含鳞翅目病毒增强子的重组表达载体在双翅目昆虫细胞中表达蛋白的应用的制作方法
本发明涉及基因工程领域,尤其是涉及一种含鳞翅目病毒增强子的重组表达载体在双翅目昆虫细胞中表达蛋白的应用。
背景技术:
昆虫细胞系是各种研究应用以及作为疫苗的病毒类颗粒(vlp)的重组蛋白异源表达的重要宿主(cherezov等,2007;imasaki等人,2011;rasmussen等人,2007;white等人,2012;metz和pijlman,2011;shrestha等人,2007;treanor等人,2011;treanor等人,2011年)。传统的在昆虫细胞系的研究应用过程中,主要是利用杆状病毒重组表达载体系统(bevs)来感染源自鳞翅目的细胞系(berger等人,2004;jarvis,2009;kost等人,2005;summers,2006)。尽管bevs的运用非常成功,其还是存在几个缺点,如杆状病毒载体的生成将可能需要长达3个星期(jarvis,2014年),而因为病毒引起的细胞裂解,蛋白生产阶段的持续时间被限制在约3-4天以内;分泌蛋白和细胞表面蛋白的通常产量往往小于10μg/ml,而细胞内蛋白质的产量已经有报道达到了100μg/ml(harrison和jarvis,2007;jarvis,2009)。可以通过稳定转染昆虫细胞系来替代使用bevs瞬时生产蛋白质的方法,但是,这种方法不仅费时,而且蛋白产量通常很低(harrison和jarvis,2007;drugmand等人,2012)。
通常使用bevs来构建重组表达载体是将目的基因人工连接于杆状病毒的晚期(late)和/或非常晚期(verylate)启动子之后,利用其强力启动子,以期能表达较大量的目标蛋白。但该方法存在非常明显的缺陷,即通常在晚期(late)或非常晚期(verylate)启动子开始表达之后,宿主细胞将在两、三天内快速死亡,无法达到长期表达的目的。
s2(drosophilaschneider2)细胞是一种果蝇细胞,是一种属于双翅目果蝇科果蝇属的昆虫细胞。由于种属隔离,应用于不同种属的重组重组表达载体通常无法通用,即在鳞翅目昆虫细胞中高效表达的重组表达载体,无法在双翅目昆虫细胞中有效表达。
技术实现要素:
基于此,有必要提供一种含鳞翅目病毒增强子的重组表达载体在双翅目昆虫细胞中表达蛋白的应用。
一种含鳞翅目病毒增强子的重组表达载体在双翅目昆虫细胞中表达蛋白的应用,所述重组表达载体具有调控单元及位于所述调控单元下游的编码单元,所述调控单元含有hr-x增强子或其功能片段或序列变体、acie-y启动子或其功能片段或序列变体、以及opie-z启动子或其功能片段或序列变体,其中,x为3或5,y为1,z为1或2。
在其中一个实施例中,所述opie-z启动子或其功能片段或序列变体位于所述acie-y启动子或其功能片段或序列变体的下游。
在其中一个实施例中,所述hr-x增强子为acmnpv中的hr-5增强子、acmnpv中的hr-3增强子或bombyxmori中的hr-3增强子。
在其中一个实施例中,所述hr-x增强子为acmnpv中的hr-5增强子,且所述hr-5增强子的序列为seqidno.1中第1位-第482位所示序列。
在其中一个实施例中,所述acie-y启动子为acmnpv中的acie-1启动子,且所述acie-1启动子的序列为seqidno.1中第487位-第1033位所示序列。
在其中一个实施例中,所述opie-z启动子为opmnpv中的opie-1启动子或opie-2启动子,且所述opie-1启动子的序列为seqidno.2中第1位-第289位所示序列,所述opie-2启动子的序列为seqidno.3中第3位-第551位所示序列。
在其中一个实施例中,所述opie-z启动子为opmnpv中的opie-2启动子。
在其中一个实施例中,所述双翅目昆虫细胞为果蝇s2细胞。
