。在此方法中任选使用的佐剂增强了所述应答(例如在辅助因子存在下增强细胞的功效或孵育的材料的共同递送)。在本发明之前已经难以或不可能工程化的操控免疫细胞的能力,允许在本发明之前不可能的治疗性和预防性疫苗应用,特别是对于目前具有挑战性的疾病(诸如癌症和hiv)。其它表现包括共同递送材料以增强细胞活力,使得细胞可以呈递抗原更长时间;同时针对多种抗原疫苗接种;疫苗接种与激活因子的递送(或暴露于激活因子)联合以提供辅助效应并增强免疫应答;和/或使用细胞裂解物作为抗原来源的快速反应疫苗。在后面的实例中(针对新的未知病原体/疾病的疫苗接种),从患者取出感染细胞或癌细胞(例如,通过组织取样或活检),并且使用上述策略将这些细胞的裂解物递送到免疫细胞。这种方法在没人事前知道抗原的身份的情况下,提高了针对与所述未知疾病相关的抗原的免疫应答。
疫苗佐剂
佐剂或免疫应答激活剂/增效剂被用于加强引发免疫细胞(例如t细胞)对疫苗抗原的应答。在一些实施方案中,在免疫细胞穿过微流体装置之后,所述免疫细胞与佐剂接触。例如,所述细胞在穿过微流体装置之后约5分钟至约2小时或其间的任何时间或时间范围内与所述佐剂接触。例如,所述细胞在穿过微流体装置之后约5分钟至约1.5小时、约5分钟至约1小时、约5分钟至约45分钟、约5分钟至约30分钟、约5分钟至约15分钟、或约5分钟至约10分钟与所述佐剂接触。在一些实施方案中,所述细胞在穿过微流体装置之后约10分钟至约2小时、约15分钟至约2小时、约30分钟至约2分钟、约45分钟至约2小时、约1小时至约2小时、或约1.5小时至约2小时与所述佐剂接触。除了经典的佐剂诸如明矾或油包水乳液(例如,弗氏不完全佐剂(freund'sincompleteadjuvant)和
单独的或与各种制剂一起的prr的天然配体或合成激动剂。prr激活刺激促炎细胞因子/趋化因子和i型ifn的产生,这增加了宿主消除病原体的能力。病原体相关分子模式(pamp)引入到疫苗制剂中改进并加速了疫苗特异性应答的诱导。与明矾或经典乳液佐剂组合使用时,pamp可用于驱动针对th1应答的免疫应答。
tlr3和rlr配体。在大多数病毒复制过程中产生的双链rna(dsrna)是有效的先天免疫诱导剂。dsrna的合成类似物(诸如聚(i:c))可用作佐剂。它们通过tlr3和rig-i/mda-5起作用,诱导il-12和i型ifn产生,促进抗原交叉呈递给mhcii类分子,以及改进细胞毒性t细胞的生成。
tlr4配体。作为tlr4配体的细菌脂多糖(lps)长期以来一直被认为是有效的佐剂,但是它们的致热活性阻止了其临床使用。毒性较小的衍生物的开发包括单磷酰脂质a(mpla)。mpla可用作佐剂并驱动针对th1应答的免疫应答。
tlr5配体。tlr5配体,细菌鞭毛蛋白是有效的t细胞抗原,并且具有作为疫苗佐剂的潜力。与其它tlr激动剂不同,鞭毛蛋白倾向于产生混合的th1和th2应答,而不是强烈的th1应答。鞭毛蛋白可以用作与抗原混合或与重组疫苗抗原融合的佐剂。
tlr7/8配体。专门用于识别单链病毒rna的这些配体也是有用的疫苗佐剂。例如,咪唑喹啉(即咪喹莫特、嘎德莫特(gardiquimod)和r848)是合成化合物,其在多个树突细胞亚群中激活tlr7/8从而导致产生ifn-α和il-12,因此促进th1应答。
tlr9配体。含有特异性cpg基序(cpgodn,诸如odn1826和odn2006)的寡脱氧核苷酸被tlr9识别。它们增强抗体产生并且驱动/促进th细胞对th1的应答并远离th2应答。
nod2配体。细菌细胞壁的片段(诸如胞壁酰二肽(mdp))是熟知的佐剂。mdp触发nod2和nlrp3炎性体的激活。
这些类别的佐剂(例如prr途径的调节剂)由于其诱导强烈的细胞介导的免疫的能力而可用于疫苗。优选的佐剂包括cpg寡脱氧核苷酸、r848、脂多糖(lps)、rhil-2、抗cd40或cd40l、il-12和/或二环核苷酸。
用于免疫疗法的工程化t细胞
本公开的某些方面涉及通过将根据所述方法修饰的免疫细胞引入患者来治疗患者的方法。在一些实施方案中,所述免疫细胞用于免疫疗法。例如,通过使多种材料能够递送到t细胞,t细胞功能被工程化为靶向目标疾病。例如,通过递送嵌合抗原受体或靶向目标抗原的tcr的dna/mrna,产生针对疾病抗原具有特异性并推进杀伤(和/或辅助)t细胞应答的t细胞。递送其它材料(诸如nf-κb、bcl-2、bcl-3、bcl-xl、cd3/cd28的上调因子、cpg、r848、pd-1的抑制因子、pdl-1的抑制因子、ctla-4的抑制因子)来增强细胞活性和存活。
例如,目前的过继t细胞转移疗法中的一个常见挑战是激活的t细胞暴露于肿瘤的免疫抑制微环境并变得衰竭/无变应性,因此使它们的功效最小化。使用所述方法的细胞内递送用于破裂免疫抑制途径[例如,通过删除免疫抑制基因ctla-4、pd-1、pd-2、pdl-1、pdl-2或sirna介导的敲低,或使用已知方法(诸如基于转录激活因子样效应物核酸酶(talen)或锌指核酸酶(zfn)的方法)的基于小分子抑制剂/抗体的抑制],并且因此允许这些t细胞在肿瘤环境中保持其高度激活的杀伤状态。此外,这种方法用于通过共同递送适当因子以驱动对所述表型的分化来诱导t细胞变成记忆细胞(因此提供更好的长期保护)。在常规的过继转移中,引入患者的t细胞由于增殖已经处于激活的半衰竭状态。本文描述的方法用于将它们的表型重新设置为初始状态。因此,细胞变得能够在转移后更多的体内增殖并且不再具有衰竭的表型。
在一些实施方案中,本公开的方法用于离体生成抗原特异性t细胞。例如,将抗原递送到免疫细胞(诸如dc),然后将负载抗原的免疫细胞与来自患者的t细胞一起培养以在体外激活它们。然后可以在再次注射到患者体内之前通过进一步的刺激来扩增这些t细胞。
耐受性的抗原呈递:
为了诱导对细胞呈递的抗原的耐受性,细胞进一步与耐受原(诸如胸腺基质淋巴细胞生成素、地塞米松、维生素d、视黄酸、雷帕霉素、阿司匹林、转化生长因子β、白介素-10或血管活性肠肽)和抗原一起接触反而可以诱导耐受性。
肿瘤和t细胞耐受性
在肿瘤微环境中,肿瘤反应性t细胞可以变得耐受。这是由于多种抑制机制,包括与肿瘤发展相关的其它细胞的耐受原活性(anderson等,jimmunol2007,178:1268-1276;probst等,natimmunol2005,6:280-286)。阻断通过检查点受体(诸如ctla-4和pd-1)的信号传导的基于抗体的药物在原发性疾病和转移性疾病中都产生了抗肿瘤应答。响应于检查点封闭的恶性肿瘤具有高突变频率,并被对癌抗原有反应性的t细胞浸润(taneja,jurol2012,188:2148-2149;brahmer等,nengljmed2012,366:2455-2465;wolchok等,nengljmed2013,369:122-133)。虽然这种方法对于某些适应症是成功的,但多种抑制性检查点表面受体的存在可能破坏目前有限的可用于免疫疗法的功能阻断抗体组的应用。此外,用多种阻断抗体系统性治疗的要求可能会增加毒性(ribas等,nengljmed2013,368:1365-1366;weber等,cancer2013,119:1675-1682),特别是组合使用时(在postow等,jclinoncol2015,33:1974-1982和gao等,oncogene2015中综述)。在本发明的一些方面,仅通过抑制肿瘤反应性t细胞中的抑制途径来实现患者结果的显著改进。在非限制性实例中,这可以通过抑制或使得在过继转移方法中使用的t细胞(诸如肿瘤浸润淋巴细胞(til)、重组tcr和嵌合抗原受体(car)t细胞)内的抑制性途径的基因敲低或敲除来实现。
shp2是普遍存在的酪氨酸磷酸酶,其在t细胞活化时,会抑制tcr信号传导并进而抑制t细胞对癌细胞的应答。通过几种免疫检查点受体通过shp2的信号传导减弱t细胞活性。激活shp2的抑制性受体包括但不限于pd-1(yokosuka等,jexpmed2012,209:1201-1217)、ctla-4(marengere等,science1996,272:1170-1173)、btla(watanabe等,natimmunol2003,4:670-679)和lair-1(lebbink等,jimmunol2004,172:5535-5543)(也在nirschl等,clincancerres2013,19:4917-4924中综述)。在一些实施方案中,肿瘤反应性t细胞中shp2的基因失活或下调(例如,使用rna干扰)提供类似于阻断几种抑制性检查点受体的信号传导的抗肿瘤应答。此类实施方案可以具有优于酪氨酸磷酸酶的药理学抑制剂葡萄糖酸锑钠(ssg)(包括shp-1和shp-2)与其它抗肿瘤免疫疗法组合的治疗用途的优点(诸如减少的副作用)(yi等,oncotarget2011,2:1155-1164;naing等,jcancer2011,2:81-89;pathak等,jimmunol2001,167:3391-3397)。shp2已经示出发挥t细胞的外在作用,并且特异性地抑制t细胞上的这个分子可以消除与其功能的系统性抑制相关的任何潜在的副作用。此外,通过靶向t细胞中单个细胞内信号传导分子的基因破裂或下调的多种抑制性信号传导途径,实现了比t细胞过继转移和系统检查点封闭的组合更大的功效。在一些实施方案中,蛋白质或核酸(例如sirna)、小分子抑制剂、抗体、转录激活因子样效应物核酸酶或锌指核酸酶被递送到免疫细胞(例如t细胞)来调控基因的表达或基因产物(诸如shp2)的活性以修饰t细胞的行为或功能。例如,shp2受损的cd8t细胞具有比未受损的细胞更高的控制肿瘤进展的效力。
使用本文所述的细胞挤压装置和方法克服了与早期方法相关的调控t细胞中的基因表达的挑战。细胞挤压装置的非限制性方面在以下中讨论:proceedingsofthenationalacademyofsciences(sharei等,procnatlacadsciusa2013,110:2082-2087)和nanoletters(lee等,nanolett2012,12:6322-6327),所述文献各自的全部内容以引用的方式并入本文。例如,细胞挤压技术可以包括微流体芯片,当细胞穿过收缩时能够使细胞迅速变形以暂时破裂细胞的膜并且能够将靶材料输送到细胞质。此外,通过消除对电场的需要(例如,在一些实施方案中,所述装置和方法不包括将细胞暴露或应用于电场)或对潜在毒性的外源性材料的需要,细胞挤压技术使细胞毒性和脱靶效应的潜力最小化。本文描述的微流体设计和处理过程能够生成用于各种癌症适应症的更有效的工程化t细胞疗法。
