肿瘤特异性富集的纳米载药体系及其制备方法与流程
本发明涉及一种含有peg和/或phpma的两亲性嵌段共聚物及其制备方法和应用,该共聚物能作为药物载体,与药物形成聚合物-药物偶联物,应用于靶向尤其是肿瘤靶向给药体系,属于药物辅料材料技术领域。
背景技术:
肿瘤一直是威胁人类健康的重大疾病,传统的治疗方法存在死亡率高,治愈率低和预后不佳等情况。然而随着纳米技术的发展,纳米载药体系能够为抗肿瘤药物带来更长的血液循环时间和更低的系统毒性,因此通过纳米药物载体对抗肿瘤药物进行输送的靶向治疗技术已经成为研究人员广泛关注的热点问题(collinsi.,natchembiol,2006,2:689-700)。
目前,大量具有不同化学结构或响应能力(温度、ph、氧化还原以及酶等)的新型纳米载药体系获得广泛的研究(gantas.,j.controlrelease,2008,126:187-204)。但是其中多数研究只关注了纳米载药体系在肿瘤组织或肿瘤细胞内部的药物释放和传递过程,而对药物在血液中的运输以及在肿瘤当中的特异性富集给予的关注略显不足。
从实际应用来看,纳米载药体系能够顺利进入肿瘤细胞需要经过血液运输、血管外渗、瘤内运输、肿瘤细胞摄取以及细胞内释放等步骤(petrakk.,drugdiscov.today,2005,10:1667-1673)。对纳米载药体系在体内的整个传输过程进行分析,会发现在整个运输环节中,纳米载药体系在血液中的长循环和有效地肿瘤富集是实现纳米载药体系各种功能的前提,只有当载药体系能够特异性富集于肿瘤组织时才能够有效发挥纳米载药体系的各种复杂功能。
聚合物-药物偶联物所构成的胶束载药体系作为一种常见的药物载体,是由两亲性共聚物在水溶液中自组装形成的具有“核-壳”结构的分子聚集 体,其尺寸一般为10-200nm(kataokak.,adv.drugdeliver.rev.,2001,47:113-131)。通过化学键合载药后,疏水性药物在水中的溶解度提高,药物穿过体内某些屏障的能力也得到增强,同时药物在体内的分布也得到极大的改善。应用某些两亲性聚合物胶束作为药物载体可以延长静脉注射类药物在人体中的循环时间。
聚乙二醇(peg)和聚n-(2’-羟基)丙基甲基丙烯酰胺(phpma)作为最常用的水溶性大分子,具有明显的优点:两者都是水溶性聚合物;具有较好的生物相容性,一般不会诱导人体发生免疫反应;大分子量的peg及phpma具有epr(增强的渗透和滞留)效应,有利于提高药物在肿瘤部位的聚集(maedah.,j.controlrelease,2000,65:271-284)。
但在利用peg和phpma制备载药聚合物胶束时,如何提高载药胶束防止蛋白非特异性吸附的能力,延长载药胶束的体内循环时间,以及提高载药胶束的肿瘤富集能力,仍是目前领域里需要解决的问题。
技术实现要素:
本发明在peg和phpma聚合物的基础上,通过调整聚合物单体的种类、聚合顺序和聚合度,合成了一类新的两亲性嵌段聚合物,由这类聚合物形成的胶束,在防止蛋白非特异性吸附、体内循环时间、肿瘤富集能力上,都有较大提高。以该聚合物作为载体,进一步结合药物、靶向分子和/或示踪分子等,能够形成具有生物相容性,高载药率,控制释放或靶向释放的给药系统或检测试剂。
本发明提供一种两亲性嵌段聚合物,所述聚合物的结构式如式ⅰ、式ⅱ或式iii所示:
式i聚合物的数均分子量为10000~100000;其中,键合药物部分(b嵌段)的分子量为4000-15000;亲水段(h嵌段)分子量为6000-96000,hpma部分占h嵌段的摩尔含量b为80-100%,magg部分占h嵌段的摩尔含量c为0-20%,azma部分占h嵌段的摩尔含量d为0-4%;并且h嵌段分子量为b嵌段分子量的1-20倍。
式i中,x和y独立地为具有可逆加成断裂链转移(raft)聚合活性、氮氧自由基聚合活性、原子转移自由基(atrp)聚合活性或开环聚合活性的引发剂基团,或者供聚合物修饰用的官能团;且当x和y同时为引发剂基团时,x与y不为同一种类型的引发剂基团。
z为-(ch2)n-,其中n为1-12的整数,例如1,2,3,4,5,6,7,8,9,10,11或12;或者z为一种可断裂化学键。
r为可键合药物的基团。
对所述式i聚合物,优选式i聚合物数均分子量为40000-100000,进一步优选为60000-98000,更优选为70000-95000,最优选为80000-90000。
优选式i聚合物的分子量分布指数d<1.3。
优选键合药物部分(b嵌段)的分子量为4500-14000,进一步优选为5000-13000,更优选为5200-8000,最优选为5500-6500。
优选亲水段(h嵌段)分子量为10000-95000,进一步优选为 40000-94000,更优选为70000-90000,最优选为75000-85000。
优选h嵌段分子量为b嵌段分子量的2-19倍,进一步优选为4-18倍,更优选为8-16倍。
优选hpma部分占h嵌段的摩尔含量为81-97%,进一步优选为82-96%,更优选为85-95%,最优选为90-95%。
优选magg部分占h嵌段的摩尔含量为1-15%,进一步优选为3-12%,最优选为5-10%。
优选azma部分占h嵌段的摩尔含量为2-4%。
优选所述引发剂基团,可以是含有一个或一个以上二硫代酯基(dithioester)的基团、三硫代碳酸酯(trithiocarbonate)、磺酸酯(xanthate)或二硫代氨基甲酸酯(dithiocarbamate)的各种基团,也可以是卤素原子,如氯原子(cl)或溴原子(br),更优选是含二硫代酯基或三硫代酯基的基团或溴原子。