一种双翅目昆虫细胞,所述双翅目昆虫细胞具有含鳞翅目病毒增强子的重组表达载体,所述含鳞翅目病毒增强子的重组表达载体是上述含鳞翅目病毒增强子的重组表达载体在双翅目昆虫细胞中表达蛋白的应用中所使用的所述含鳞翅目病毒增强子的重组表达载体。
一种目的蛋白的制备方法,包括如下步骤:
将含鳞翅目病毒增强子的重组表达载体转染双翅目昆虫细胞,所述含鳞翅目病毒增强子的重组表达载体为上述含鳞翅目病毒增强子的重组表达载体在双翅目昆虫细胞中表达蛋白的应用中所使用的所述含鳞翅目病毒增强子的重组表达载体;
培养所述双翅目昆虫细胞,使目的蛋白表达,收集表达的目的蛋白,即得。
本发明通过将含鳞翅目病毒增强子的重组表达载体应用在双翅目昆虫细胞中,可以显著提高导入双翅目昆虫细胞中的编码单元的表达水平,使各种异源基因或重组基因得到大量表达。
附图说明
图1为piex-x载体的结构示意图;
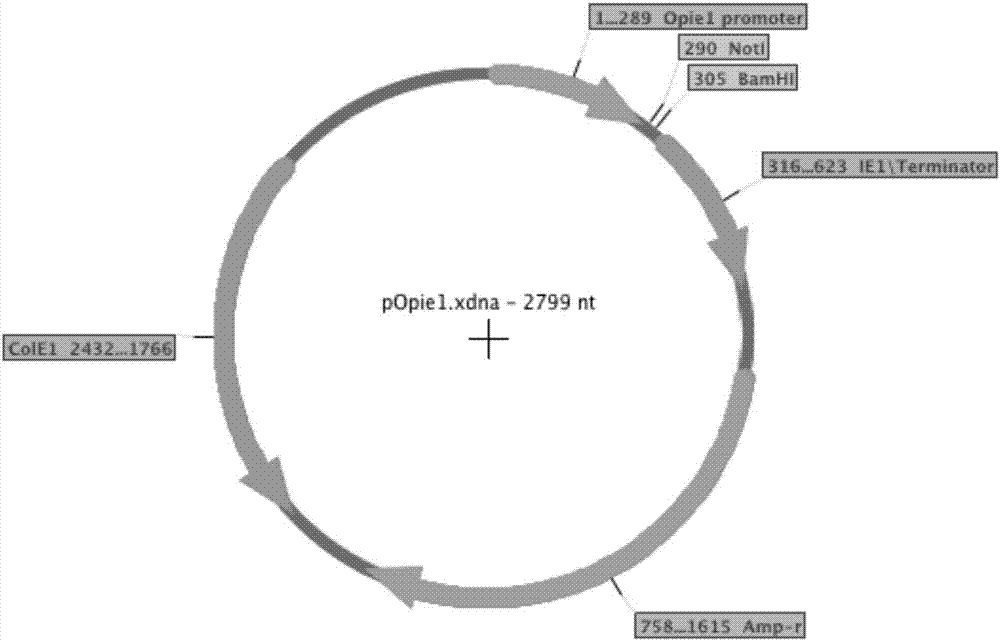
图2为popie1载体的结构示意图;
图3为popie2载体的结构示意图;
图4为phropie1载体的结构示意图;
图5为phropie2载体的结构示意图;
图6为在瞬时表达过程中,转染不同重组表达载体后的egfp阳性细胞比率;
图7为在瞬时表达过程中,转染不同重组表达载体后egfp的相对荧光单位;
图8为在稳定表达中,转染不同重组表达载体后egfp的相对荧光单位;
图9为在瞬时表达过程中,转染不同重组表达载体后tnfr-fc单位产量;
图10为在瞬时表达过程中,在不同培养体系中,转染不同重组表达载体后的tnfr-fc单位产量随时间的变化曲线;
图11为在稳定表达中,转染不同重组表达载体后的tnfr-fc单位产量随时间的变化曲线。
具体实施方式
为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
本发明提供了一种含鳞翅目病毒增强子的重组表达载体在双翅目昆虫细胞中表达蛋白的应用。重组表达载体具有调控单元及位于调控单元下游的编码单元。调控单元含有hr-x增强子或其功能片段或序列变体、acie-y启动子或其功能片段或序列变体、以及opie-z启动子或其功能片段或序列变体。编码单元含有编码目的蛋白的基因序列,如各种重组或异源基因。
本发明的hr-x增强子或其功能片段或序列变体、acie-y启动子或其功能片段或序列变体与opie-z启动子或其功能片段或序列变体依次连接,且opie-z启动子或其功能片段或序列变体位于acie-y启动子或其功能片段或序列变体的下游。hr-x增强子或其功能片段或序列变体可以位于acie-y启动子或其功能片段或序列变体的上游,或者位于opie-z启动子或其功能片段或序列变体的下游。
在本实施例中,hr-x增强子为acmnpv中的hr-5增强子、acmnpv中的hr-3增强子或bombyxmori中的hr-3增强子;hr-x增强子优选为acmnpv中的hr-5增强子,hr-5增强子的序列为seqidno.1中第1位-第482位所示序列。acie-y启动子为acmnpv中的acie-1启动子,acie-1启动子的序列为seqidno.1中第487位-第1033位所示序列。opie-z启动子为opmnpv中的opie-1启动子或opie-2启动子,opie-1启动子的序列为seqidno.2中第1位-第289位所示序列,opie-2启动子的序列为seqidno.3中第3位-第551位所示序列;opie-z启动子优选为opmnpv中的opie-2启动子。
“启动子”定义为一种通过引导rna聚合酶连接到dna并启动rna合成来介导转录起始的dna序列。“增强子”指能强化转录起始的一段dna序列。增强子(enhancer)指增加同其连锁的基因转录频率的dna序列。