用于免疫抑制的工程化t细胞
自身免疫疾病通常涉及损伤健康组织的自身反应性免疫细胞。使用细胞内递送foxp3(和/或其它因子)到t细胞来生成调节性t细胞以抵抗自身免疫。生成这些treg用于广泛的系统性免疫抑制或用来将自身抗原特异性t细胞重编程为treg表型。后者导致treg归巢到与保持自身免疫并诱导局部抑制细胞活性的细胞相同的靶位点。
自扩增rna(sarna)
自扩增rna提供独特的能力,因为它们不仅表达了目标蛋白质,而且还在没有整合入宿主基因组的风险的情况下在细胞质中复制它们的序列。因此,将自扩增rna引入免疫细胞可用于工程化免疫细胞功能。一些具体的表现包括神风免疫细胞、蛋白质递送的替代方案或细胞功能的连续调整,其各自描述如下。
神风免疫细胞
对于与上述那些类似的疫苗接种或免疫抑制策略,将编码目标抗原的sarna递送到归巢到所需位置的免疫细胞。例如,将编码sarna的癌抗原递送到t细胞(或b细胞、单核细胞、dc或巨噬细胞)以及将那些细胞注射到患者体内,导致t细胞中抗原的快速产生以及由于sarna的快速复制的这些t细胞的最终死亡。这些负载靶抗原的濒死的t细胞的破裂模拟感染并导致材料被靶组织中先天细胞摄取,并准备针对靶抗原的疫苗应答。根据靶组织、佐剂的存在和患者的整体炎症状态,此策略也用于诱导耐受性。为了诱导耐受性,将上述神风细胞工程化以释放耐受原因子和抗原或者与耐受原因子(诸如tgf-β和il-10)接触。例如,可以将神风t细胞或b细胞负载过表达所需抗原同时还表达耐受原因子(例如tgf-β和il-10的分泌)的mrna、dna或sarna。然后,这些细胞将在经历死亡之前迁移到淋巴样器官,这将使抗原和耐受原因子释放到环境中并有助于诱导针对靶抗原的耐受性。这将有助于消除自身反应性效应t细胞并刺激能够防止自身反应的treg的产生。在一个非限制性实例中,使用类风湿性关节炎抗原来采用此方法。
蛋白质递送的替代方案
sarna、mrna或表达载体(例如质粒),在疫苗治疗期间提供相对于蛋白质递送脉冲的连续蛋白质产生。
细胞功能的连续调控
使用t细胞过继转移疗法作为实例,通过使用编码免疫抑制途径的抑制剂的sarna来完成更好的t细胞疗法的开发。sarna也用于表达刺激性蛋白质以维持t细胞活化和/或抗凋亡蛋白的高状态来延长存活。抑制抑制剂的非限制性实例包括阻断pd-1、pd-l1、ctla-4的任何材料或其它检查点抑制剂。通过表达il-2、nf-kb、il-7、il-15、il-12等,可以维持高t细胞活性,并且蛋白质如bcl2和bclx1可以帮助延长存活。
免疫细胞功能的重编程
免疫细胞被用作自体细胞的来源。细胞被重编程以进行许多疾病治疗功能。例如,在关节炎的情况下,将材料递送到t细胞以诱导其表达联合归巢表型以及编程可以缓解症状的因子(诸如il-4、il-6、il-10、il-13、il-11、tgfb、视黄酸和检查点刺激剂)的释放,或者在帕金森病的情况下,将材料递送到t细胞或b细胞以诱导其表达脑归巢受体以及分泌改进患者结果的因子。关于关节炎,除去促炎细胞因子也是有用的,例如对il-2具有高亲和力的t细胞将吸收自身反应性效应细胞所需的细胞因子。
使用以下材料和方法来生成本文所述的数据。
细胞挤压微流体装置
细胞挤压平台由三个主要部件组成:a)硅和玻璃微流体芯片,其包括平行的多个通道,每个通道包括至少一个收缩点,b)贮存器系统,其与芯片连接并允许一个贮存器负载/收集细胞悬浮液,c)压力调节系统,其用于对贮存器加压并促进流体流过所述芯片。在典型的工作流程(图1a)中,将靶递送材料与所需的细胞(悬浮液)混合并将它们负载到贮存器中。然后将压力管连接到贮存器,并且将室加压到所需水平以引发流体流动。处理后,细胞可以从输出贮存器处收集并在所需温度下孵育一段时间,例如至少1、2、3、4、5分钟或更多,以确保在进一步加工之前适当的膜恢复。
细胞挤压装置和相关的操作设备均从美国sqzbiotechnologies获得。根据制造商协议组装和使用装置。sharei,a.,n.cho,s.mao,e.jackson,r.poceviciute,a.adamo,j.zoldan,r.langer和k.f.jensen.2013.cellsqueezingasarobust,microfluidicintracellulardeliveryplatform.journalofvisualizedexperiments:jove:e50980.
例如,将个体细胞挤压装置和相关的贮存器系统保存在70%乙醇中以维持无菌性。对于每个实验,将所需的细胞挤压装置连接到贮存器,并且在使用细胞样品之前使用70ulpbs冲洗所述系统。
在递送实验期间,将靶细胞、装置+贮存器和收集板保持在冰上(t细胞和b细胞)或在室温下(树突细胞)。将细胞(在pbs或培养基中2×106-1×107个细胞/ml的浓度下)与靶递送材料在添加到流体贮存器之前以所需浓度混合。连接压力管,将系统设定在所需的操作压力下,并且通过对包含样品的贮存器加压来引发流动。穿过芯片后,从收集贮存器处收集细胞并转移到96孔板。任选地重复此过程。为了使堵塞最小化,芯片中的流动方向在样品之间交替。允许样品在处理后在添加培养基之前在冰上孵育5分钟,并将样品转移用于进一步加工。
cart细胞
通过修饰t细胞以表达识别癌症特异性抗原的嵌合抗原受体(car),可以引发细胞识别和杀伤肿瘤细胞,否则其将逃避免疫检测。所述过程涉及提取患者的t细胞,用car的基因转染它们,然后将所述转染的细胞再灌注到患者体内。
这些人工t细胞受体(也被称为嵌合t细胞受体、嵌合免疫受体或car)是工程化受体,其将任意的特异性移植到免疫效应细胞上。通常,这些受体用于将单克隆抗体的特异性移植到t细胞上。在本发明之前,通常通过逆转录病毒载体促进核酸编码序列的转移。本文描述的方法不利用或包括病毒载体。在不需要病毒载体的情况下,使用利用所述装置的细胞挤压将编码序列或蛋白质car递送到免疫细胞(诸如t细胞)的细胞溶质。
对于治疗性应用,患者的t细胞从外周血获得(并任选地富集或纯化)并修饰以表达对于特定癌症相关抗原具有特异性的人造(嵌合)受体。修饰后,t细胞识别并杀伤癌症。例如,示例性car识别在b细胞-血液恶性肿瘤中表达的抗原cd19。在t细胞被修饰以表达car之后,将修饰的t细胞再灌注到患者体内。工程化细胞识别并杀伤癌性细胞。这种治疗已被用于all、非霍奇金氏淋巴瘤和慢性淋巴细胞白血病(cll),并且适用于任何类型的癌症疗法,包括血源性癌症诸如白血病、b细胞恶性肿瘤(例如急性淋巴细胞白血病(all)和慢性淋巴细胞白血病)以及实体癌症。本文所述的细胞加工方法表示生成cart细胞的优良过程。
自体t细胞表达car蛋白,其赋予工程化t细胞识别肿瘤细胞上特异性抗原的能力。此类肿瘤相关抗原已经被鉴定,并且是本领域已知的(见下表)。然后将工程化的cart细胞在实验室中扩增,然后将扩增的cart细胞群灌注到患者体内。t细胞在患者体内繁殖,并且识别、结合并杀伤其表面上携带肿瘤相关抗原的癌细胞。任选地,免疫检查点抑制剂如程序性死亡-1(pd-1)抑制剂或配体抑制剂(pd-l1)和/或抗细胞毒性t淋巴细胞抗原4抗ctla4药物可以与cart细胞组合。
肿瘤的治疗
使用如上所述处理以将化合物或组合物引入细胞溶质中的免疫细胞来治疗肿瘤,例如通过引起肿瘤特异性t细胞介导的免疫应答来杀伤或抑制肿瘤的增殖。该方法适用于任何肿瘤类型,因为所述装置和方法将肿瘤特异性/肿瘤相关抗原或其混合物(例如肿瘤活检细胞裂解物制品)引入免疫细胞中。例如,肿瘤类型包括在美国人群中普遍存在的膀胱癌、乳腺癌、结直肠癌、子宫内膜癌、肾癌、白血病、肺癌、黑色素瘤、非霍奇金氏淋巴瘤、胰腺癌、前列腺癌、甲状腺癌。(americancancersociety:cancerfactsandfigures,2015,atlanta,ga:americancancersociety,2015。网上可获得。)使用加工的免疫细胞治疗的其它肿瘤类型包括脑(成胶质细胞瘤)、肝(肝细胞癌)以及转移性癌,所述转移性癌发生在与原发肿瘤的部位或组织不同的身体内解剖部位或组织中。
在优选的实施方案中,肿瘤为胰腺癌、卵巢癌、黑色素瘤、肺癌、神经胶质瘤或成胶质细胞瘤肿瘤。在一些实施方案中,靶向具体患者的肿瘤。例如,来自患者的肿瘤裂解物可以用作使用细胞挤压递送到免疫细胞的抗原。
肿瘤细胞抗原
将纯化的肿瘤相关抗原或肿瘤细胞裂解物(抗原的不均匀混合物)用作使用细胞挤压递送到免疫细胞的抗原。肿瘤细胞裂解物由从受试者获得的肿瘤细胞系或肿瘤活检组织来产生,所述受试者已被诊断患有肿瘤和/或被预定用于治疗病理性恶性肿瘤。肿瘤细胞裂解物的产生是本领域已知的。此类肿瘤裂解物制品适用于基于细胞挤压递送到免疫细胞的细胞溶质。
例如,从受试者切除肿瘤组织,并将组织切碎。加工肿瘤细胞,例如分级、富集或纯化/分离。在非实体肿瘤、血源性肿瘤(原发性或转移性)或细胞系、细胞富集)的情况下,细胞从体液(例如外周血)中获得,并且任选地富集或纯化以从非肿瘤细胞中浓缩肿瘤细胞。加工肿瘤细胞(或富含肿瘤细胞的细胞群)以获得肿瘤细胞裂解物(例如,如hatfield等,jimmunother.2008sep;31(7):620-632中所述。)例如,细胞经受冻融循环(例如,1、2、3、4、5、10或更多个冻融循环)。任选地,步骤包括除去固体碎片并过滤(例如0.2微米过滤器),以获得患者特异性肿瘤细胞抗原的混合物。
在非限制性实例中,通过使用液氮的多次(例如4次)连续冻融循环来裂解细胞。然后将细胞超声处理约10、15或20秒,然后在1000g下离心以除去不溶性碎片。然后将上清液用于裂解物递送。任选地,在递送到免疫细胞之前,从裂解物中除去脂质和/或核酸。任选地在递送之前添加佐剂以增大apc功能。
在一些实施方案中,除去常见的内源性蛋白质(诸如肌动蛋白),以便选择性地增加裂解物中癌抗原的比例。在一些实施方式中,将温和的表面活性剂添加到裂解的细胞组合物中以帮助防止蛋白质形成可能难以用于细胞呈递的递送或加工的聚集体。
自体肿瘤细胞裂解物也可以使用可商购获得的装置/方法(例如,gentlemacstm解离器(miltenyibiotecgmbh))来生成。
肿瘤相关抗原是本领域已知的,并且此类抗原可以被生物化学纯化、重组表达和/或以其它方式纯化/分离。此类抗原的实例示于下表中。
