高糖基化人生长激素融合蛋白及其制备方法与用途与流程
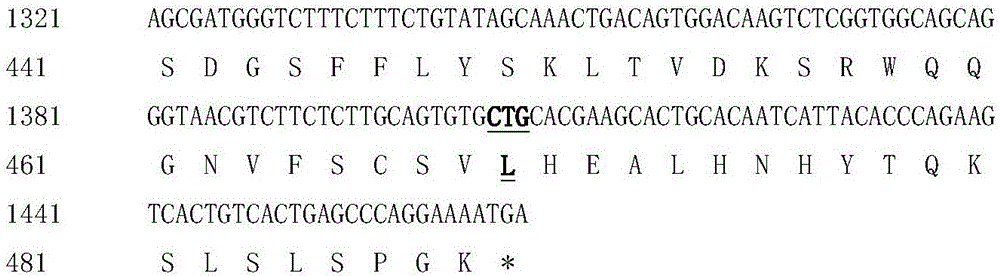
本发明涉及融合蛋白领域,更具体地,本发明涉及一种高糖基化人生长激素融合蛋白及其制备方法和用途。
背景技术:
人生长激素(human growth hormone,hGH)是由脑垂体前叶嗜酸性细胞分泌的一种蛋白质激素,由191个氨基酸组成,其生理功能主要是促进物质代谢和生长发育。hGH通过促生长因子或胰岛素样生长因子介导作用于软骨组织,增加骨骼长度,从而达到促进人体的线性生长。最近几年hGH更多的功能被发现,包括促进骨骼肌和心肌细胞生长、促进蛋白质合成、调节免疫系统功能及增强免疫防御能力等作用。hGH的大规模临床应用也从最初的生长激素缺乏的儿童矮小症的预防和治疗,拓展到烧伤、急性胰腺炎、老年人延缓衰老等领域,其临床适应证还在不断扩展中。
儿童和成人在生长激素缺乏的情况下,可以通过外源补充生长激素来进行治疗。天然hGH的体内清除半衰期约为0.3小时,目前商业化的重组人生长激素的体内清除半衰期约为2-3小时,注射后很快就会通过肝脏、肾脏清除,所以需要每天皮下注射给药,给患者带来很多痛苦。此外,长期注射重组人生长激素在少数病人体内产生抗体而影响疗效。因此,需要一种技术在延长hGH体内循环半衰期的同时还可以保持较高的活性。构建重组长效、高活性、降低的免疫原性及较高的生物利用度的人生长激素成为下一代药物开发的首要目标。
为了稳定蛋白质,防止酶解和肾脏的清除作用,可采用诸如聚乙二醇(PEG)修饰(Sada et al.,J.Ferment Bioeng 71:137-139,1991)、糖基化修饰改造(美国专利号US 7,217,689)和与其他蛋白质融合(WO 93/15199)的方法以促进体内吸收、防止被降解和肾脏清除。PEG修饰的蛋白能够防止水解,不会引起严重副作用,然而PEG与目标蛋白偶联属于非特异性共价结合,这种随机偶联不同程度的阻碍目标蛋白与其受体之间的相互作用而导致其体内活性降低。增加糖基化可以增加目标蛋白的分子量,特别对于处在肾脏滤过临界线的蛋白质(~30KD),糖基化可以降低蛋白质对蛋白酶水解的敏感性。韩国专利申请公开号KR10-2013-0029713公开了通过在α-1抗胰蛋白酶上引入至少一个突变加入N-糖基,尝试增加体内半衰期的方法。蛋白质的糖基化修饰有两类,包括O-糖链和N-糖链。然而,糖链的附着和添加可引起生理活性蛋白质失活,可额外附着糖链的生理活性蛋白质的位点的选择面也非常窄,因此向蛋白质引入额外的糖基化位点的糖基化工程技术还未广泛使用。
改善蛋白药代动力学和体内稳定性的另一种方法是将生理活性蛋白的基因连接在一个具有高稳定性的蛋白的表达基因上,通过基因重组技术以产生融合蛋白。Fc融合蛋白是利用基因工程等技术将某种具有生物学活性的功能蛋白分子(可溶性配体、受体或其他需要延长半衰期的生物活性物质)与Fc片段融合而产生的新型重组蛋白。该类融合蛋白不仅保留了功能蛋白分子的生物学活性,还能延长融合蛋白的半衰期并提高其稳定性,Fc片段的链间二硫键有利于融合分子形成二聚体,从而增强配体结合能力和提高生物活性;Fc片段的引入还有利于提高融合分子在哺乳动物细胞内的表达水平。因此制造含有与人IgG蛋白的Fc区域相连的融合蛋白,将有助于延长药物的循环半衰期和/或增加它的生物活性。专利CN102875683公开了一种长效重组人生长激素的Fc融合蛋白,在hGH和人IgG Fc变体中增加了一段柔性肽接头来降低空间位阻效应,预期目标是使其血清半衰期延长,生物活性增加,从而改善了药代动力学和药效。