本发明所述的序列变体是指与相应的天然序列具有70%以上同源性,优选80%以上,更优选90%以上,更优选95%以上,更进一步优选98%以上同源性的序列。例如,本发明优选采用的acie-1启动子也可以是acie-1启动子的功能性片段或其功能性序列变体。因此,具有介导转录起始功能或能介导转录起始从而能瞬时或稳定地驱动重组或异源产物基因表达的acie-1启动子的任何序列变体或片段均可用作acie-1启动子。acie-1启动子的“序列变体”包括含有碱基插入、删除或点突变的天然acie-1序列,可用本领域熟知方法产生,如引物-定向pcr、“易错pcr”、重叠dna片段的被称为‘基因重组’的pcr-重装,或先在体内随机诱变细胞克隆,再经文库转染和在双翅目昆虫细胞(如果蝇s2细胞)中进行功能性选择来产生。例如,随机诱变或通过烷化化学试剂或uv辐射来实现。任选地,可以采用宿主菌的天然突变菌株。acie-1启动子功能性序列变体的例子是含有转录起始位点的经基因工程改造而设置了合适的限制性位点用于插入重组产物基因的启动子序列。opie-z启动子序列变体同理。本发明优选采用的hr-5增强子也可以是其功能性片段或功能性序列变体。因此,具有acie-1启动子和opie-2启动子转录活性功能或能增强acie-1启动子和opie-2启动子转录活性的hr-5增强子的任何序列变体或片段均可用作hr-5增强子。hr-5增强子的“序列变体”包括含有碱基插入、删除或点突变的天然hr-5序列。
本发明内容中,术语“异源编码序列”、“异源基因序列”、“异源基因”、“重组基因”、“感兴趣的基因”和“转基因”可互换使用。这些术语用于dna序列时指编码重组或异源基因产物的dna序列。所述异源基因序列在宿主细胞中天然不存在而且来自不同种的生物。可使本发明的重组或异源基因产物在昆虫细胞中表达和大量收集。所述基因产物也可以是肽或多肽,可以是任何感兴趣的蛋白质,如治疗蛋白质(如白介素)或酶或多聚蛋白质的亚单元(如抗体或其片段)。所述重组产物的基因可包含信号序列,其编码的信号肽可使宿主生产细胞表达的多肽分泌出来。因此,在本发明的进一步优选实施方式中,产物蛋白是为分泌蛋白质。更优选地,所述产物蛋白是抗体或工程改造的抗体或其片段,最优选是人肿瘤坏死因子受体-fc融合蛋白(tnfr-fc)或免疫球蛋白g(igg)。
为了将表达载体诱导入本发明的昆虫宿主细胞中,如果对给定的宿主细胞类型合适,可采用任何转染技术,例如本领域熟知的技术,如电穿孔、磷酸钙共沉淀、deas-葡萄糖转染、脂转染、应该注意,可用本发明载体转染的昆虫宿主细胞应是可瞬时或稳定转染的细胞系。因此,本发明的昆虫表达载体可维持于游离体形式或可稳定地整合进昆虫宿主细胞的染色体组中。
瞬时转染的特征是对含有选择标记的载体不施加任何选择压力。来源于瞬时转染的细胞群或一批细胞包括插入了外来dna并表达的细胞和没有插入外来dna的细胞的混合细胞群。在转染后通常持续20-50小时的瞬时表达实验中,转染的载体维持为游离型元件尚未整合入染色体中,即转染的dna,经常没有整合入宿主细胞染色体中。宿主细胞倾向于丢失转染的dna。培养瞬时转染细胞群时细胞群中的转染细胞过度生长。因此在紧接转染后的时间内表达最强而随时间推移减弱。优选理解本发明的瞬时转染子是能在转染后在没有选择压力的细胞培养中维持表达120个小时的细胞。
用载体转染昆虫宿主细胞或昆虫宿主细胞系的方法是本领域熟知的。在本发明的方法中,可采用适合于给定类型宿主细胞的任何转染技术,例如本领域熟知的技术,如电穿孔、磷酸钙共沉淀、deae-葡聚糖转染、脂转染。本发明的宿主细胞可用此表达载体稳定或瞬时转染。
术语“宿主细胞”或“宿主细胞系”指能够培养生长并表达所需蛋白质重组产物的任何细胞,具体指昆虫细胞。本发明方法中所采用昆虫宿主细胞优选果蝇s2等昆虫细胞。
昆虫细胞系的合适培养液和培养方法是本领域熟知的。可采用实验室烧瓶培养或低密度细胞培养可满足特定类型细胞需要的标准细胞培养液。
在本发明一优选实施方式中,所采用的细胞培养液不含胎小牛血清(fcs或fbs),因此称为“无血清”。