(buonaguro等,clinvaccineimmunol.2011jan;18(1):23-34。)
其它纯化的肿瘤抗原是本领域已知的,例如,由oliviergires和barbaraseliger编的tumor-associatedantigens.2009,wiley-vchverlaggmbh&co.,kgaa,weinheim中所述的。
病毒抗原
使用细胞挤压可以将病毒相关的抗原递送到免疫细胞。病毒相关抗原是本领域已知的,并且此类抗原可以被生物化学纯化、重组表达和/或以其它方式纯化/分离。病毒抗原的实例示于下表中。
细菌抗原
使用细胞挤压也可以将细菌抗原递送到免疫细胞。在优选的实施方案中,细菌抗原与以下细胞内细菌相关联:诸如支原体属(mycoplasmasp.)、分枝杆菌属(mycobacteriumsp.)(例如,结核分枝杆菌(m.tuberculosis))或单核细胞增生李斯特菌(listeriamonocytogenes)。
使用以下材料和方法来生成本文所述的数据。
小鼠免疫细胞分离
使用已知方法从野生型c57bl6/j小鼠的脾脏分离t细胞和b细胞,例如来自stemcelltechnologies(vancouver,canada)的基于制造商的说明书(阴性选择技术)的细胞特异性分离试剂盒。在腹膜内注射1ml巯基醋酸盐溶液后3天,从野生型c57bl6/j小鼠的腹膜腔中分离单核细胞/巨噬细胞。根据制造商的说明书,使用来自stemcelltechnologies(vancouver,canada)的cd11b阳性选择试剂盒纯化细胞。将细胞在含谷氨酰胺的rpmi1640培养基中培养,所述培养基含有10%胎牛血清、1%抗生素/抗真菌剂、0.5%β-巯基乙醇、1%非必需氨基酸、1mm丙酮酸钠和10mmhepes缓冲液(所有均来自(lifetechnologies,ny,usa))。
人原代t细胞和单核细胞来源的树突细胞
使用已知方法分离人pbmc,例如来自全血的ficoll-paque(gehealthcare,uppsala,sweden)密度梯度离心。使用cd14和cd4磁性微珠(macsmiltenyibiotec,auburn,ca)从pbmc的cd14阴性部分分离cd4+t细胞。将t细胞在rpmi1640培养基(cellgro,manassas,va)中培养以在无细胞激活的情况下维持细胞活力,所述培养基含有10%人血清(ab)(gemcell,westsacramento,ca)、100u/ml青霉素和硫酸链霉素100μg/ml(h10培养基)且增补有5ng/mlrhil-15(r&dsystems,minneapolis,mn)。人单核细胞来源的树突细胞(mddc)使用抗cd14磁性微珠(macsmiltenyibiotec)由选自外周血单核细胞的cd14阳性单核细胞制备,并用100ng/ml白细胞介素-4和50ng/ml粒细胞巨噬细胞集落刺激因子(r&dsystems)培养6天。
细胞转染
人cd45sirna:有义链5’-af488cuggcugaauuucagagcadtdt-3’(seqidno:1),人cd4sirna:有义链5’-gaucaagagacuccucagudtdt-3’(seqidno:2)(alnylam,cambridge,ma);vifsirna:有义链5’-cagauggcaggugaugauugt-3’,(seqidno:3),gagsirna:有义链5’-gauuguacugagagacaggcu-3’(seqidno:4)(huang等,2013,naturebiotechnology31:350-356);(genepharma,shanghai,china);对照乱序sirna:5’-gccaagcaccgaaguaaauuu-3’(seqidno:5),人dc-signsirna:有义链5’-ggaacuggcacgacuccauuu-3’(seqidno:6)(dharmacon,thermoscientific,pittsburgh,pa)。
核转染
在所述的电穿孔实验中,根据制造商的建议使用amaxa核转染仪ii(lonzainc.,allendale,nj)。人t细胞实验使用人未刺激的t细胞高活力的u-014的程序与人t细胞试剂盒进行。对于人mddc,我们使用人树突细胞u-002的程序与mddc的人树突细胞试剂盒。简单来说,将2×106个细胞悬浮在100μl具有200pmolsirna的核转染溶液中,并由机器进行核转染。为了测试蛋白质递送,我们使用0.02mg/ml的apc-标记的小鼠igg1(cl.mopc-21,biolegend)进行细胞挤压和核转染实验。我们还使用0.2mg/ml的3kda瀑布蓝标记的葡聚糖和70kd荧光素标记的葡聚糖(invitrogen)。
调节性t细胞
使用已知方法分离和扩增调节性t细胞(treg)。例如,cd4+t细胞富集的pbmc使用cd4+t细胞rosettesep富集试剂盒(sigma-aldrich和stemcelltechnologies)通过密度离心从健康个体的外周血中分离,并用抗cd3-pe-cy7、cd4-fitc、d25-apc和cd127-pe标记。将cd3+cd4+cd25+cd127低treg在facsaria细胞分选仪(bdbiosciences)上分选,用抗cd3/抗cd28包被的微珠(invitrogen)刺激,并用il-2(300u/ml)培养。
对于sirna递送,在培养的第7天,将treg洗涤并以1.0×107个细胞/ml单独重悬于x-vivo15(lonza)培养基中。每个条件使用1.0×106个细胞。在1μm下用100psi的30-4芯片设计使用cd4sirna(5’-gaucaagagacuccucagu-3’(seqidno:7),alnylam)和对照sirna(sigenome非靶向sirna汇集物#1,thermofisher)。
sirna递送后2天,将细胞用
流式细胞术
将小鼠细胞用以下抗体染色:抗cd8-太平洋蓝、抗cd4-apc、抗cd11b-pe(cl.m1/70)、抗cd11c-apc。使用碘化丙啶来排除死细胞。数据使用facscantoii、lsrii或lsrfortessa(bdbiosciences)获取,并使用flowjo(treestar,ashland,or)进行分析。
将人细胞用以下抗体染色:来自biolegend(sandiego,ca)的抗cd3-apc(c1.okt3)、抗cd45ra-pe-cy7(cl.hi100)和抗cd4-af488(c1.okt4)以及抗dc-sign-apc(c1.9e9a8)(r&dsystems,minneapolis,mn)。使用sytoxblue和7-aad(7-氨基放线菌素d)死染染料(invitrogen)排除死细胞。数据使用facscantoii(bdbiosciences)获取,并使用flowjo(treestar,ashland,or)进行分析。
hiv感染和细胞内p24抗原染色
使用10-4芯片用5μmsirna处理原代cd4+t细胞。为了敲低cd4,在感染前48小时递送sirna,而在感染前24小时递送靶向病毒基因vif和gag的sirna。然后用5μg/ml植物血球凝集素(pha)刺激细胞过夜,并用hiviiib(400ng/mlp24)以2×105个细胞/孔在96孔板中用hiviiib感染细胞。hiviiib从美国国立卫生研究院艾滋病试剂项目(nihaidsreagentprogram)获得,并且如前所述制备病毒原液(18)。通过添加5μg/ml的聚凝胺以及在1200xg下离心接种(spinoculation)在37℃下持续2小时来增强感染(19)。24小时后,使用抗p24kc57-fitc抗体(beckmancoulter,fullerton,ca)和用于细胞透化作用的fix&perm试剂盒(invitrogen)进行细胞内p24抗原染色,并通过流式细胞术分析。
定量rt-pcr
使用rneasy迷你试剂盒(qiagen)从t细胞中分离总rna,并使用superscriptiii和随机六聚体(invitrogen)合成拷贝dna。使用ssofastevagreensupemix和bio-radcfx96实时pcr系统(bio-radlaboratories,hercules,ca)进行实时pcr。引物如下:gapdh正向:5’-agccacatcgctcagacac-3’(seqidno:8),gapdh反向:5’-gcccaatacgaccaaatcc-3’(seqidno:9),cd4正向:5’-ggcagtgtctgctgagtgac-3’(seqidno:10),cd4反向:5’-gaccatgtgggcagaacct-3’(seqidno:11)。
统计分析
使用graphpadprism4软件(graphpadsoftware,sandiego,ca),当比较多组时,进行单因素方差分析(anova)与bonferroni多重比较检验,或者当比较2组时,进行双尾学生t检验。*、**和***指示使用bonferroni多重比较检验时p值低于0.05、0.01和0.001,而###指示使用双尾学生t检验时p值低于0.001。除非另外指明,否则数据表示为平均值±1标准偏差。
通过机械膜破裂的递送
测试的微流体装置含有具有不同收缩长度(10-50μm)、宽度(4-9μm)和每个通道不同的收缩数(1-5个收缩)的45-75个平行的微流体通道。(sharei等,2013,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica110:2082-2087;sharei等,2014,integrativebiology:quantitativebiosciencesfromnanotomacro6:470-475)。
操作微流体芯片所需的系统包括将流体贮存器固定到硅和玻璃装置的安装部件,以及控制用来驱动流体通过系统的气体压力的压力调节系统。在一些实施方案中,所述装置包括注射器或压力源以诱导流体通过微流体通道。