优选所述聚合物修饰用官能团可以是氨基、巯基、羧基、羟基、氰基、叠氮基、酰胺基、肼基等。
优选n为2,3或4,更优选为2,即z为-ch2-ch2-。
优选可断裂化学键是环境响应性化学键,例如:氧化还原响应性化学键、氧化响应性化学键、ph响应性化学键、酶响应性化学键,包括但不限于二硫键(-s-s-、-ch2-s-s-ch2-)(还原响应性)、-ch2-o-c(ch3)2-o-ch2-(ph响应性)、gflg四肽(gly-phe-leu-gly)以及plgiag六肽(pro-leu-gly-ile-ala-gly)(酶响应性)、-s-(氧化响应性)等。
优选r基团为如下结构i-v之一:
在本发明的一个实施例中,式i聚合物为ch3(ch2)11sc(=s)s-pbhmagg6k-s-s-p(hpma-co-magg-co-azma)80k-br。
式ⅱ或式iii的聚合物,数均分子量为10000~80000;其中,peg部分的分子量为2000-10000;键合药物部分(b嵌段)的分子量为4000-15000;亲水段(h嵌段)分子量为4000-74000,hpma部分占h嵌段的摩尔含量b为80-100%,magg部分占h嵌段的摩尔含量c为0-20%,azma部分占h嵌段的摩尔含量d为0-4%;并且h嵌段分子量为b嵌段分子量的1-20倍。
式ii或式iii中,x为具有可逆加成断裂链转移(raft)聚合活性、氮 氧自由基聚合活性、原子转移自由基(atrp)聚合活性或开环聚合活性的引发剂基团,或者供聚合物修饰用的官能团。
r为可键合药物的基团。
对所述式ii或式iii聚合物,优选式ii或式iii聚合物的数均分子量为30000-80000,进一步优选为40000-80000,例如可以为40000-41000,42000-45000,46000-50000,60000-65000,70000-75000,或者76000-80000等等。
优选式ii或式iii聚合物的分子量分布指数d<1.3。
优选peg部分的分子量为3000-9000,进一步优选为3500-8000,更优选为4000-7000,最优选为4500-5500。
优选键合药物部分(b嵌段)的分子量为5000-14000,进一步优选为5000-12000,更优选为5000-10000,例如可以为:5000,5500,6000,6500,7000,7500,8000,8500,9000,9500,10000等等。
优选亲水段(h嵌段)分子量为10000-73000,进一步优选为20000-72000,更优选为25000-70000,例如可以为:25000,30000,35000,40000,45000,50000,55000,60000,65000等等。
优选h嵌段分子量为b嵌段分子量的2-19倍,进一步优选为3-18倍,更优选为6-16倍。
优选hpma部分占h嵌段的摩尔含量为81-97%,进一步优选为82-96%,更优选为85-95%,最优选为90-95%。
优选magg部分占h嵌段的摩尔含量为1-15%,进一步优选为3-12%,最优选为5-10%。
优选azma部分占h嵌段的摩尔含量为2-4%。
优选所述引发剂基团可以是含有一个或一个以上二硫代酯基(dithioester)的基团、三硫代碳酸酯(trithiocarbonate)、磺酸酯(xanthate)或二硫代氨基甲酸酯(dithiocarbamate)的各种基团,也可以是卤素原子,如氯原子(cl)或溴原子(br),更优选是含二硫代酯基或三硫代酯基的基团或溴原子。
优选所述聚合物修饰用官能团可以是氨基、巯基、羧基、羟基、氰基、 叠氮基、酰胺基、肼基等。
优选r基团为如下结构i-v之一:
在本发明的实施例中,式ii或式iii聚合物为:peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3、peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3、peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3、peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3、peg5k-b-p(hpma-co-magg-co-azma)60k-b-pbhmagg9k-sc(=s)s(ch2)11ch3。
在本发明中,hpma代表:
magg代表:
azma代表:
式i、式ii和式iii的聚合物整体为嵌段共聚物,而在hpma、magg(drobnikj.,macromol.chem.phys.,1976,177:2833-2848)和azma(mortenf.e.,polym.chem.-uk,2013,51:5091-5099)链段间为无规共聚物。