然而,由于hGH的活性位点主要在C端,C末端融合的其他蛋白大大影响了hGH的活性及功能,CN102875683专利中显示二聚化的hGH-L-vFc的摩尔比活性为1.2nM,其活性相较重组hGH仍不够理想。根据本发明研究发现,单纯的延长柔性肽接头,增加Fc与hGH C端的空间距离并不能解决问题,需要其它方法来解决融合配体对hGH活性影响的难题。本发明旨在寻找新的融合方法,进一步提高融合蛋白的血清半衰期及体内活性。
CTP是一段来自人绒毛膜促性腺激素(hCG)的β-亚基羧基末端的短肽。四种与生殖相关的多肽类激素,促卵泡激素(FSH)、黄体生成素(LH)、促甲状腺素(TSH)和绒毛膜促性腺激素(hCG)含有相同的α-亚基和各自特异的β-亚基。与其它三种激素相比,hCG体内半衰期明显延长,这主要来源于其β-亚基上特有的羧基末端肽(CTP)(Fares FA et al.Proc Natl Acad Sci USA.89:4304–4308,1992)。CTP含有37个氨基酸残基,它具有4个O-糖基化位点,终端是唾液酸残基。CTP可以增加蛋白质的糖基化水平,提高目标蛋白的活性,同时带负电、高度唾液酸化的CTP能够抵抗肾脏对其的清除作用,从而延长蛋白在体内的半衰期。美国专利13,195,931公开了一种通过在hGH上连接绒毛膜促性腺激素羧基末端肽(CTP)的方法,使生长激素循环半衰期仅延长了约6倍,但并未提及几种hGH/CTP嵌合蛋白体外活性的大小;体内药效显示融合1个或2个CTP分子的嵌合蛋白较重组hGH的效果差,仅CTP-hGH-CTP-CTP这种嵌合了3个CTP分子的嵌合蛋白药效略高于重组hGH。本发明人创造性地将具有多个O-糖基位点的CTP多肽作为连接肽的一部分,用于连接hGH和Fc片段,而不是作为融合配体发挥作用,因它具有的天然糖基化位点,不仅能使融合蛋白的半衰期进一步延长,生物利用度提高,同时还大大降低了融合配体Fc对hGH的位阻效应,使其保持了较高的生物学活性。
综合以上内容,目前已经有许多蛋白融合方法被构建用于研制长效蛋白类药物,然而迄今尚没有活性及半衰期非常令人满意的长效hGH制剂。因此,本领域迫切需要开发长效、高活性、纯化步骤简单、便于工业化生产的长效hGH。
技术实现要素:
本发明为了解决重组hGH血清半衰期短及生物活性差的问题,设计并制备了一种高糖基化的长效hGH融合蛋白。本发明人经过长期而深入的研究,首次设计了独特的肽接头来降低融合分子间的空间位阻,由hGH的C端与Fc连接组成的融合蛋白,中间以柔性肽和刚性肽连接。出乎意料地,此融合蛋白比以前本发明人研制的hGH-L-vFc的体外活性提高了约1倍(参见,中国专利号CN102875683)。此外,还取得了预料不到的技术效果,即所述融合蛋白的生物利用度也大幅提高。
在本发明的第一方面,提供了一种重组hGH融合蛋白,所述融合蛋白从N端到C端依次含有人生长激素(hGH)、柔性肽接头(L)、人绒毛膜促性腺激素β-亚基羧基末端刚性肽(CTP)和人免疫球蛋白Fc片段;其中,Fc片段优选人IgG Fc变体(表示为vFc)。
其中,所述柔性肽接头优选非免疫原性的,并且在hGH和Fc之间产生足够的距离,使相互之间的位阻效应降至最低。较佳地,使用含有以下2个或多个氨基酸构成的柔性肽接头:Gly(G)、Ser(S)、Ala(A)和Thr(T)。优选地,所述柔性肽接头包含G和S残基。连接肽的长度对融合蛋白的活性非常重要。对本发明而言,优选地,所述柔性肽接头氨基酸组成的结构通式为(GS)a(GGS)b(GGGS)c(GGGGS)d,其中a,b,c和d是大于或等于0的整数,且a+b+c+d≥1。