操作程序示于图1a中。当细胞流过微流体通道(图1b)时,它们到达通道中的收缩点(比待处理的大多数细胞的直径小约50%),这导致细胞的快速机械变形或挤压(靶细胞的收缩点的示例性尺寸如下:t细胞(静息:7-8μm,激活(7-15μm)、巨噬细胞(静息和激活:10-30μm)、树突细胞(静息和激活:10-30μm)。当通道收缩尺寸适当时,所述变形会暂时破裂细胞膜(例如,变形过程需要0.1μs-1ms,但膜破裂可保持开放达5分钟)。如果存在于周围缓冲液中的大分子足够小以运送通过膜破裂,那么它们进入到细胞细胞溶质。在约5分钟内,膜恢复其完整性,并且由细胞吸收的大分子仍被捕获在细胞细胞溶质中。先前研究将收缩长度(l)、宽度(w)和流体速度(v,注意流体速度由操作压力确定)鉴定为影响递送效率和细胞活力的重要参数。(sharei等,2013,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica110:2082-2087)。
在不同的流动条件下测试了16个不同收缩设计的库。测试的装置设计库。第一个数字指示收缩长度,前面加上破折号的随后的数字指示收缩宽度。如果有多个相同的串联收缩,则由'x'指示,后面是收缩的数量。例如,10-5-4-5含有具有5μm、4μm和5μm宽度的3个10μm长的串联收缩。10-4x5含有5个10μm长的串联收缩,每个具有4μm的宽度。
这些变量包括压力(以改变流速)和温度的变化以优化大分子递送并使细胞毒性最小化。发现测试的所有缓冲液(pbs、pbs+2%血清、完全培养基和全人血)与系统相容,这指示任何生理上相容的流体或溶液都适于悬浮通过递送装置和过程的细胞。
还对于鼠和人t细胞测试了30-5x5、10-4x2、10-5-4-5、10-6-4-6、30-5-4-5和10-4x5设计,但没有一个优于30-4的性能。
表2:递送参数及其对性能的影响
在一些实施方案中,所述装置包括约5μm至约50μm或其间的任何长度或长度范围的收缩长度。例如,收缩长度范围为约5μm至约40μm、约5μm至约30μm、约5μm至约20μm或约5μm至约10μm。在一些实施方案中,收缩长度范围为约10μm至约50μm、约20μm至约50μm、约30μm至约50μm或约40μm至约50μm。在一些实施方案中,收缩深度范围为约2um至约200um或其间的任何深度或深度范围。例如,收缩深度范围为约2μm至约150μm、约2μm至约100μm、约2μm至约50μm、约2μm至约25μm、约2μm至约15μm或约2μm至约10μm。在一些实施方案中,收缩深度范围为约10μm至约200μm、约25μm至约200μm、约50μm至约200μm、约100μm至约200μm或约150μm至约200μm。在一些实施方案中,收缩的入口部分或出口部分的角度的范围为约0度至约90度或其间的任何角度或角度范围。例如,所述角度为约5、约10、约15、约20、约30、约40、约50、约60、约70、约80、或约90度或更大。在一些实施方案中,压力范围为约50psi至约200psi或其间的任何压力或压力范围。例如,所述压力范围为约50psi至约150psi、约50psi至约125psi、约50psi至约100psi或约50psi至约75psi。在一些实施方案中,所述压力范围为约75psi至约200psi、约100psi至约200psi、约125psi至约200psi、约150psi至约200psi或约175psi至约200psi。在一些实施方案中,所述装置包括约2μm至约10μm之间或其间的任何宽度或宽度范围的收缩宽度。例如,收缩宽度可以为约3μm、约4μm、约5μm、约6μm或约7μm中的任一个。
递送到原代小鼠细胞
为了评估此平台能够细胞内递送到原代免疫细胞的潜力,在荧光标记的葡聚糖(3kda,和70kda,和2,000kda)和抗体(约150kda)的存在下,将小鼠t细胞、b细胞和单核细胞/巨噬细胞穿过微流体装置。这些材料分别被选为小分子、多糖和蛋白质的模型。每个细胞类型测试至少四种装置设计和两种操作压力。收缩尺寸的最初选择由细胞系、原代成纤维细胞和胚胎干细胞的工作指导。(sharei等,2013,proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica110:2082-2087)。具体地说,收缩宽度被选择为靶细胞的平均直径的约50%。使用30-4设计(即收缩具有30μm长度和4μm宽度)的递送对于淋巴细胞和髓样细胞是最有效的(图2a-c)。
通过流式细胞仪在处理后1-2小时测量生物分子的递送(图2a-c和图6)和细胞活力(图7)。图2a-c示出了抗体递送的代表性直方图(a),独立实验中3kda葡聚糖、70kda葡聚糖和抗体的递送效率(b)和代表性的中值强度数据(c)。递送效率(定义为荧光高于背景的活细胞的百分比)和中值荧光强度被用作主要的递送量度。分别将3kda葡聚糖递送到66.2±13.4%的t细胞、67.5±13.9%的b细胞和80.8±3.46%的髓样细胞。如所预期的,更大的70kda葡聚糖和ab的摄取效率低于3kda葡聚糖的摄取,这表明较小的分子被更有效地递送。与未处理的细胞相比,t细胞但不是b细胞或髓样细胞的细胞活力在某种程度上受损。用装置处理的b细胞和髓样细胞的活力变化与未处理的或无装置细胞没有显著差异。由于葡聚糖和抗体较大的尺寸,骨髓来源的树突细胞在有限的活力损失的情况下使用较大的6μm的收缩宽度来吸收它们(图8)。葡聚糖(3kda和70kda)和抗体的同时递送示出了这些分子的递送是成比例的,即接受抗体的细胞也接受相当数量的葡聚糖分子(图9)。
递送到人免疫细胞
为了检查这种方法对人免疫细胞的适用性,测试了对于t细胞的4-6μm和对于单核细胞来源的树突细胞(mddc)的6-9μm的收缩宽度范围的装置设计。不同生物分子的抗体递送和效率数据的代表性直方图示于图3a中。最有效的设计将3kda葡聚糖递送到70%±9%的t细胞(4μm收缩尺寸)和60%±4.5%的mddc(7μm收缩尺寸)(图3a)。70kda的葡聚糖被递送到71%±11%的t细胞和40%±14%的mddc,并且蛋白质(抗体)被递送到65%±15%的t细胞和38%±7%的mddc。递送荧光标记的sirna(cd45rasirna–alexa-fluor-488)产生相似的结果(图12、13)。
为了测试蛋白质敲低,将针对人cd4或cd45ra的sirna递送至血液来源的t细胞,并且使用5μm、1μm和0.2μmsirna在处理后72小时观察到蛋白质水平的剂量依赖性敲低,而对照sirna没有显著效应(图3b,图12、13)。在处理后48小时通过qrt-pcr测量时,cd4mrna也降低(图14)。还测试了用5μmcd4sirna处理的t细胞中cd4敲低的耐久性。敲低持续约10天(图14)。靶向树突细胞标志物dc-sign的sirna的递送还导致了使用不同装置设计的mddc中dc-sign蛋白表达的显著的序列特异性敲低。不同装置设计中的敲低水平与葡聚糖/ab递送效率有关。来自cd4t细胞和mddc的独立实验的蛋白质表达和编译的敲低数据的代表性直方图示于图3b中。还发现所述方法适用于人调节性t细胞(图3c)、b细胞和单核细胞(图16、17、18)。
为了比较微流体装置与核转染(基于电穿孔的核酸递送方法)的性能,通过细胞挤压平台或核转染机平行用sirna平行处理人cd4t细胞,并且72小时后通过流式细胞术测量蛋白质敲低。尽管cd4敲低的程度在两者之间类似(图3d),但核转染后t细胞活力显著低于用微流体装置处理的细胞(p<0.05)。比较两种平台递送标记的葡聚糖和蛋白质的效率。在mddc的情况下,变形装置与核转染的性能的比较产生与t细胞相似的结果。具体地说,最好的微流体装置(对于t细胞为30-4,对于mddc为10-6)在细胞活力和递送中展示出优点(图18)。两种技术之间的sirna敲低效率的比较也指示,细胞挤压引起较少的脱靶效应(图19)并改进长期活力。
原代人t细胞中hiv-1感染的抑制
进行研究以确定微流体方法是否可用于通过递送靶向病毒基因的sirna来抑制人原代cd4+t细胞中的hiv感染和复制。先前示出抑制病毒复制的针对vif和gag的sirna在感染hiv之前24小时被递送到细胞。使用靶向hiv受体cd4的sirna作为阳性对照。感染前48小时递送cd4sirna以确保表面cd4水平的降低,因此抑制感染。hiv复制通过细胞内p24抗原染色来确定并通过流式细胞术测量。独立感染实验的代表性直方图和编译结果示于图4a-b中。在用vif和gag靶向的sirna分别单独处理和组合处理的t细胞中观察到p24抗原的显著降低(p<0.01)。所述抑制效应大于cd4敲低诱导的抑制效应(p<0.05)(图4a-b)。
使用细胞形状改变装置离体细胞溶质递送化合物到免疫细胞
在本发明之前,细胞内递送大分子到免疫细胞是重要的挑战。这里示出的结果证明了无载体膜破裂技术将小分子以及大的大分子递送到真核细胞诸如小鼠和人免疫细胞的效用。结果令人惊奇并且与临床使用特别相关,因为免疫细胞对于将组合物递送到这些细胞的细胞质的其它技术是顽抗的。本文所述的数据证明:(i)递送多种生物学相关的大分子(多糖、蛋白质和核酸)的能力;(ii)在临床最相关的免疫细胞亚群(t细胞、b细胞、dc、单核细胞/巨噬细胞)中的功效(图2a-c和图3a-d);(iii)对载体材料和电场的独立性,因此克服与胞吞截留和电穿孔水平毒性相关的一些挑战;和(iv)多类大分子同时递送到靶细胞(图2c、9、12、13)。
这些令人惊奇和显著的优点能够实现先前未预见到的免疫细胞操纵和临床应用。例如,此系统充当将基于肽或蛋白质的治疗剂递送到淋巴细胞的平台,其中现有方法(例如,电穿孔)功效有限。系统的同时传递特征也可用于平行地筛选多种治疗候选物以加速筛选过程并潜在地识别补偿效应。另外,使用类似尺寸的标记分子作为示踪剂,允许独立地监测未标记的靶材料的递送效率。也可以使用此系统递送其它成像剂(诸如量子点),因此促进活细胞中的分子相互作用的直接观察来获得生物过程的更深的理解。
在非限制性实例中,递送量子点或磁性纳米颗粒以促进转移的免疫细胞的体内成像。例如,mri可以检测负载磁性颗粒的过继转移的t细胞的定位。