对于优选的r基团,其中结构ⅰ为bhmagg(bredihhina.,tetrahedron,2008,64:6788-6793)含有boc基团保护的肼键,可以用来键合如阿霉素(doxorubicin)、表阿霉素(epirubicin)、吡柔比星(pirarubicin)等含有羰基的化疗药物。结构ⅱ与结构ⅲ(nickyc.,chem.commun.,2013,49:7534-7536)都是以hema为基础通过酯键来键合药物,两种结构的区别在于结构ⅲ包含二硫键,会更有利于药物的释放。结构ⅳ(kopecekj.,uspatent,5037,883)则是以gflg为基础通过酯键键合药物。可通过酯键链接的药物包括但不限于喜树碱(camptothecin)、紫杉醇(paclitaxel)、鬼臼毒素(podophyllotoxin)和7-乙基-10-羟基喜树碱(sn38)等。结构v可通过羟基键合带有羟基或羧基的化合物,例如具有光热性质的近红外染料分子(ir825)或者螯合锌离子的原卟啉分子(protoporphyrinix)等,可分别用于光热治疗和光动力治疗过程。
结构i-v的r基团键合药物之后相应的结构式如下:
其中drug代表药物分子。
由于不同药物的化学结构及抑瘤机理不同,负载不同药物后本发明聚合物形成的载药胶束的治疗效果也不同。
本发明针对之前的共聚物体内循环时间较短且肿瘤富集量较低的缺陷,通过将b嵌段和h嵌段单体分别聚合构建两亲性嵌段共聚物,在此基础上调节p、b和h嵌段的分子量和嵌段间排列顺序,使形成的新聚合物相比以往的以peg和hmpa为成分的聚合物,在防止蛋白非特异性吸附、延长体内循环时间、提高肿瘤富集能力上都得到很大改善。
在利用本发明聚合物作为药物载体负载药物时,可以将药物分子通过 共价键直接连接到聚合物b嵌段的r基团上,除此之外,还可以利用聚合物形成胶束核层疏水段与药物分子的疏水相互作用或者静电作用,在聚合物胶束形成的过程中自动将药物包覆到胶束中,即利用非共价键作用负载药物,或者两种方式结合使用。
能通过疏水作用或静电作用包覆到胶束中,或者通过共价键连接到本发明聚合物上的药物,都适用于本发明,包括小分子化合物、寡肽或蛋白质、dna或rna等,优选为能用于肿瘤治疗的药物,更优选为含有羰基、羧基和/或羟基的肿瘤治疗药物,例如阿霉素、表阿霉素、吡柔比星、喜树碱、紫杉醇、鬼臼毒素、7-乙基-10-羟基喜树碱、protoporphyrinix、ir825等。
相较于没有响应性化学键的药物载体而言,基于式i中聚合物的带有响应性化学键的药物载体能够在肿瘤细胞内外环境的刺激下发生化学结构变化,从而刺激肿瘤细胞内吞并促进抗肿瘤药物在细胞内的释放,会有更好的应用前景。例如,含有可断裂键(如二硫键)链接主链的聚合物在形成胶束进入肿瘤细胞后,由于细胞内环境中还原性比胞外环境高,二硫键易断裂,从而使胶束解体,进而促进药物的释放。
此外,所述式i、式ii或式iii的聚合物可以利用其上的羧基、叠氮基等通过共价键连接将功能分子键合到聚合物h段、b段或者端基上,所述功能分子包括靶向分子和示踪分子。
因此,本发明还保护如下的化合物:式i-药物分子偶联物,式ii-药物分子偶联物,式iii-药物分子偶联物。其中式i、式ii和式iii是如前所述的式i、式ii和式iii的聚合物分子,药物分子通过共价键结合在聚合物分子b嵌段的r基团上。优选结合药物后的r基团转换为前述的结构i’-v’。
本发明还保护如下的化合物:式i-功能分子偶联物,式ii-功能分子偶联物,式iii-功能分子偶联物。其中式i、式ii和式iii是如前所述的式i、式ii和式iii的聚合物分子,功能分子通过共价键结合在聚合物b嵌段的r基团上、h嵌段的magg末端羧基上、h嵌段的azma末端叠氮基上、和/或聚合物端基上,所述功能分子为靶向分子和/或示踪分子。
进一步,本发明还保护如下的化合物:药物分子-式i-功能分子偶联物,药物分子-式ii-功能分子偶联物,药物分子-式iii-功能分子偶联物。其中式i、式ii和式iii是如前所述的式i、式ii和式iii的聚合物分子,药物分子通过共价键结合在聚合物分子b嵌段的r基团上,功能分子通过共价键结合在聚合物h嵌段的magg末端羧基上、h嵌段的azma末端叠氮基上、和/或聚合物端基上,所述功能分子为靶向分子和/或示踪分子。
所述药物分子能通过共价键连接到本发明聚合物r基团上,包括小分子化合物、寡肽或蛋白质、dna或rna等,优选为含有羰基、羧基和/或羟基的药物,更优选为含有羰基、羧基和/或羟基的肿瘤治疗药物,例如阿霉素、表阿霉素、吡柔比星、喜树碱、紫杉醇、鬼臼毒素、7-乙基-10-羟基喜树碱、protoporphyrinix、ir825等。
所述靶向分子,可以是例如环状rgd、叶酸、低密度脂蛋白、核酸适配体和穿膜肽等,能够提高载药体系的肿瘤富集能力和进入肿瘤的效率;可以是生物活性因子,如egf(上皮细胞生长因子)、vegf(血管上皮细胞生长因子)和gpcr(g蛋白偶联受体配体)等;也可以是抗体,例如单克隆抗体,比如贝伐单抗、曲妥珠单抗等。利用配体和受体、抗体与抗原之间的特异性结合,使由上述化合物形成的载药体系具有对细胞分子水平上的识别能力,提高靶向作用。
引入示踪分子,可以实现实时跟踪监测的目的。所述示踪分子包括:同位素示踪剂、酶标示踪剂、荧光标记示踪剂、自旋标记示踪剂等,例如罗丹明、cy3、cy3.5、cy5、cy5.5、cy7、异硫氰酸荧光素等。
在本发明的一类实施方式中,共价结合药物分子的本发明聚合物的结构,即,式i-药物分子偶联物,式ii-药物分子偶联物,式iii-药物分子偶联物的结构如下:
进一步,优选上述结构式中药物分子可以是阿霉素,阿霉素通过阿霉素上的羰基与b嵌段hmagg上的肼基形成腙键相连接。