在涉及表达和收集重组产物蛋白质的方法中,可采用常规的方法例如在工业化反应器中培养昆虫宿主细胞。然后进行常规的下游加工。
收集方法,即从细胞、细胞培养物或细胞培养液中分离和/或纯化给定蛋白质的方法是为本领域熟知的。例如,可通过用盐或有机溶剂进行分级沉淀、离子交换层析、凝胶层析、hplc、亲合层析等,分离和/或纯化生物材料中的蛋白质。
本发明另一个方面涉及利用opie-2启动子来提高昆虫宿主细胞表达重组或异源基因产物的acie-1启动子片段和hr-5增强子活性的方法。本发明的此方法是,用分子克隆方法将opie-2启动子插入现有载体分子中acie-1启动子片段和hr-5增强子序列与编码所需蛋白的基因序列之间,使acie-1启动子片段和hr-5增强子及opie-2启动子操作性连接于异源基因序列而显著增强acie-1启动子片段和hr-5增强子驱动所需异源基因产物的表达。分子克隆方法是本领域熟知的。然后,用如此构建的在acie-1启动子片段和hr-5增强子与位于下游编码所需蛋白质的异源dna序列之间含有opie-2启动子的载体转染昆虫宿主细胞如果蝇s2细胞。然后,在适当条件下培养转染子使细胞生长和/或增殖并表达/产生重组蛋白。最后,收集即分离和纯化产生的重组蛋白。通过在本发明的方法中利用所述opie-2启动子,可以显著增强acie-1启动子片段和hr-5增强子驱动重组蛋白的表达效率而获得高水平表达的重组蛋白。因此,本发明可能改进现有的重组基因表达的表达载体的表达效率。
本发明还提供了一种宿主细胞。该宿主细胞具有含鳞翅目病毒增强子的重组表达载体,宿主细胞为双翅目昆虫细胞,含鳞翅目病毒增强子的重组表达载体具有调控单元及位于调控单元下游的编码单元,调控单元含有hr-x增强子或其功能片段或序列变体、acie-y启动子或其功能片段或序列变体、以及opie-z启动子或其功能片段或序列变体。宿主细胞优选为果蝇s2细胞。
本发明还提供了一种目的蛋白的制备方法,包括如下步骤:
将含鳞翅目病毒增强子的重组表达载体转染双翅目昆虫细胞,含鳞翅目病毒增强子的重组表达载体具有调控单元及位于调控单元下游的用于编码目的蛋白的编码单元,调控单元含有hr-x增强子或其功能片段或序列变体、acie-y启动子或其功能片段或序列变体、以及opie-z启动子或其功能片段或序列变体;双翅目昆虫细胞优选为果蝇s2细胞;
培养双翅目昆虫细胞,使目的蛋白表达,收集表达的目的蛋白,即得。
本发明通过将含鳞翅目病毒增强子的重组表达载体应用在双翅目昆虫细胞中,可以显著提高导入双翅目昆虫细胞中的编码单元的表达水平,使各种异源基因或重组基因得到大量表达。并且该重组表达载体在双翅目昆虫细胞中可以保持长期的表达,不会造成宿主细胞的死亡。
以下为具体实施例部分:
实施例1宿主细胞
s2细胞:来自果蝇的drosophilaschneider2细胞,已适应悬浮培养和无蛋白质培养液。
s2细胞的增殖:s2细胞从tcmetrix公司(epalinges,瑞士)获得,并在sf900iisfm培养基中(lifetechnologies公司,巴塞尔,瑞士)进行悬浮培养。细胞在tubespin生物反应器50(ts50)或在tubespin生物反应器600(ts600)(tpp,trasadingen,瑞士)中培养,其各自培养基的量分别为5-10ml或300ml。细胞每周传代2次,传代细胞密度为1×106细胞/ml。所有培养物在isf1-x振荡培养器(kühnerag,birsfelden,瑞士)中保持在28℃温度下,摇晃速度为180rpm,摇动直径5厘米。细胞密度和活性通过台盼蓝排除法使用血细胞计数器测定。
实施例2质粒准备