递送性能对系统参数(诸如收缩形状、温度、操作压力和缓冲液组成)的依赖性被定制为将化合物/组合物递送到免疫细胞或免疫细胞混合物(例如全血中的细胞)。例如,用于免疫细胞的示例性装置参数包括(t细胞,30-4静息,10-4激活)、b细胞(30-4静息,10-4激活)、巨噬细胞(10-6)、树突细胞(10-6)以及混合物(诸如白细胞级分/血沉棕黄色层细胞(buffycoatcells)或全血细胞)。对于全部的收缩设计的范围为2-10μm宽,0.1-90μm长)。因此,可以通过使用为其它细胞类型建立的设计规则开发装置架构和操作协议来优化靶淋巴细胞和髓样细胞群中的平台的性能。例如,可以通过制造具有更长更窄的收缩的装置来增加递送效率。通过每个设备包括更多的平行通道或增加操作压力,也可以增加目前处于100,000-1,000,000个/秒的系统的通过量。在一些实施方案中,增加通道深度或平行添加另外的通道来增加通过量。操作压力可能因装置的设计而变化。在某些实施方案中,压力为1psi至1000psi。
vif和gag敲除研究证明了此方法改变细胞表型并影响疾病相关生物的过程(诸如病毒(例如hiv)复制)的潜力(图4a、b)。此结果不仅证明了这种方法疾病治疗和研究疾病机制(例如病毒复制对具体基因的依赖性)的效用,而且还证明了在不使用潜在毒性递送载体的情况下通过靶向具体宿主细胞功能/途径来使临床使用的免疫细胞工程化。通过挤压递送的蛋白质转录因子(例如irf5)可用于增加人pdc中ifn-α的产生,因此证明通过挤压递送的蛋白质也是功能性的并且能够影响细胞表型。因此,此细胞内递送系统可用于具有超出细胞外抗体和基于细胞因子的方法的能力的免疫细胞工程化。
本文描述的数据示出了无病毒载体的微流体方法对免疫细胞的细胞溶质递送的惊奇效力,所述免疫细胞(在本发明之前)难以工程化并且难以实现化合物/组合物的细胞溶质递送。所述数据还证明了这种方法促进多种大分子(包括多糖、蛋白质和核酸)递送到t细胞、b细胞、髓样细胞(cd11b+)和树突细胞的能力。在靶向五种不同基因的基于sirna的敲低研究中验证了递送材料的功能。最后,hiv感染研究强调了此方法改变细胞表型并影响病毒复制用于抑制传染病的效用。本文所述的无载体递送系统提供了安全、可靠和有效的方法,用于工程化免疫细胞的功能和/或改变免疫细胞的表型、功能、激活状态。
用于未鉴定病原体的快速应答疫苗系统
传染病原体对士兵和平民一样构成严重威胁。为了防止潜在的生物攻击和大流行病毒的发展,必须开发稳健的快速应答疫苗接种能力。使用最先进的技术,即使可以由科学家分离和表征强毒株,开发有效的疫苗也可能需要数年和数十亿美元。尽管几十年的研究,许多致命的病原体(诸如埃博拉和hiv)仍然不能被当代的疫苗接种方法解决。本发明通过使用本文所述的无载体细胞内递送平台直接工程化个体的免疫细胞来提供用于对抗病原体的快速应答的多靶向的个体化的保护的方法和装置。所述方法可以在识别新感染个体的数小时内有效地疫苗接种局部危险人群,并且具有优于先前方法的几个优点,如下表3所示。
表3
抗原蛋白直接递送到个体apc(例如,b细胞、dc和重编程的t细胞)的细胞质消除了鉴定和开发减毒病毒载体的需要,同时通过诱导对抗病毒逃逸所必需的多靶向免疫应答提供更有效的保护。可紧急移动的疫苗接种平台在数小时内而不是数月/数年内处理发病。
当个体怀疑有病原体时,首先取感染组织进行活检。含有病原体的这种组织被裂解,使得分子组分(即抗原)保持完整,同时由于裂解过程使病原体失活。然后将此抗原混合物通过抽取健康个体的血液并在重新进入血流之前驱动他们的细胞通过微流体细胞内递送装置(3)递送到所述健康个体的免疫细胞。此递送过程将病原体相关抗原引入血液中驻留的apc的细胞质中,所述apc包括t细胞、b细胞、树突细胞和单核细胞。然后所述抗原片段由mhc-i呈递,并驱动疾病特异性细胞毒性t细胞ctl的激活以提供保护。裂解可以在获得感染的活检(从单次活检中为多个健康患者生产剂量)的1-2小时内完成,并且随后接种健康患者将需要<5分钟每人。(图5)此外,如果病原体已被表征(例如在hiv或埃博拉的情况下),可以用化学定义的合成肽作为抗原来源来替代细胞裂解物的使用,因此消除了围绕使用裂解物的潜在的安全性问题。
当常规的昂贵的多年开发过程可能导致大量的生命损失时,快速应答疫苗平台提供了针对多种生物威胁的一线防御。这种接种方法也适用于肿瘤和传染病治疗。
用于b细胞中的细胞内抗原负载的微流体挤压
抗原呈递细胞(apc)是从组织中捕获外来的或自身的蛋白质和肽的免疫细胞(包括树突细胞、巨噬细胞、b细胞)的不同亚群,并且激活适应性免疫细胞以生成针对这些抗原的炎症或耐受原性免疫应答。蛋白质通过其周围环境的液相取样或受体介导的外来微生物或死细胞碎片的摄入而在体内被apc摄入。摄入的蛋白质被降解成肽片段(抗原),所述肽片段被加工并与共刺激信号一起呈递给t细胞,这说明了基于apc所接受的特异性信号和所呈递的抗原的初始t细胞激活。由于在t细胞激活中的这种关键作用,负载抗原并离体激活的纯化apc可用于在培养物中扩增功能t细胞(例如,用于过继t细胞疗法)或用作体内有效的细胞疫苗。使用微流体系统,apc的离体操作被示出作为生成具体类型的免疫的替代方法是有效的,特别是在疾病(诸如癌症和hiv)中的细胞毒性t淋巴细胞(ctl),其中病原细胞的靶向杀伤是至关重要的并且内源性apc功能被积极抑制。尽管是有前途的临床前研究,基于细胞的疫苗的临床转译仍受到多种限制因素的阻碍。用于将抗原递送到免疫细胞的细胞溶质的微流体装置和相关方法解决了早期方法的许多问题和缺点。
先前的基于细胞的疫苗的临床研究已经集中在树突细胞(dc),即所谓的“专业”apc,因为它们引发ctl的效率以及它们高活性的细胞外蛋白质摄取和抗原加工能力。然而,作为临床使用的平台,dc受到人血液中相对缺乏、复杂亚群不均匀性、寿命短和无法增殖的限制。这些挑战导致其它细胞类型也被考虑用于基于细胞的apc疫苗,包括巨噬细胞和b细胞。因为它们作为淋巴细胞的独特性质及其克服dc的许多限制因素的潜力,所以b细胞是此目的的理想细胞群。例如,b细胞在血液循环中丰富(高达50万个细胞/ml血液),可以在细胞激活时增殖,并且在静脉内施用时有效归巢到次级淋巴样器官。
b细胞作为apc的这些优点被b细胞获取和加工用于引发t细胞的抗原的能力的限制因素抵消。b细胞表达基因重排的b细胞受体(bcr),所述b细胞受体在与其靶抗原结合时促进抗原摄取和b细胞激活。虽然b细胞能够通过其bcr内化抗原并引发原代t细胞应答,但是与有效地从它们的周围胞饮和吞噬抗原的巨噬细胞和dc相比,它们对非特异性抗原(即抗原未被它们的bcr识别)的摄取较差。此外,ctl的引发通过由i类mhc分子的肽呈递发生,i类mhc分子通常仅负载位于细胞溶质中(i类mhc加工机械主要驻留其中)的抗原。相比之下,通过bcr吸收到内吞溶酶体中的蛋白质倾向于被定向到呈递给cd4+t细胞的mhcii类呈递途径。或者,b细胞和其它专业apc可以通过交叉呈递使i类mhc分子负载肽,这是通过蛋白酶体加工或液泡蛋白降解由胞吞抗原产生i类肽-mhc复合物的过程,但是此过程通常是非常低效率的。
尽管已经开发了增加b细胞中抗原摄取和交叉呈递的方法,但是这些策略在很大程度上依赖于靶向用于胞吞摄取的特异性受体,激活与流体相蛋白暴露组合的b细胞以增加非特异性胞吞作用,递送抗原作为免疫刺激复合物,或生成融合蛋白以定向b细胞功能。这些方法受到以下事实的限制:抗原摄取与由通过靶向受体的信号传导介导的b细胞状态的其它变化相耦合,这意味着不能单独调节抗原负载和b细胞激活。例如,已经示出静息b细胞对初始的cd8+t细胞具有耐受原性,即治疗自身免疫的潜在有用的性质,并且b细胞的激活在这种应用中将是有问题的。用dna、rna33或编码抗原的病毒载体转染b细胞也示出了希望,但是受到众多问题的限制,所述问题诸如电穿孔毒性、病毒载体包装能力、转导效率、稳定性和抗载体免疫等。本文所述的方法提供了对早期方法的这些缺点的解决方案。
通过暂时的质膜破裂/扰动(perturbation)将全蛋白(即未加工的抗原)直接细胞溶质递送到活的b细胞中,在b细胞穿过微流体装置的微量通道中的收缩(机械破裂)时完成。使用明确定义的和本领域公认的模型抗原卵白蛋白(ova),通过此方法递送全蛋白使得甚至静息的b细胞也能在体外和体内引起效应ctl的稳健引发。这种通过mhci类的全蛋白递送和抗原呈递的方法是b细胞中的第一种抗原递送方法,所述方法将抗原负载与b细胞激活过程分开,这允许将这两个过程单独定制用于免疫原性疫苗或耐受原性疫苗。细胞挤压提供了替代的模块化平台,所述平台引发用于体外ctl扩增的自体b细胞并且促进基于b细胞的疫苗的开发。
使用以下材料和方法来生成与抗原呈递有关的数据。
试剂。tritc和瀑布蓝标记的3kda葡聚糖购自lifetechnologies。fitc标记的40kda葡聚糖购自chondrex。模型抗原、低内毒素卵白蛋白蛋白购自worthingtonbiochemicalcorporation。cpgodn1826(cpgb)、cpgodn2395(cpgc)和lps大肠杆菌k12(lps)购自invivogen。多聚体/megacd40l购自adipogen和enzolifesciences。
细胞分离。分离/纯化或富集免疫细胞或免疫细胞亚群的方法和程序是本领域熟知的。例如,对于人,外周血单核细胞从通过静脉穿刺取的全血和使用标准协议分离的免疫细胞亚群(例如b细胞、t细胞、树突细胞、巨噬细胞)获得。骨髓也被用作免疫细胞的来源。
对于本文所述实例中的b细胞分离,从小鼠收获脾脏并通过70μm细胞过滤器捣碎。裂解红细胞,并且按照制造商的说明书,用b细胞分离试剂盒、小鼠(miltenyibiotec)从细胞悬浮液中分离静息的b细胞。分离后,如通过流式细胞术测量的,b细胞悬浮液>95%b220+。对于cd8+t细胞分离,收获脾脏和腹股沟淋巴结并捣碎。红细胞裂解后,根据制造商的说明书用cd8a+t细胞分离试剂盒、小鼠(millenyibiotec)分离cd8a+t细胞。用cd4+t细胞分离试剂盒、小鼠(millenyibiotec)在脾脏和腹股沟淋巴结悬浮液进行cd4+t细胞分离。如通过cd8a或cd4染色和流式细胞术所测量的,t细胞始终>90%纯度。所有细胞培养在增补有1μl/ml的55mmβ-巯基乙醇的t细胞培养基(含10%fbs、青霉素-链霉素、1x丙酮酸钠的rpmi)中进行。