在本发明另一优选实施方式中,共价结合了阿霉素的本发明聚合物为:peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3;peg5k-b-p(hpma-co-magg-co-azma)60k-b-pbhmagg9k-sc(=s)s(ch2)11ch3;ch3(ch2)11sc(=s)s-pbhmagg6k-s-s-p(hpma-co-magg-co-azma)80k-br;
其中阿霉素通过阿霉素上的羰基与b嵌段hmagg上的肼基形成腙键连接在上述聚合物上。
在本发明的一类实施方式中,式i-功能分子偶联物,式ii-功能分子偶联物,式iii-功能分子偶联物为式i-环状rgd,式ii-环状rgd,式iii-环状rgd,其中环状rgd通过共价键连接在聚合物h嵌段magg末端羧基上。
在本发明的另一类实施方式中,结合了荧光物质cy7的本发明聚合物结构式,即,式i-功能分子偶联物,式ii-功能分子偶联物,式iii-功能分子偶联物的结构分别如下:
进一步的,r基团优选选自前述结构i-v之一,偶联药物后r基团相应转换为前述结构i’-v’之一。
在本发明另一优选实施方式中,共价结合了cy7的本发明聚合物为:peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3;peg5k-b-p(hpma-co-magg-co-azma)60k-b-pbhmagg9k-sc(=s)s(ch2)11ch3;ch3(ch2)11sc(=s)s-pbhmagg6k-s-s-p(hpma-co-magg-co-azma)80k-br;其中cy7通过三唑基团与h嵌段azma的末端共价连接。
在本发明另一类优选的实施方式中,药物分子-式i-功能分子偶联物,药物分子-式ii-功能分子偶联物,药物分子-式iii-功能分子偶联物的结构如下:
优选上述结构式中药物分子可以是阿霉素,阿霉素通过阿霉素上的羰基与b嵌段hmagg上的肼基形成腙键相连接。
在本发明另一优选实施方式中,共价结合了阿霉素和cy7的本发明聚合物为:
peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3;peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3;peg5k-b-p(hpma-co-magg-co-azma)60k-b-pbhmagg9k-sc(=s)s(ch2)11ch3;ch3(ch2)11sc(=s)s-pbhmagg6k-s-s-p(hpma-co-magg-co-azma)80k-br;其中阿霉素通过阿霉素上的羰基与b嵌段hmagg上的肼基形成腙键连接在上述聚合物上,cy7通过三唑基团与h嵌段azma的末端共价连接。
进一步,本发明提供式i、式ii或式iii的聚合物的制备方法。
嵌段共聚物的合成方法有多种,如自由基聚合、阴离子聚合、阳离子聚合、开环歧化聚合、活性自由基聚合等,或者将已经制备好的嵌段以共价键偶联,或者对嵌段聚合物进行化学改性。本领域中用于合成嵌段共聚物的方法,都能用于本发明聚合物的制备。
优选制备本发明聚合物的方法为活性自由基聚合和开环聚合,特别是可逆加成-断裂链转移聚合(raft)、氮氧自由基聚合、原子转移自由基(atrp)聚合和开环聚合,这些方法能有效控制聚合物分子量分布。
在本发明优选的实施方案中,采用可逆加成-断裂转移聚合(raft)方法,通过对聚合物中各组分的组成比以及聚合物的分子量等因素的调节,得到聚合物组成可调、分子量可控、分子量分布窄(d<1.3)的本发明聚合物。
例如:
式i聚合物的制备方法可优选采用下述步骤:
步骤1:在n,n’-二环己基碳二亚胺(dcc)和4-二甲氨基吡啶(dmap)的催化作用下,制备x和y之间通过-z-连接的异端官能化引发剂。
x、y和-z-的定义如前所述。
优选所述异端官能化引发剂,一端为raft反应链转移剂、另一端为atrp反应引发剂,或者一端为氮氧自由基活性(nmp)、另一端为引发开环聚合(rop)反应的引发剂。
步骤2:在氩气或氮气气氛下的有机相介质中,利用步骤1制得的异端官能化引发剂与相关单体magg、hpma、azma等进行聚合反应合成h嵌段,再在此基础上进一步与b嵌段的单体进行聚合;或者利用步骤1制得的异端官能化引发剂与b嵌段单体进行聚合反应合成b嵌段,再在此基础上进一步与h嵌段的单体进行聚合。
若需将链末端基团x或y转化为其它可修饰用官能团,可以进一步含有步骤3,将链末端基团x或y转化为其它可修饰用官能团:以raft链转移剂为例,首先利用硼氢化钠脱除链末端二硫代酯或三硫代酯基团,使得端基暴露出巯基,然后采用含有双键的小分子功能化试剂与巯基进行michael加成反应以得到所需端基官能化的聚合物。对于端基为溴原子的聚合物而言,其转化可通过叠氮钠直接反应转化为叠氮基团以进行后续修饰。
通过调整聚合先后顺序可以调控聚合物的拓扑结构。
步骤1中所述异端官能化引发剂两端的引发分子结构为不同聚合类型的引发剂,通过不同聚合方法实现-z-两端不同聚合物嵌段的合成。
优选步骤1中所述反应的反应温度为室温,反应时间为72-96小时。
步骤1所述反应在溶剂中进行,所述溶剂优选为甲苯、二氯甲烷、三氯甲烷或四氢呋喃。
优选步骤2中所述异端官能化引发剂与所述聚合物单体的摩尔比为1:(10-1000)。
优选步骤2中所述聚合反应的反应温度为65-70℃,反应时间为24-48小时。
优选步骤2中有机相介质为dmso或dmf。