piex-xegfp和piex-tnfr-fc(如表1所示)分别携带绿色荧光蛋白和人tnfr-fc的基因,并且受autographacalifornicamulticapsidnucleopolyhedrovirus(acmnpv)同源区5(hr-5)增强子以及立早1(acie-1,简写ie-1)启动子的控制(shen等人,2014年)。使用pib/v5-his(lifetechnologies公司)为模板,orgyiapseudotsugatamulticapsidnuclearpolyhedrosisvirus(opmnpv)立早1(opie-1)和2(opie-2)的启动子通过使用特异性寡核苷酸引物(表2)的pcr进行放大。单独的聚合酶链反应产物可被xmaⅰ和notⅰ消化,并且被亚克隆至被同样两种限制性内切酶消化后的piex–xegfp,分别产生popie1-egfp和popie2-egfp(表2)。对于这两个质粒,acmnpvhr-5增强子以及piex-xegfp的iel启动子可分别由opie-1或opie-2启动子取代。特异性寡核苷酸引物(表2)被用来放大来自pib/v5-his的opie-1和opie-2,并且在用nhei和noti来进行限制性酶切以后,聚合酶链反应产物被亚克隆进piex–xegfp用以分别构造phropie1-egfp和phropie2-egfp。对于这两个载体来说,acmnpvhr-5增强子被保留,而acmnpvie1启动子被opie-1或是opie-2的任意一个替换。tnfr-fc基因通过noti和bamhi对piex-xtnfr-fc进行限制性酶切获得,并且tnfr-fc基因用于取代经过noti和bamhi消化的上述两个质粒中的egfp基因。其所得的质粒分别命名为popie1-tnfr-fc、popie2-tnfr-fc、phropie1-tnfr-fc和phropie2-tnfr-fc(表1)。利用市售试剂盒(qiagengmbh,希尔登,德国)并按厂家使用说明进行质粒提纯。
表1质粒列表
表2引物序列列表
实施例3载体构建
在s2细胞的转染中,本实施例使用携带acmnpvie-1启动子和hr-5增强子的表达载体。为了研究其他启动子,本实施例分别构建了带有opie-1、opie-2启动子的表达载体。此外,携带opie-1启动子与acmnpvhr-5增强子的载体,以及携带opie-2启动子与acmnpvhr-5增强子的载体也被构建。载体携带egfp基因或tnfr-fc的基因的任何一个基因,并由相同的起始载体piex-x(shen等人,2014)构建。
载体piex-xegfp和piex-xtnfr-fc的产生:
piex-xegfp和piex-xtnfr-fc(表1)分别携带绿色荧光蛋白和人tnfr-fc的基因,并且受autographacalifornicamulticapsidnucleopolyhedrovirus(acmnpv)同源区5(hr-5)增强子以及立早1(ie1)启动子的控制。piex-xegfp和piex-xtnfr-fc两个质粒分别为在序列为seqidno.1的piex-10基本载体的noti和bamhi限制位点之间插入egfp基因和tnfr-fc基因获得。
载体popie1-egfp和popie2-egfp的产生:
使用pib/v5-his(lifetechnologies公司)为模板,orgyiapseudotsugatamulticapsidnuclearpolyhedrosisvirus(opmnpv)立早1(opie-1)和2(opie-2)的启动子通过使用特异性寡核苷酸引物(表2)的pcr进行放大。单独的聚合酶链反应产物可被xmaⅰ和notⅰ消化,并且被亚克隆至被同样两种限制性内切酶消化后的piex–xegfp,分别产生popie1-egfp和popie2-egfp(表2)。该两个质粒分别为在序列为seqidno.2的popie1载体和序列为seqidno.3的popie2载体的noti和bamhi限制位点之间插入egfp基因。
载体phropie1-egfp和phropie2-egfp的产生:
特异性寡核苷酸引物(表1)被用来放大来自pib/v5-his的opie-1和opie-2,并且在用nhei和noti来进行限制性酶切以后,聚合酶链反应产物被亚克隆进piex–xegfp用以分别构造phropie1-egfp和phropie2-egfp。对于这两个载体来说,acmnpvhr5增强子被保留,而acmnpvie1启动子被opie-1或是opie-2的任意一个替换。该两个质粒分别为在序列为seqidno.4的phropie1载体和序列为seqidno.5的phropie2载体的noti和bamhi限制位点之间插入egfp基因。
载体popie1-tnfr-fc,popie2-tnfr-fc,phropie1-tnfr-fc和phr-tnfr-fc的产生:
tnfr-fc基因通过noti和bamhi对piex-xtnfr-fc进行限制性酶切获得,并且tnfr-fc基因用于取代经过noti和bamhi消化的上述两个质粒中的egfp基因。其所得的质粒分别命名为popie1-tnfr-fc,popie2-tnfr-fc,phropie1-tnfr-fc和phropie2-tnfr-fc(表1)。这四个质粒分别为在序列为seqidno.2的popie1载体、序列为seqidno.3的popie2载体、序列为seqidno.4的phropie1载体和序列为seqidno.5的phropie2载体的noti和bamhi限制位点之间插入tnfr-fc基因。
实施例4转染
线性pei(polysciences,eppenlheim,德国)配制为ph7.0,浓度1mg/ml的水溶液,经过过滤除菌并保存在-20℃。对于小规模转染,将细胞离心并重悬于含培养基的ts50管中。dna和pei直接加入细胞培养液中,或预混合在去离子水中再加入培养基。在不同的细胞密度下进行转染并用新鲜培养基稀释。所有的细胞都在28℃180rpm摇晃培养。
对于300ml的转染,将细胞离心并重悬于含培养基的ts600管中。dna和pei预混合在去离子水中再加入培养基。所有的细胞都在28℃180rpm摇晃培养。
实施例5蛋白质定量
egfp阳性细胞的百分比通过guavaeasycyte流式细胞仪(guavatechnologies,hayward,美国)测定。激发光和发射光分别为488nm和532nm。egfp的荧光定量通过tecansaphireii荧光光度计(tecan,maennedorf,瑞士)测定,激发光和发射光分别为485nm和515nm。
tnfr-fc的浓度通过elisa方法确定。使用羊抗人fcγ抗体进行包被,并通过碱性磷酸酶共聚羊抗人iggγ链来检测tnf-fc。加入底物后,在405nm和490nm下用读板器检测吸收值。
使用streamlinerproteina(ge,uppsala,瑞典)纯化重组tnfr-fc,并使用dc蛋白检测(biorad,reinach,瑞士)方法进行检测。
瞬时表达的egfp阳性细胞比例和相对荧光单位(rfu)分别见图6、图7;稳定表达的egfp相对荧光单位(rfu)见图8。
转染后的细胞在细胞培养摇床(kuhner,瑞士)内悬浮培养,转速180rpm,温度28℃。转染后48小时培养基中的tnfr-fc浓度通过所述的夹心酶联免疫分析(sandwichelisa)测定(matasci等2011)。在瞬时表达中,采用了hr5增强子和opie-1启动子和hr5增强子和opie-2启动子的表达载体其tnfr-fc表达产量比无hr5增强子的表达载体有明显增加(图9、图10),
而在稳定表达中,采用了hr5增强子和opie-1启动子和hr5增强子和opie-2启动子的表达载体tnfr-fc产量更高,而长期稳定性也有更好的保证。每周取样通过夹心酶联免疫分析(sandwichelisa)测定tnfr-fc产量,结果见图11。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!