通过细胞挤压的蛋白质递送。使用细胞挤压、微流体装置和压力系统(sqzbiotech)进行全蛋白抗原递送到静息b细胞;所使用的芯片设计包括30-4×1、10-4×1和30-5×5,其中x-y×z表示xμm长和yμm直径的尺寸的z个顺序收缩通道。b细胞以5×106个细胞/ml悬浮在具有100μg/ml卵白蛋白、0.3mg/mltritc或者太平洋蓝标记的3kda葡聚糖或0.3mg/mlfitc标记的40kda葡聚糖的培养基中,并放在冰上。微流体芯片和支架也放在冰水浴中,直到冷却。将细胞悬浮液在120psi下以200μl等分试样通过装置送出。胞吞对照b细胞在具有ova的培养基中相同地制备,但没有通过微流体装置。负载抗原后,将细胞在室温下静置5分钟,并用pbs洗涤两次。为了评估递送效率,通过流式细胞术(荧光标记的组合物的检测)测量抗原或其它递送的摄取。
体外细胞培养、活化和增殖测定。为了表征机械化(mechano-porated)b细胞的体外激活,通过sqz装置的细胞和胞吞对照细胞以5×105个细胞/ml在具有5μmcpgb、5μmcpgc或100ng/mllps的96孔u底板中孵育。在24小时和48小时进行流式细胞术来测量cd86、cd40、cd69、mhci类和mhcii类的细胞表面水平。对于体外增殖测定,将纯化的siinfekl卵白蛋白肽特异性的ot-icd8+t细胞(mhc1类限制)或ot-iicd4+t细胞(mhcii类限制)以107个细胞/ml悬浮并且用cfse(5μm,lifetechnologies)标记10分钟。一次洗涤后,将t细胞和b细胞以1:0.8比例涂铺在96孔u底板中的200μlt细胞培养基中。将cpgb、cpgc或lps添加到适当的孔中,并且将抗cd3/cd28免疫磁珠(lifetechnologies)以1个磁珠/t细胞添加到阳性对照孔中。用于细胞因子分析的上清液收集和评估t细胞增殖的流式细胞术在第2天和第4天进行。
体内增殖测定。在第1天,将106个用cfse(5μm)标记的ot-ithy1.1cd8+静息t细胞眼眶后(retro-orbitally)(r.o.)注射到c57bl/6小鼠体内。第二天(第0天),用1-300万个cd45.1+b细胞眼眶后注射动物,所述b细胞在前一天使用微流体sqz装置已经负载有ova并且与5μmcpgb孵育过夜,或者在临注射前通过机械破裂负载有ova并且不暴露于任何tlr配体。在第4天将动物进行尸检,并收获其脾脏、腹股沟淋巴结和颈淋巴结。通过细胞过滤器捣碎器官。将单细胞悬浮液与小鼠抗cd16/cd32(ebioscience)孵育以减少非特异性抗体结合,并用抗cd8-apc、抗-b220-pe-cy7、抗cd45.1-percp-cy5.5和抗thy1.1-apc-cy7染色。使用已知方法进行流式细胞术来确定细胞数量和/或cfse稀释度。
机械破裂(微流体细胞挤压)能够快速并有效地递送大分子
使用本文所述的装置来完成用于使b细胞负载抗原的机械破裂过程。活细胞穿过硅装置中的平行微流体通道;在每个通道中,1个或更多个收缩在穿过所述装置的细胞的膜中产生暂时的孔或扰动。存在于周围流体中的大分子货物在此暂时的扰动/膜破裂期间扩散到细胞中,导致细胞内负载。机械破裂对于促进将大分子细胞溶质递送到各种细胞类型(包括原代鼠b细胞)中是有效的;用30μm长度和5μm宽度尺寸的5个连续收缩实现了有效的递送。可以改变装置设计(将平行收缩通道数量增加至75、进入区域更长、可逆性等),推进定制用于蛋白质递送到b细胞和随后的抗原呈递的机械破裂参数。试验的优化实验示出,在120psi下运行的30-4×1微流体芯片(每个为30μm长和4μm直径的通道1个收缩)是用于在5×106个b细胞/ml的浓缩离子下具有高效率递送和高细胞活力的鼠b细胞的机械破坏的有效的芯片设计。本文描述了其它配置,例如30-5x5相对于30-4x1。在此压力下细胞以约100万个细胞/秒的速度流过装置。与胞吞作用相比,微流体挤压推进了葡聚糖摄取的极大增强,且3kda和40kda葡聚糖的内化分别增加约65倍和约25倍。这表示两种葡聚糖递送到所有细胞的75-90%;相比之下,少于10%的静息b细胞胞吞可检测量的货物。机械破解后恢复的细胞的活力为约95%,与胞吞作用对照相似。可以穿过这些一次性微流体芯片的细胞的最大数量的容量范围为每个装置1-500万个细胞;然而,装置运行多个等分的细胞(每次运行100万个细胞)直到堵塞,并且在给定实验中运行通过每个单独装置的细胞的最大数量通常显著超过500万个细胞。从第一次运行到装置堵塞,递送的细胞的百分比可变性很低,这指示装置内可变性很小。同一实验阶段内的多个装置的递送性能(装置间可变性)非常一致。认识到一些应用可能需要更高数量的b细胞,即微流体装置在不同细胞密度下的功效。细胞内葡聚糖递送的效率在很大程度上独立于高达至少50×106个/ml细胞的细胞浓度,这指示稳健的可扩展性的潜力。
示出以增强抗原呈递细胞起作用的处理的b细胞的示意图示于图30中。
用全蛋白挤压的多克隆b细胞扩增体外分泌效应细胞因子的抗原特异性cd8+t细胞
为了确定机械破裂是否可以促进蛋白质递送到细胞溶质i类mhc抗原加工和呈递机制,我们利用上述最佳条件通过在周围培养基中存在过量ova的情况下将细胞穿过微流体装置将模型蛋白卵白蛋白(ova)递送到静息的纯化多克隆b细胞中。然后,在存在或不存在cpg作为b细胞激活刺激的情况下,将挤压的b细胞与cfse标记的ot-icd8+t细胞共培养,所述t细胞携带针对mhci类限制性ova肽siinfekl具有特异性的转基因t细胞受体。通过流式细胞术分析ot-it细胞中的cfse稀释度,以评估t细胞响应于b细胞呈递抗原的增殖/扩增。发现通过细胞挤压递送全蛋白到b细胞能够在体外实现稳健的mhci类抗原呈递和抗原特异性cd8+t细胞引发。如本文所述的细胞挤压主要将抗原定向到细胞溶质而不是发生mhcii类负载的内体区室。与此相一致,数据指示,共培养4天后,静息b细胞和cpg激活的机械化b细胞均不能扩增ova特异性ot-iicd4+t细胞。通过在共培养的第2天和第4天测量效应分子的分泌来测定b细胞引发的cd8+t细胞群的功能性。通过挤压负载抗原的b细胞(无论是静息的还是激活的)引发t细胞分泌大量的颗粒酶b、ifn-γ和tnf-α,而通过胞吞作用的负载抗原的b细胞产生基础水平的细胞因子。
挤压的b细胞在体内引发抗原特异性cd8+t细胞
此外,体外抗原特异性t细胞扩增平台,如上所述处理的b细胞可以作为用作细胞疫苗的树突细胞的替代物使用。为了评价体内性能,将表达thy1.1作为同源标志物的cfse标记的ot-icd8+t细胞作为抗原呈递的报告子过继转移到受体小鼠体内。一天后,在机械破裂介导的抗原负载之后立即注射静息的b细胞,或者注射随后在体外用cpg激活24小时的机械破裂负载的b细胞。在用cpg激活之后立即或24小时之后通过胞吞作用负载抗原的静息的b细胞被用作对照。b细胞转移后4天,处死小鼠,并且通过流式细胞术分析脾脏和腹股沟淋巴结的ot-i增殖。与体外结果一致,机械化b细胞能够引起过继转移的ot-it细胞的分裂,而胞吞作用对照仅示出基础分裂。cpg激活的和静息的挤压的b细胞在脾脏中均引起ot-i增殖(分别比较sqz相对于胞吞作用,分为注射ot-it细胞的约45%和约35%,且p<0.001和p=0.001)。相比胞吞作用b细胞,cpg激活的和静息的挤压的b细胞也在淋巴结中引起了同样增强的ot-i增殖(分别比较sqz与静息和cpg的胞吞作用,由cpgb细胞和静息的挤压的b细胞分为注射ot-it细胞的约40%和约35%,p<0.001)。胞吞作用对照示出了淋巴结内约4%的基线分配。这些结果表明,通过微流体机械破裂负载的b细胞可用于细胞疫苗。
通过评价应答cd8+t细胞的激活状态的实验进一步证实所述结果(图29)。
因此,如本文所述的负载特异性抗原的b细胞可用作基于细胞的疫苗的apc并且用作扩增抗原特异性t细胞的自体试剂,具有优于树突细胞的显著优点,特别是关于它们在外周血中大量随时可用以及它们在培养中进一步大量扩增的能力。在本发明之前,用于在b细胞中有效地负载抗原的方法,特别是对于i类mhc呈递的方法,已经成为作为apc在体外或体内使用的多克隆b细胞的开发的主要障碍。本文描述的装置和方法代表了使用基于微流体的机械变形和被动扩散导致蛋白质抗原直接负载到静息或激活的b细胞的细胞溶质中以及来源于天然全蛋白的肽的mhci类呈递的简单方法。机械破裂所赋予的优点是众多的,包括不需要加工、工程化或修饰的天然蛋白质的递送,所述过程的快速性质,相对较高的递送效率和功能结果,以及将材料递送至与细胞生物学分开(诸如编程刺激或受体靶向)的静息细胞的能力。
将天然全蛋白或多肽直接递送到细胞溶质用于加工和mhci类呈递的能力能够在天然抗原加工之后实现多种肽的无偏差呈递。微流体细胞挤压装置还能够递送蛋白质的混合物,包括肿瘤裂解物(例如肿瘤活检裂解物)或其它复合蛋白质来源。蛋白质独立于细胞激活状态被递送用于mhci类加工的上述证明是此方法的主要优点,并且所述方法能够分开蛋白质负载和细胞编程,促进各组分的独立调控。
示例性设计含有75个平行收缩通道;然而,通道数量的增加或平行操作多个装置显著地改进了通过量。通过这种方法实现的易用性和快速加工(例如,对于总实验时间,约1×106个细胞/s的速度流过装置<2小时)为临床转译提供了益处,诸如减少制备基于细胞的治疗剂所需的时间和资源。
使用b细胞作为apc,与基于dc的方法相比,制备单剂量细胞疫苗所需的患者血液量大大降低。避免通过细胞摄取过程(如胞吞作用或胞饮作用)负载蛋白质所需的时间。体外结果证明了b-apc作为扩增效应ctl的替代平台的显著潜力,并且挤压的b细胞也在体内作为有效的apc起作用。所述方法也可用于共同负载mhci类和ii类抗原呈递途径以生成cd4+t细胞辅助。
通过b类细胞的i类限制性抗原加工和呈递
因此,通过细胞挤压加工b细胞来生成自体抗原呈递细胞(apc)以在体外和体内均引发抗原特异性t细胞。这种微尺度细胞挤压加工在质膜中产生暂时的扰动或孔,使得能够通过机械破裂将全蛋白从周围培养基中细胞内递送到b细胞内。静息和激活的b细胞加工和呈递抗原都是通过机械破裂专门递送到抗原特异性cd8+t细胞,而不是cd4+t细胞。挤压的b细胞在体外引发并扩增大量效应cd8+t细胞,所述效应cd8+t细胞产生对细胞溶解功能至关重要的效应细胞因子(包括颗粒酶b和干扰素γ)。挤压的负载抗原的b细胞也能够在过继转移到小鼠时在体内引发抗原特异性cd8+t细胞。这些数据证明机械破裂/膜扰动是用于b细胞抗原负载、抗原特异性cd8+t细胞引发和将b细胞激活与抗原摄取分开的有用且高效率的方法。
t细胞作为免疫刺激或耐受性的抗原呈递细胞