式ii或式iii聚合物的制备方法优选可以包括下述步骤:
步骤1:在n,n’-二环己基碳二亚胺(dcc)和4-二甲氨基吡啶(dmap)的催化作用下,使末端基团为羟基或氨基的peg与含羧基的可逆加成断裂链转移试剂进行反应,得到peg大分子链转移剂;
步骤2:在氩气或氮气气氛下的有机相介质中,以4,4’-偶氮二(4-腈基戊酸)(v501)为引发剂,使所述peg大分子链转移剂与式ii或式iii聚合物中的单体进行活性自由基聚合反应(raft聚合),可以先与h嵌段的单体进行聚合反应合成h嵌段,在此基础上进一步与b嵌段的单体进行聚合;也可以先与b嵌段单体进行聚合反应合成b嵌段,在此基础上进一步与h嵌段的单体进行聚合。
若需将链末端基团x转化为其它可修饰用官能团,可以进一步含有步骤3,将链末端基团x转化为其它可修饰用官能团:以raft链转移剂为例,首先利用硼氢化钠脱除链末端二硫代酯或三硫代酯基团,使得端基暴露出巯基,然后采用含有双键的小分子功能化试剂与巯基进行michael加成反应以得到所需端基官能化的聚合物。
其中,步骤1中所述末端基团为羟基或氨基的peg,优选一末端基团为羟基或氨基、另一末端基团为不易水解的基团的peg;优选所述不易水解的基团为甲基、乙基、叔丁基。
优选步骤1中所述含羧基的可逆加成断裂链转移试剂为十二烷基三硫代碳酸酯衍生物,例如2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸、4-(十二烷基三硫代碳酸酯基)-4-氰基戊酸等。
优选步骤1中所述反应的反应温度为室温,反应时间为72-96小时。
优选步骤1所述反应在无水溶剂中进行,所述溶剂可为甲苯、二氯甲烷、三氯甲烷或四氢呋喃。
优选步骤1所述末端基团为羟基或氨基的peg与含羧基的可逆加成断裂链转移试剂的摩尔比为1:(3-5)。
优选步骤2中所述有机相介质为dmso或dmf。
优选步骤2中所述peg大分子链转移剂与所述聚合物单体的摩尔比可为1:(10-1000)。
优选步骤2所述聚合反应的反应温度为65-70℃,反应时间为24-48小时。
在利用共价键将药物连入式i、式ii或式iii的聚合物时,可以根据药物结构以及聚合物上可键合药物的基团,选择适当的化学反应。
以阿霉素(dox)为例,以键合药物的基团r为结构i为例,连接步骤可以如下:首先将聚合物溶于三氟乙酸中,室温搅拌10-60分钟,优选30分钟,脱除bhmagg侧链的叔丁氧羰基(boc),暴露出肼键;然后将脱除boc基团的聚合物溶于dmso中,在乙酸的催化下加入1-10倍摩尔量,优选2-4倍摩尔量的dox,肼键与dox的羰基形成对酸敏感的腙键,反应条件为20-40℃,优选30℃,避光搅拌48-72小时。
此外,可以采用本领域中常规的化学反应,在本发明的聚合物上引入功能分子。不作为对本发明保护范围的限制,例举两种引入方式。
第一种是通过magg的端羧基引入功能分子。
以引入主动靶向分子环状rgd为例,合成步骤可以如下:首先将聚合物溶解于dmf中,加入5-20倍摩尔量,优选9-12倍摩尔量的edc·hcl与nhs搅拌,室温下活化12-48h,优选24h;在丙酮中沉淀,收集沉淀聚合物后再用dmf溶解,加入1-10倍摩尔量,优选2-4倍摩尔量的环状rgd及少量三乙胺室温下反应36-72h,优选48h,产物在水溶液中透析纯化。
第二种方法是通过对聚合物主链端基的修饰使其转化为可反应基团,再引入功能分子。
例如,在低温下加入正丁基锂,再冲入干燥的二氧化碳,将br转化为羧基,然后在端基引入功能分子。此外,端基br还可以通过与叠氮钠反应转化为叠氮基团,通过点击化学反应引入功能分子。
例如花氰染料cy7的引入方法可以如下:将聚合物与cy7溶于dmso 中,在氩气或氮气气氛下冻融2-5次,优选3次;然后在冷冻状态及氩气或氮气保护下加入硫酸铜及l-抗坏血酸钠,再次抽真空除氧,然后溶解后在30-50℃,优选40℃,避光搅拌36-72h,优选48h。
在端基引入功能分子对胶束的粒径影响较小,同时胶束的形成方式使功能分子能更多的暴露在胶束表面,从而能更好地与细胞表面接触发挥其生物功能。本领域技术人员可以根据需要,选择引入方式。
除上述h段引入功能分子的方法之外,还可以利用本领域中常用的反应方法,在b段引入功能分子,如此一来,聚合物在体内循环时,功能分子被保护于胶束内部,对聚合物的循环性能产生更小的影响。当到达肿瘤部位后,h段在肿瘤微环境响应下脱除,使得功能分子重新暴露出来实现其功能。
进一步,本发明提供所述式i、式ii或式iii的聚合物在制备药物载体或检测试剂中的应用,以及所述式i、式ii或式iii的聚合物作为药物载体或检测试剂的应用,以及式i、式ii或式iii的聚合物作为药物载体在制备药物中的应用。
所述药物载体优选是靶向药物载体,更优选是抗肿瘤药物靶向药物载体。
所述检测试剂优选为体内肿瘤检测试剂,更优选为体内早期肿瘤检测试剂。
在制备药物载体或作为药物载体制备药物应用时,可以将药物分子通过共价键和/或非共价键的方式负载到本发明聚合物上。药物分子通过共价键负载到本发明聚合物上的方式是,通过前述的化学反应,将药物分子共价连接到聚合物b嵌段的r基团上。药物分子通过非共价键负载到本发明聚合物上的方式是,利用聚合物形成胶束核层疏水段与药物分子的疏水相互作用或者静电作用,在聚合物胶束形成的过程中自动将药物包覆到胶束中。药物分子还可以通过所述的共价键和非共价键这两种负载方式结合使用,负载到本发明聚合物上。