本发明的一些实施例中,所述肽接头选自如下序列:
(a)L1:GSGGGSGGGGSGGGGS;
(b)L2:GSGGGGSGGGGSGGGGSGGGGSGGGGS;
(c)L3:GSGGGSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGS;
(d)L4:GGGGSGGGGSGGGGSGGGGS。
其中,所述人绒毛膜促性腺激素β-亚基羧基末端肽刚性肽(CTP)选自由人绒毛膜促性腺激素β亚基羧基末端第113至145位氨基酸所组成的全长序列或其片段(以下简称CTP),具体地,所述CTP刚性肽包含SEQ ID NO:1或其截短的序列。
优选地,所述CTP刚性肽包含至少2个糖基化位点;例如,本发明的一优选实施例中,所述CTP包含2个糖基化位点,示例性地,所述CTP包含SEQ ID NO:1N端的10个氨基酸,即SSSS*KAPPPS*(*代表糖基化位点);或所述CTP包含SEQ ID NO:1C端的14个氨基酸,即S*RLPGPS*DTPILPQ;又如,另一实施例中,所述CTP包含3个糖基化位点,示例性地,所述CTP包含SEQ ID NO:1N端的16个氨基酸,即SSSS*KAPPPS*LPSPS*R;再如,另一些实施例中,所述CTP包含4个糖基化位点,示例性地,所述CTP序列包含28、29、30、31、32或33个氨基酸并开始于人绒毛膜促性腺激素β亚基的第113、114、115、116、117或118位,终止于第145位。具体地,所述CTP包含SEQ ID NO:1N端的28个氨基酸,即SSSS*KAPPPS*LPSPS*RLPGPS*DTPILPQ。每种可能性都代表本发明的独立实施方式。
在另一些实施例中,本发明提供的CTP刚性单元与天然CTP氨基酸序列至少70%同源;在另一些实施例中,本发明提供的CTP刚性单元与天然CTP氨基酸序列至少80%同源;在另一些实施例中,本发明提供的CTP刚性单元与天然CTP氨基酸序列至少90%同源;在另一些实施例中,本发明提供的CTP刚性单元与天然CTP氨基酸序列至少95%同源。
优选地,本发明的实施例中,所述CTP刚性肽可优选地包含如下序列单元:
(i)CTP1:SSSSKAPPPSLPSPSRLPGPSDTPILPQ;
(ii)CTP2:PRFQDSSSSKAPPPSLPSPSRLPGPSDTPILPQ;
(iii)CTP3:SSSSKAPPPS;
(iv)CTP4:SRLPGPSDTPILPQ。
本发明所述融合蛋白还可以包含2个或2个以上的上述CTP刚性肽序列单元,如本发明的一实施例中,所述CTP包含2个CTP3单元:SSSSKAPPPSSSSSKAPPPS(CTP3-CTP3,或表示为(CTP3)2)。
其中,延长半衰期部分优选自免疫球蛋白IgG、IgM、IgA Fc片段;更优选自人IgG1、IgG2、IgG3或IgG4及其变体的Fc片段;进一步地,所述人IgG Fc变体包含位于野生型人IgG Fc中的至少一种氨基酸修饰,且变体具有降低的效应子功能(ADCC和/或CDC效应)和/或与新生儿受体FcRn的结合亲和力增强。进一步地,人IgG Fc变体可选自下组:
(a)vFcγ1:含有Leu234Val、Leu235Ala和Pro331Ser突变的人IgG1绞链区、CH2和CH3区域(如SEQ ID NO:4所示氨基酸序列);