在本发明之前,出于免疫刺激或耐受性目的的抗原呈递通常被认为是被称为抗原呈递细胞(例如b细胞、树突细胞和巨噬细胞)的细胞的选择亚群的范围,并且不包括t细胞,因为他们被认为缺乏在促进免疫刺激或耐受性的情况下加工和呈递抗原的必要机制。所述装置和挤压介导的细胞的抗原负载导致了惊人的发现,将抗原蛋白直接递送到t细胞的细胞溶质赋予了此类加工细胞抗原呈递表型。
所述方法在体内以前面未报道的方式调控t细胞免疫应答。使用细胞挤压装置平台将模型抗原卵白蛋白递送至鼠原代t细胞,并将所述细胞注射到宿主体内以测量cd8+t细胞应答。负载抗原的t细胞能够生成如通过流式细胞术测量的大量的cd8+t细胞应答,这指示通过外部机制(例如细胞挤压)负载抗原的t细胞确实可以在其表面上呈递表位,与其它t细胞交流并生成可测量的免疫应答。此应答显著大于对照,并且与使用专业apc(诸如树突细胞)的实验中观察到的应答相当。
这一发现是预期不到的,并且对免疫学和免疫治疗领域具有重要意义,因为它证明了t细胞作为用于治疗和研究应用的有效抗原呈递者的用途。在本发明之前,dc是唯一被广泛接受用于此用途的细胞,并且如上所述,细胞挤压方法可用于出于此目的的快速、有效和大大增强了的b细胞的生产。因为t细胞比dc更丰富且更容易获得,因此它们提供了更大的临床功效并且促进了对当前昂贵的难以生产的细胞疫苗应用的更广泛的影响。因此,t细胞现在可以用作抗原呈递细胞,用于呈递任何类型的抗原、目的(耐受原性与免疫刺激)、注射途径、临床以及研究应用。
使用细胞挤压方法用整个的未加工的抗原处理的小鼠t细胞作为抗原呈递细胞起作用(图29)。从小鼠脾脏中分离鼠t细胞,并使用示例性的10-3和30-4装置配置将ova以(250、50、10ug/mlova)的ova浓度递送到rpmi中的t细胞。然后用ot-1t细胞(siinfekl肽表位特异性)培养这些t细胞,并评估激活标志物cd25和cd69。结果证明,使用细胞挤压将整个的未处理的抗原(例如全长卵白蛋白)细胞溶质递送到其中的t细胞,令人惊奇且有效地起作用,以将抗原呈递给表位特异性效应cd8+t细胞并激活表位特异性效应cd8+t细胞。
另外的数据证明了dq-ova(dqtm卵白蛋白,目录号d-12053,molecularprobes,inc.)递送到从血液分离的原代人t细胞(图28a和b)。如果蛋白质被加工,则dqova是化学缀合形式的卵白蛋白蛋白,其在fitc通道上发荧光,但如果蛋白质未被细胞加工,则不会发荧光。因此,对于装置处理的情况fitc信号的出现指示细胞处理了dq-ova抗原,还支持了制造抗原呈递t细胞(t细胞作为apc)在除了上述小鼠系统之外的人体系统中起作用的观察。在一些实验中,共同递送在太平洋蓝通道上发荧光的3kda葡聚糖染料。结果示出了cd4t细胞和cd8t细胞之间ova加工方面的差异以及相对于胞吞作用对照的显著改进。这些数据指示,当抗原被直接递送到t细胞的细胞溶质中时,抗原加工由人t细胞进行。dq-ova抗原改变其荧光性质的数据示出抗原正在人t细胞中被加工,并且因此由组织相容性抗原呈递给t效应细胞。
体内也证明了表位特异性t细胞的激活。图26示出了体内cfse增殖测定(抗原特异性cd8+t细胞的增殖)的结果。当cd8t细胞在小鼠中被激活并增殖时,它们稀释cfse染料并具有较低的荧光强度。在这种情况下,通过装置或阳性对照处理的供体t细胞在受体小鼠中产生了显著更大的cd8t细胞的激活和增殖。胞吞作用对照示出了最小效应。
还在体内证明了,抗原特异性ot-it细胞在小鼠中响应于用抗原处理的野生型t细胞的疫苗接种的增殖(图27)。通过cfse染色测量t细胞增殖应答。随着细胞增殖,染色剂被稀释;因此,越低的强度指示越大的应答,较高的强度峰指示无/少应答。每列代表使用来源于同一只小鼠的淋巴结和脾脏的实验的重复。每列实验涉及3只小鼠(总计n=9只小鼠)。
以下描述的材料和方法用于生成有关t细胞作为上述免疫刺激或耐受性的抗原呈递细胞的数据。实验时间表。使用以下实验时间表:
第0天:将cfse/celltraceviolet标记的ot-1cd45.1t细胞注射入初始b6宿主
第1天:将抗原递送到t细胞apcs(tc-apc),将tc-apc和对照apc注射入b6宿主
第5/6天:从免疫小鼠收获脾脏和淋巴结,通过流式细胞术分析t细胞增殖
t细胞分离(用于过继转移)。材料:100umfalcon细胞过滤器;70umfalcon细胞过滤器;氯化铵钾(ack)裂解缓冲液;cd8a+t细胞分离试剂盒(miltenyibiotec);macs细胞分离柱。t细胞培养基(具有10%fbs、青霉素-链霉素、1x丙酮酸钠、50umb-巯基乙醇的rpmi);macs缓冲液(pbs中0.5%bsa、2mmedta)。方法:在c57bl/6j对照下从ot-1cd45.1rag2-/-小鼠中收获脾脏和皮肤引流淋巴结,并通过湿的100um细胞过滤器捣碎到50mlfalcon管中。将过滤器用macs缓冲液洗涤,然后以500rcf自旋沉积4分钟。吸出上清液,然后添加3ml的ack裂解缓冲液以裂解红细胞。将反应用12ml的macs缓冲液淬灭,然后以500rcf自旋沉积4分钟。将细胞沉淀物以40ulmacs缓冲液/1×107个细胞重悬,然后通过70um细胞过滤器过滤到50mlfalcon管中。每1×107个细胞添加10ul的cd8a+t细胞抗体混合物,并在冰上孵育10分钟。添加30ulmacs缓冲液/1×107个细胞,然后每1×107个细胞添加20ul链霉亲和素珠粒,并在冰上孵育15分钟。将细胞以800rcf自旋沉积5分钟并吸出上清液,然后重悬于3mlmacs缓冲液中。通过将3mlmacs缓冲液穿过,然后将70um细胞过滤器上的3ml细胞悬浮液穿过在15mlfalcon管中制备细胞分离柱。将柱子用3mlmacs缓冲液洗涤3次,将汇集的流通液以500rcf自旋沉积4分钟。吸出上清液,并且将富集的cd8+t细胞悬浮于t细胞培养基中。通过染色cd8a和流式细胞仪分析检查到>90%的纯度。
celltraceviolet/cfse标记。材料:dmso中的cfse(lifetechnologies,grandisland,ny);dmso中的celltraceviolet(lifetechnologies,grandisland,ny);纯化的ot-icd45.1cd8+t细胞;无菌pbs;无菌胎牛血清。方法:如上所述富集ot-1cd45.1cd8+t细胞,并悬浮于15mlfalcon管中的2.5ml温的pbs中。将dmso中的celltraceviolet或cfse溶解于10um的温的pbs中,然后将2.5ml添加到细胞悬浮液中(终浓度5um)。将细胞置于37℃温水浴中10分钟,然后添加7mlpbs以淬灭反应。使用无菌玻璃移液管将2mlfbs层压到底部,然后以500rcf自旋沉积4分钟。吸出上清液,并且再次在10mlpbs中洗涤细胞,然后以500rcf自旋沉积并以2×107个细胞/100ulpbs重悬。
t细胞分离(作为tc-apc)。材料:100umfalcon细胞过滤器;70umfalcon细胞过滤器;ack裂解缓冲液;pant细胞分离试剂盒(miltenyibiotec,sandiego,ca);macs细胞分离柱;t细胞培养基(具有10%fbs、青霉素-链霉素、1x丙酮酸钠、50umb-巯基乙醇的rpmi);macs缓冲液(pbs中0.5%bsa、2mmedta)。方法:从c57bl/6j小鼠中收获脾脏和皮肤引流淋巴结,并通过湿的100um细胞过滤器捣碎到50mlfalcon管中。将过滤器用macs缓冲液洗涤,然后以500rcf自旋沉积4分钟。吸出上清液,然后添加3ml的ack裂解缓冲液以裂解红细胞。将反应用12ml的macs缓冲液淬灭,然后以500rcf自旋沉积4分钟。将细胞沉淀物以40ulmacs缓冲液/1×107个细胞重悬,然后通过70um细胞过滤器过滤到50mlfalcon管中。每1×107个细胞添加20ul的pant细胞抗体混合物,并在冰上孵育10分钟。添加30ulmacs缓冲液/1×107个细胞,然后每1×107个细胞添加20ul链霉亲和素珠,并在冰上孵育15分钟。将细胞以800rcf自旋沉积5分钟并吸出上清液,然后重悬于3mlmacs缓冲液中。通过将3mlmacs缓冲液穿过,然后将70um细胞过滤器上的3ml细胞悬浮液穿过在15mlfalcon管中制备细胞分离柱。将柱子用3mlmacs缓冲液洗涤3次,将汇集的流通液以500rcf自旋沉积4分钟。吸出上清液,并且将富集的t细胞悬浮于t细胞培养基中。通过染色cd3e和流式细胞仪分析检查到>90%的纯度。
t细胞抗原递送和体外激活。材料:纯化t细胞;细胞挤压30-4芯片;细胞挤压装置;脂多糖(lps);t细胞培养基(具有10%fbs、青霉素-链霉素、1x丙酮酸钠、50umb-巯基乙醇的rpmi);ova蛋白;siinfekl肽。方法:将纯化的t细胞在具有1ug/mllps的t细胞培养基中在冰上孵育30分钟。将t细胞在t细胞培养基中洗涤2次,并以500rcf自旋沉积4分钟,然后与0.01mg/ml-0.1mg/mlova或1ug/mlsiinfekl在冰上温育。细胞挤压设备根据标准协议装配30-4芯片,然后在100psi下将200μlt细胞培养基通过每个芯片。一半的t细胞在100psi下穿过细胞挤压装置,而另一半被留在冰上(胞吞情况)。将流通液收集在96孔板中并在冰上放置15分钟用于使细胞膜修复。将细胞以350rcf自旋沉积10分钟,并且轻弹出去上清液。在注射入宿主之前,将细胞在1ug/mllps中重激活10分钟。
患者免疫细胞的富集
细胞挤压也可用于离体富集或扩增患者来源的免疫细胞,用于随后转移回患者。例如,抗原通过机械破裂被递送到抗原呈递细胞(树突细胞、b细胞、t细胞、巨噬细胞)的细胞溶质。这些抗原呈递细胞在mhc/hla异二聚体的情况下在其表面上加工并呈递的加工的抗原以刺激和扩增t细胞。然后将扩增的t细胞群再灌注到患者体内以增大治疗性免疫应答。例如,抗原是增大抗肿瘤应答的肿瘤抗原(或裂解物)。或者,抗原或细菌或病毒抗原(或其片段或减毒或杀死的细菌细胞或病毒颗粒)以增大微生物传染病。在另一个实例中,抗原是与耐受原(如上所述)一起呈递的自身抗原,以耐受并随后扩增耐受免疫细胞群,用于再灌注回患者体内来下调异常(例如自身免疫)应答。
t细胞作为抗原呈递细胞的用途