在制备或作为抗肿瘤靶向药物载体或肿瘤检测试剂应用时,可以将本 发明所述式i、式ii或式iii的聚合物与肿瘤靶向特异性分子反应,制备得到共价结合靶向分子的聚合物。所述靶向分子包括但不限于环状rgd、叶酸、低密度脂蛋白、穿膜肽、核酸适配体、egf、vegf和单克隆抗体等与肿瘤细胞或血管具有特异性识别能力的功能性分子。
在制备或作为检测试剂使用时,在聚合物中引入示踪分子,便于跟踪和检测。所述示踪分子例如:同位素示踪剂、酶标示踪剂、荧光标记示踪剂、自旋标记示踪剂等。
所述功能分子(包括靶向分子和示踪分子)通过共价键,键合到所述式i、式ii或式iii的聚合物的h段、b段或者端基上。例如,可以通过magg的端羧基共价结合功能分子。
也可以通过对聚合物主链端基的修饰使其转化为可反应基团,再引入功能分子。例如,将聚合物主链端基上的br转化为羧基,而后再共价结合功能分子;或者端基br还可以通过与叠氮钠反应转化为叠氮基团,通过点击化学反应引入功能分子。
在端基引入功能分子对胶束的粒径影响较小,同时胶束的形成方式使功能分子能更多的暴露在胶束表面,从而能更好地与细胞表面接触发挥其生物功能。在b段引入功能分子,聚合物在体内循环时,功能分子被保护于胶束内部,对聚合物的循环性能产生更小的影响,当到达肿瘤部位后,h段在肿瘤微环境响应下脱除,使得功能分子重新暴露出来实现其功能。本领域技术人员可以根据需要,选择在h段、b段或者端基上引入功能分子及相应的引入方式。
进一步,本发明提供一种药物组合物或检测试剂,所述药物组合物或检测试剂含有本发明所述的式i、式ii或式iii的聚合物,或者本发明的式i-药物分子偶联物,式ii-药物分子偶联物,或式iii-药物分子偶联物,或者本发明的式i-功能分子偶联物,式ii-功能分子偶联物或式iii-功能分子偶联物,或者本发明的药物分子-式i-功能分子偶联物,药物分子-式ii-功能分子偶联物或药物分子-式iii-功能分子偶联物。
所述药物组合物中,药物分子共价和/或非共价结合在本发明所述的式 i、式ii或式iii的聚合物上。其中共价结合是,药物分子共价连接在聚合物b嵌段的r基团上。非共价结合是利用聚合物形成胶束核层疏水段与药物分子的疏水相互作用或者静电作用,在聚合物胶束形成的过程中自动将药物包覆到胶束中。药物分子可以通过所述的共价结合和非共价结合两种方式一同作用负载到本发明聚合物上。
为提高药物的靶向性,可以进一步将靶向分子共价结合到本发明所述式i、式ii或式iii的聚合物上。所述靶向分子包括但不限于环状rgd、叶酸、低密度脂蛋白、穿膜肽、核酸适配体、egf、vegf和单克隆抗体等与生物体内组织、细胞具有特异性识别能力的功能性分子。
为便于跟踪和检测药物在体内的行为,可以进一步将示踪分子共价结合到本发明所述式i、式ii或式iii的聚合物上。所述示踪分子例如:同位素示踪剂、酶标示踪剂、荧光标记示踪剂、自旋标记示踪剂等。
所述检测试剂中,将靶向分子和示踪分子分别共价结合到本发明所述式i、式ii或式iii的聚合物上。通过靶向分子结合目标靶,经由示踪分子的显示作用,实现检测目标靶是否存在的效果。所述靶向分子包括但不限于环状rgd、叶酸、低密度脂蛋白、穿膜肽、核酸适配体、egf、vegf和单克隆抗体等与目标组织或细胞具有特异性识别能力的功能性分子。所述示踪分子例如:同位素示踪剂、酶标示踪剂、荧光标记示踪剂、自旋标记示踪剂等。
上述靶向分子和示踪分子通过共价键,键合到所述式i、式ii或式iii的聚合物的h段、b段或者端基上。例如,可以通过magg的端羧基共价结合靶向分子或示踪分子。
也可以通过对聚合物主链端基的修饰使其转化为可反应基团,再引入靶向分子和示踪分子。例如,将聚合物主链端基上的br转化为羧基,而后再共价结合靶向分子或示踪分子;或者端基br还可以通过与叠氮钠反应转化为叠氮基团,通过点击化学反应引入靶向分子或示踪分子。
本发明的优点:
1、本发明通过将b嵌段和h嵌段单体分别聚合构建两亲性嵌段共聚物, 在此基础上调节p、b和h嵌段的分子量和嵌段间排列顺序,使本发明式i、式ii和式iii的聚合物在水溶液中发生自组装形成尺寸较小的胶束,所述胶束有效逃脱网状内皮系统(res)的摄取。此外,所得胶束尺寸大于肾脏排出阈值,在体内拥有很长的血液循环时间。
2、所形成的胶束体系通过epr效应富集于肿瘤组织部位,在肿瘤组织局部高浓度条件下亲水链段之间相互作用,进一步发生聚集形成尺寸较大的多胶束聚集体,从而有效地滞留在肿瘤组织内部,延长了在肿瘤内部的滞留时间。
3、由本发明聚合物形成的胶束表面被长链peg或phpma聚合物所覆盖,因此胶束能够很好地防止蛋白非特异性吸附。
4、在更优选的方案中,通过在聚合物的主链或侧链引入环境响应性化学键,所得胶束聚集体能够在肿瘤外基质、肿瘤细胞质、溶酶体或内涵体中响应性释放其所负载的抗肿瘤药物。
5、在更优选的方案中,通过在所述聚合物中进一步引入主动靶向分子或者细胞穿膜物质,可以提高肿瘤细胞对聚合物的摄取,进一步提高聚合物在肿瘤组织的富集量。
附图说明
图1聚合物载体peg-b-pbhmagg-b-p(hpma-co-magg-co-azma)的合成路线
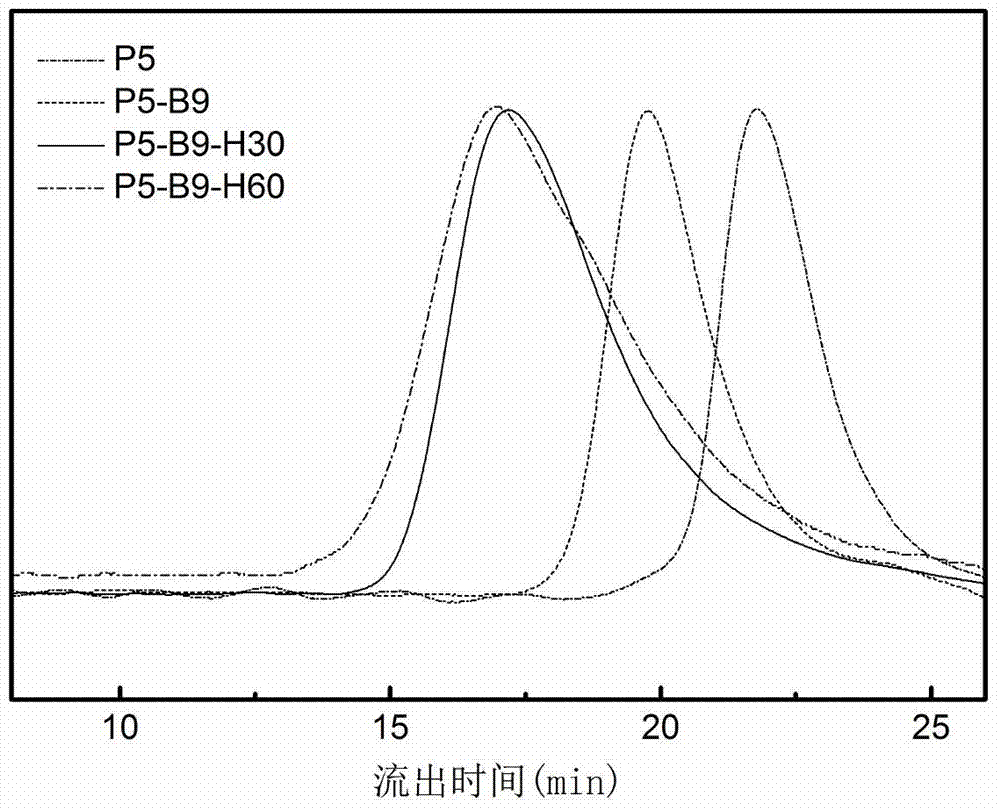
图2聚合物的gpc曲线
图3动态光散射(dls)测定p5-h60-b9胶束粒径结果
图4p5-h60-b9胶束聚集现象的透射电镜(tem)照片
图5载药胶束在生物体内代谢的活体成像结果
图6载药胶束在生物体内代谢的光声成像结果
图7载药胶束在生物体内对肿瘤的抑制效果图,图中为各实验动物体内肿瘤块。
图8给药后小鼠体重变化曲线
具体实施方式
本发明中聚合物的数均分子量及其分布用凝胶渗透色谱(gpc)和1h核磁共振波谱(1hnmr)来共同表征。
下述实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和生物材料,如无特殊说明,均可从商业途径获得。
实施例1
peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3的制备
合成路线如图1所示。
(1)将数均分子量为5000的羟基和甲基封端的peg10g溶于40ml甲苯,与甲苯共沸除水两次后,加入4-十二烷基三硫代碳酸酯基-4-腈基戊酸3.23g,dmap0.1g,加入70ml甲苯溶解,待完全溶解后加入dcc1.98g,室温搅拌下反应90小时。反应结束后抽滤除去环己基脲(dcu),然后将滤液倾入过量乙醚中,收集沉淀,所得沉淀溶于少量甲苯,再用乙醚沉淀,如此重复3-5次,直至乙醚无色。将沉淀物在40℃真空干燥24小时,所得产物即为peg的大分子链转移剂。
(2)取此大分子链转移剂0.3g,bhmagg0.6g,4,4’-偶氮二(4-腈基戊酸)2mg,dmso2ml,于真空反应管内,在冷冻下抽真空,然后恢复至室温,充入氩气,将此冷冻-溶化-充氩气过程重复三次,最后在氩气保护下将反应管置于70℃油浴中反应。终止反应时将反应管迅速放入液氮中,然后通过乙醚沉淀得到产物peg5k-b-pbhmagg9k。
(3)再取上一步产物0.1g,hpma0.36g,magg0.04g,azma0.01g,4,4’-偶氮二(4-腈基戊酸)2mg,dmso2ml,于真空反应管内,重复上述反应步骤,反应48h后通过丙酮沉淀获得标题终产物。该产物的分子量和组成可以通过gpc和1hnmr来共同确认。其gpc曲线如图2所示。
下文中将该聚合物简称为p5-b9-h60。
实施例2
peg5k-b-pbhmagg9k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3的制备
采用与实施例1相同的反应路线和反应条件,调整hpma、magg和azma的加入量为0.18g、0.02g以及0.005g,聚合制备得到标题终产物。
下文中将该聚合物简称为p5-b9-h30。
实施例3
peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)60k-sc(=s)s(ch2)11ch3的制备
采用与实施例1相同的反应路线和反应条件,调整bhmagg加入量为0.35g,聚合制备得到标题终产物。
下文中将该聚合物简称为p5-b5-h60。
实施例4
peg5k-b-pbhmagg5k-b-p(hpma-co-magg-co-azma)30k-sc(=s)s(ch2)11ch3的制备
采用与实施例1相同的反应路线和反应条件,调整bhmagg加入量为0.35g,hpma为0.18g,magg为0.02g,azma为0.005g,聚合制备得到标题终产物。
下文中将该聚合物简称为p5-b5-h30。
实施例5
peg5k-b-p(hpma-co-magg-co-azma)60k-b-pbhmagg9k-sc(=s)s(ch2)11ch3的制备
调换实施例1的步骤(2)和步骤(3)的反应顺序,采用与实施例1对应相同的反应条件,聚合制备得到标题终产物。
下文中将该聚合物简称为p5-h60-b9。
实施例6
ch3(ch2)11sc(=s)s-pbhmagg6k-s-s-p(hpma-co-magg-co-azma)80k-br的制备
小分子链转移剂cta-s-s-br的合成分为两步,首先利用1.54g(10mmol)双(2-羟基乙基)二硫醚与2.27g(10mmol)二溴异丁酰溴在1.01g(10mmol)三乙胺催化下反应合成一端为溴另一端为羟基的小分子引发剂,产物通过柱层析(乙酸乙酯/石油醚=4/1)分离纯化,旋蒸除去有机溶剂后得到无色或淡黄色油状液体2g(产率66%)。所得产物进一步与2.66g(6.6mmol)4-十二烷基三硫代碳酸酯基-4-腈基戊酸在1.6gdcc和50mgdmap催化下进行酯化反应,反应完毕后过滤除去dcu,产物经柱层析分离纯化,最终获得一端为raft链转移剂,另一端为溴原子的双功能化小分子引发剂。
采用实施例1相同的反应条件,通过raft合成pbhmagg6k。
嵌段p(hpma-co-magg-co-azma)通过atrp进行合成。首先取大分子链转移剂pbhmagg6k0.06g,hpma0.7g,magg0.07g,azma0.03g,配位剂1,4,8,11-四甲基-l,4,8,ll-四氮杂环四癸烷(me4cyclam)20mg,dmso3ml,于真空反应管内,在冷冻下抽真空,然后恢复至室温,充入氩气,将此冷冻-溶化-充氩气过程重复两次,然后冷冻样品,在氩气保护下加入18mgcubr,再次冷冻抽真空,最后在氩气保护下将反应管置于70℃油浴中反应。后续产物处理同实施例1。
下文中将该聚合物简称为b6-h80。
实施例7、制备负载dox的聚合物
分别取100mg实施例1-6制备的聚合物各溶于5ml三氟乙酸中,室温搅拌30分钟后旋蒸除去三氟乙酸,产物溶于10ml无水dmso中,加入三倍摩尔量的dox和催化量的冰醋酸,37℃搅拌反应48-72小时,反应完毕后在水中透析出去游离的dox,分别制备获得实施例1-6的各聚合物负载dox的产物。