基于使用抗体治疗剂激活患者t细胞的癌症免疫疗法已经在具有高突变频率和预先存在的对肿瘤相关抗原(taa)的t细胞应答的那些适应症中示出了成功(topalian等,cancercell2015;27:450-461;hodi等,nengljmed2010;363:711-723;topalian等,cell2015;161:185-186)。因为许多癌症具有低突变频率和低的自发性t细胞应答速度,所以使用癌症疫苗可能有助于加强t细胞对taa的应答(melief等,jclininvest2015;125:3401-3412)。癌症疫苗具有作为单一疗法(ly等,cancerres2010;70:8339-8346;kantoff等,nengljmed2010;363:411-422)、与检查点封闭疗法组合(agarwalla等jimmunother2012;35:385-389)或与过继t细胞疗法组合(lou等,cancerres2004;64:6783-6790)使用的潜力。
在临床前环境下,使用专业抗原呈递细胞(apc)进行癌症疫苗接种的策略已经证明抗原致敏的树突细胞(dc)可以生产抗肿瘤的保护性免疫(celluzzi等,jexpmed1996;183:283-287;mayordomo等,natmed1995;1:1297-1302;7489412;flamand等,eurjimmunol1994;24:605-610)。在各种类型的癌症中进行的临床研究(nestle等,natmed1998;4:328-332;hu等,cancerres1996;56:2479-2483;hsu等,natmed1996;2:52-58;reichardt等,blood1999;93:2411-2419;morse等,clincancerres1999;5:1331-1338;yu等,cancerres2001;61:842-847)表明,单核细胞来源的dc能够在人体中引发抗原特异性免疫,然而临床应答较低。例如,第一个fda批准的基于dc的癌症疫苗(sipuleucel-t,provenge,dendreon,seattle,wa)在激素难治性前列腺癌患者中证明了四个月的延长的存活(kantoff等,nengljmed2010;363:411-422;higano等,cancer2009;115:3670-3679)。在解释为什么这种疫苗具有如此低的临床益处的假说中,用于使sipuleucel-tdc负载抗原的程序可能主要导致了mhcii类抗原呈递,而不是具有mhci类和ii类呈递。因此,低细胞毒性t细胞活性可能在此产品的较差功效中起作用。
开发的用于将taa递送到dc的方法利用细胞挤压平台在mhci类和ii类组织相容性分子上进行最佳抗原呈递。通过解决现有抗原负载技术的限制因素,本文所述的方法在体内导致更强的细胞毒性和辅助t细胞应答。所述平台包括微流体芯片,当所述细胞穿过收缩时能够使细胞迅速变形以暂时破裂细胞的膜并且能够将靶材料输送到细胞质。通过消除对电场或外源性增强子或载体材料的需要,所述方法使细胞毒性和脱靶效应的可能性最小化。
靶向病毒抗原或新抗原
由于可以诱导自身抗原的中枢耐受性,所以治疗性疫苗接种现在优选靶向新抗原(gubin等,jclininvest2015;125:3413-3421;gubin等,nature2014;515:577-581;yadav等,nature2014;515:572-576;quakkelaar等,advimmunol2012;114:77-106;castle等,cancerres2012;72:1081-1091)和病毒抗原(melief等,jclininvest2015;125:3401-3412;quakkelaar等,advimmunol2012;114:77-106)。
为了鉴定和评价抗原,hpv16的癌基因e6和e7的九种合成长重叠肽(slp)的混合物(hpv16-slp)被用于评价免疫应答,所述混合物代表了这两种癌蛋白的整个长度并在临床前模型以及患有癌症前期的患者中展示出应答。细胞挤压平台用于使dc负载hpv16-slp混合物,其允许肽被引导到mhci类和ii类呈递途径中,并且因此诱导cd4t细胞和cd8t细胞的抗原特异性扩增。因此,此方法推进了克服hla特异性短肽的限制因素并且能够进行更有效的治疗的多表位应答。
将hpv16-slp细胞挤压到人和小鼠dc。测试将hpv16-slp混合物递送到人和小鼠dc的不同的细胞挤压条件。评价具有不同的收缩长度、收缩宽度、收缩数量以及接近收缩的角度的芯片设计。关于加工参数,研究了细胞浓度、递送介质、递送介质中的肽浓度、加工温度、操作压力和穿过芯片的次数。优化加工的实验方法还包括hpv16-slp肽混合物的荧光标记,以解释递送到细胞中的肽的量。此外,在i类和ii类的情况下,通过流式细胞术检测所呈递的具有tcr特异性四聚体的肽,解释了肽的最佳加工和呈递。将此方法与用于交叉呈递的负载hla特异性i类短肽的dc进行比较。
此研究确定了使人和鼠dc有效地负载hpv16-slp肽抗原或抗原混合物(例如病毒抗原的混合物)的芯片设计和递送条件,例如包括有效地加工长肽和由apc(例如树突细胞)负载抗原的条件。
hpv16-slp挤压的dc扩增抗原特异性t细胞的体外和体内评估。
测试了在几种条件下负载肽混合物的dc扩增抗原特异性t细胞的能力的功能。例如,通过用hpv16-slp和cpg免疫小鼠来诱导抗原特异性t细胞。加强后8天,从免疫小鼠的脾脏中分离t细胞并用于与负载hpv16-slp的dc共培养,以确定负载细胞触发抗原特异性cd4t细胞和cd8t细胞扩增并产生细胞因子的能力。为了测试人负载的dc,使用与hpv16反应的人t细胞克隆在几种条件下与负载hpv16-slp混合物的人dc共培养。评估抗原特异性t细胞扩增和细胞因子产生。在体外测试是否人和/或小鼠负载dc是功能性的并且能够扩增抗原特异性t细胞。负载hla特异性i类短肽的dc被用作对照。匹配hla的t细胞被用作应答者。
此后,并且为了测试基于细胞挤压dc的疫苗在体内生成对抗hpv-16的抗肿瘤应答的有效性,将小鼠疫苗接种负载hpv16-slp的dc并用tc1肿瘤攻击(具有hpve6和e7以及c-h-ras癌基因的c57bl/6小鼠的逆转录病毒转导的肺成纤维细胞)。读出攻击的小鼠的存活。
这些研究示出,用细胞挤压技术生成的基于dc的疫苗在体内触发了保护性抗肿瘤t细胞应答和/或抗病毒应答。
其它实施方案
本文引用的专利和科学文献建立了本领域技术人员可获得的知识。本文中引用的所有美国专利和公开或未公开的美国专利申请均以引用方式并入本文。本文中引用的所有公开的外国专利和专利申请均以引用方式并入本文。本文中引用的所有其它公开的参考文献、文档、手稿和科学文献均以引用方式并入本文。虽然本发明已参考其优选实施方案进行特别示出和描述,但是本领域技术人员应了解,可在不脱离由所附权利要求书所涵盖的本发明的范围的情况下对其中的形式和细节做出各种改变。
序列表
<110>massachusettsinstituteoftechnology
presidentandfellowsofharvardcollege
<120>递送生物分子至免疫细胞
<130>38172-508001wo
<150>62/073,548
<151>2014-10-31
<160>11
<170>patentin3.5版
<210>1
<211>21
<212>dna
<213>人工序列
<220>
<223>人cd45sirna有义链
<400>1
cuggcugaauuucagagcatt21
<210>2
<211>21
<212>dna
<213>人工序列
<220>
<223>人cd4sirna有义链
<400>2
gaucaagagacuccucagutt21
<210>3
<211>21
<212>dna
<213>人工序列
<220>
<223>vifsirna有义链
<400>3
cagauggcaggugaugauugt21
<210>4
<211>21
<212>rna
<213>人工序列
<220>
<223>gagsirna有义链
<400>4
gauuguacugagagacaggcu21
<210>5
<211>21
<212>rna
<213>人工序列
<220>
<223>对照乱序sirna
<400>5
gccaagcaccgaaguaaauuu21
<210>6
<211>21
<212>rna
<213>人工序列
<220>
<223>人dc-signsirna有义链
<400>6
ggaacuggcacgacuccauuu21
<210>7
<211>19
<212>rna
<213>人工序列
<220>
<223>cd4sirna
<400>7
gaucaagagacuccucagu19
<210>8
<211>19
<212>dna
<213>人工序列
<220>
<223>gapdh正向
<400>8
agccacatcgctcagacac19
<210>9
<211>19
<212>dna
<213>人工序列
<220>
<223>gapdh反向
<400>9
gcccaatacgaccaaatcc19
<210>10
<211>20
<212>dna
<213>人工序列
<220>
<223>cd4正向
<400>10
ggcagtgtctgctgagtgac20
<210>11
<211>19
<212>dna
<213>人工序列
<220>
<223>cd4反向
<400>11
gaccatgtgggcagaacct19
- 还没有人留言评论。精彩留言会获得点赞!
- 一种基于荧光PCR检测ESR1基因突变的引物及探针的制造方法与工艺
- 一种用于检测乳制品中荧光假单胞菌的分子标记及其应用的制造方法与工艺
- 一种用于检测水产品中副溶血弧菌的分子标记及其应用的制造方法与工艺
- 一种用于检测草莓灰霉病菌的分子标记及其应用的制造方法与工艺
- 一种用于检测B族链球菌四环素耐药基因的分子标记及其应用的制造方法与工艺
- 一种用于检测粪肠球菌庆大霉素耐药基因的分子标记及其应用的制造方法与工艺
- 一种用于检测奶牛乳腺炎性金黄色葡萄球菌青霉素耐药基因的分子标记及其应用的制造方法与工艺
- 与甘蓝叶片表皮毛有无紧密相关的分子标记及其应用的制造方法与工艺
- 检测黄杉大小蠹的引物、试剂盒及检测方法与流程
- 一种用于紫斑牡丹与中原牡丹的分子鉴定方法与流程