实施例8、制备cy7标记的聚合物
分别取实施例7制备的各负载dox聚合物50mg、配位剂n,n,n',n,'n”- 五甲基二亚乙基三胺(pmdeta)3mg与3mgcy7溶于dmso中,在氩气气氛下冻融3次;然后在冷冻状态及氩气保护下加入4.3mg五水硫酸铜及3.4mgl-抗坏血酸钠,再次抽真空除氧,溶解后在40℃避光搅拌48h。反应完毕后在水中透析除去未反应cy7和催化剂,分别制备获得实施例1-6的各聚合物负载dox的cy7标记产物。
实施例9、胶束的制备及胶束粒径的测定
分别取实施例8所制备的聚合物10mg,溶于1mldmso中,然后缓慢滴入10mlpbs缓冲液中,搅拌速度100rpm,然后透析三天以除去有机溶剂得到胶束溶液,置于4℃冰箱中保存备用。
胶束的粒径通过动态光散射(dls)进行测定,dls在英国马尔文(malvern)公司的zetasizernanozs上测定。取1.2ml胶束溶液于石英比色皿中,放入仪器中于25℃稳定2min后测量三次取平均值。dls测定的胶束粒径结果以图3所示的p5-h60-b9的胶束为代表予以展示。
同时还利用透射电镜(tem)对胶束进行观察。首先将20μl的胶束样品(0.2mg/ml)滴在铜网支撑膜上,室温过夜干燥后在铜网表面滴加5μl磷钨酸染色液,干燥后即得到tem样品。透射电镜在jeol公司的jem-2200fs上测定,加速电压为200kv。在tem中可以明显的观察到胶束的聚集现象,以图4所示的p5-h60-b9的胶束为代表予以展示。
从图3可见,本发明的两亲性嵌段聚合物形成胶束的粒径主要在5-40nm之间,由于亲水链段间的相互作用,所形成的小胶束发生进一步聚集形成多胶束聚集体,如图4所示。
实施例10、载药胶束在生物体内分布的活体成像实验
在此实施例中,使用负载小鼠乳腺癌(4t1)肿瘤细胞的balb/c小鼠作为动物模型。为了获得荷瘤鼠,在小鼠背部通过皮下注射注入1×106个提前培养好的4t1细胞。经过10天左右的生长,当固体瘤体积达到50-100mm3时,小鼠即可用于动物实验。
在此实施例中,3只balb/c荷瘤鼠作为一组进行载药胶束生物体内活体 成像实验。分别将实施例8所制备的p5-b9-h60、p5-b5-h60、p5-b5-h30、p5-h60-b9、b6-h80载药胶束通过尾静脉注射进入小鼠体内,小鼠在1h、3h、6h、9h、12h、24h和48h时通过异氟烷进行麻醉后,放入
成像结果如图5所示。从整体活体成像结果可见,随着时间的延长,载药胶束在体内的分布逐渐由全身的血液循环慢慢聚集到肿瘤上。48h后脏器的活体成像结果显示,载药胶束在肿瘤中的富集度最高,其次是在血管组织丰富的器官—肝脏、肾脏、肺、脾有存留,其中以肝脏最多,血液中也仍有胶束的显像。由此可见,本发明的载药胶束在体内拥有很长的血液循环时间,并且能够通过epr效应富集于肿瘤组织部位。
实施例11、载药胶束在生物体内分布的光声成像实验
在此实施例中,使用的荷瘤鼠获得方法与实施例10相同。将实施例8所制备的载药胶束通过尾静脉注射进入小鼠体内,小鼠在6h、12h和24h时通过异氟烷进行麻醉后,放入实时多光谱光声成像仪(itheramedicalgmbh,neuherberg,germany)中进行扫描,选取680、720、750、770、800、830六个检测波长,扫描的4个截面厚度均为0.3mm,可以实时检测到肝脏、脾脏、肾脏、肿瘤和血管中的荧光信号。
成像结果如图6所示。从图中可见,随着时间的延长,载药胶束在体内的分布逐渐由全身的血液循环慢慢聚集到肿瘤上。12-24h的图像中,肿瘤中的荧光信号逐渐增强,说明载药胶束富集到肿瘤中。由此可见,本发明的载药胶束能够通过epr效应富集于肿瘤组织部位。此外,24h的图像中,肝脏、脾脏、肾脏和血管中仍然能检测到荧光信号,说明载药胶束在体内循环保留时间很长。
实施例12、载药胶束在生物体内对肿瘤的抑制效果的测定
在此实施例中,使用的荷瘤鼠获得方法与实施例10相同。每5只balb/c 荷瘤鼠作为一组进行肿瘤的抑制实验。分别取100μl实施例7所制备的p5-b9-h60、p5-b5-h60、p5-h60-b9、b6-h80各载药胶束通过尾静脉注射进入小鼠体内,给药量为5mg/kg小鼠体重(以dox量计),每4天给药一次,共给药4次。同时选择注射生理盐水和游离dox溶液(5mg/kg体重)的两组实验作为对照组,给药时间相同。在第17天实验结束后,通过颈椎错位处死小鼠,解剖小鼠获得小鼠肿瘤及主要脏器,通过切片进行病理分析,肿瘤的抑制效果可以直观通过肉眼观察得出结论,如图7所示。
从图7中可见,载药胶束组和游离dox溶液组的肿瘤体积小于生理盐水对照组的,其中b6-h80、p5-h60-b9、p5-b9-h60组的肿瘤体积又明显小于生理盐水对照组的,而部分载药胶束组的肿瘤体积又小于游离dox溶液组的。同时从图8中可见,游离dox组小鼠体重发生明显下降,表明游离dox毒性较大,而载药胶束组小鼠体重与生理盐水组差别不大,表明用本发明的聚合物作为药物载体给予dox能有效降低dox的毒性,同时也佐证了载药胶束的肿瘤富集和响应性释药能力。综上所述,将dox负载到本发明的载药胶束上,不会影响dox的药物活性,而通过载药胶束的靶向给药,实现药物的肿瘤内富集,在抑制肿瘤生长的同时还能明显降低药物的毒副作用。
- 还没有人留言评论。精彩留言会获得点赞!