(b)vFcγ2-1:含有Pro331Ser突变的人IgG2绞链区、CH2和CH3区域(如SEQ ID NO:5所示氨基酸序列);
(c)vFcγ2-2:含有Pro331Ser、Thr250Gln和Met428Leu突变的人IgG2绞链区、CH2和CH3区域(如SEQ ID NO:6所示氨基酸序列);
(d)vFcγ4:含有Ser228Pro和Leu235Ala突变的人IgG4绞链区、CH2和CH3区域(如SEQ ID NO:7所示氨基酸序列)。
较优地,所述hGH-L-CTP-vFc融合蛋白的氨基酸序列如SEQ ID NO:2所示。
在本发明的第二方面,提供了一种编码本发明第一方面所述重组hGH-L-CTP-vFc融合蛋白的DNA分子,优选地,所述的DNA序列具有SEQ ID NO:3所示的核苷酸序列。
根据本发明的第三方面,提供一种载体,该载体包含本发明第二方面所述的DNA序列。
根据本发明的第四方面,提供一种宿主细胞,该宿主细胞包含本发明第三方面所述载体,或者转染了上述载体。
在本发明的具体实施方式中,宿主细胞是CHO的衍生细胞株DXB-11。
优选地,所述宿主细胞在其生长培养基中在每24小时内,产生超过30μg/百万个细胞的如本发明第一方面所述的重组hGH-L-CTP-vFc融合蛋白。
在本发明的第五方面,提供了一种制备本发明第一方面所述重组hGH-L-CTP-vFc融合蛋白的方法,所述方法包括:
(a)将编码融合蛋白的DNA引入CHO细胞,生成CHO衍生细胞系;
(b)筛选步骤(a)中每24小时期间内,表达超过30μg/106(百万)个细胞的高产量细胞株;
(c)培养步骤(b)筛选到的细胞株,表达融合蛋白;
(d)收获步骤(c)得到的发酵液,纯化融合蛋白。
在本发明的第六方面,本发明提供了一种药物组合物,所述药物组合物包括药学上可接受的载体、赋形剂或稀释剂,以及有效量的所述hGH-L-CTP-vFc融合蛋白。
再一方面,本发明提供了所述hGH-L-CTP-vFc融合蛋白在用于治疗因内源性生长激素分泌不足造成的生长发育障碍,以及特纳综合症引起的体型矮小、慢性肾功能衰竭、Prader-Willi综合症或特发性体型矮小等病症。
综上,本发明融合蛋白结构具有以下特点:1、融合蛋白采用的融合配体人IgG Fc变体是非裂解性的,降低了与FcγRs及Clq结合而触发的效应子功能;2、本发明制备的融合蛋白无论在发酵、纯化过程以及储存过程中均具有良好的稳定性;3、通过L-CTP连接hGH与Fc变体,CTP刚性肽不仅能进一步延长其体内半衰期;同时借助多个糖基化侧链的阻隔作用,增加融合分子间的空间距离,促使hGH和Fc段各自折叠形成正确的三维构象而互不影响各自的生物活性,与hGH-L-vFc相比,hGH-L-CTP-vFc的生物活性大幅度提高;4、融合蛋白具有降低的免疫原性,使其诱导中和抗体产生的风险降低;5、与hGH-L-vFc相比,hGH-L-CTP-vFc具有提高的生物利用度,血药浓度波动小,安全性提高,改善耐受性;具有延长的体内循环半衰期,可降低注射频率,提高患者依从性;6、与重组hGH相比,hGH-L-CTP-vFc纯化步骤简单、纯化效率高。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
发明详述:
IgG Fc变体
非裂解性Fc变体
Fc元件来源于免疫球蛋白IgG的恒定区Fc片段,它在消灭病原体的免疫防御中起重要作用。Fc介导的IgG的效应子功能发挥通过两种机制:(1)与细胞表面Fc受体(FcγRs)结合,由吞噬作用或裂解作用或杀伤细胞通过抗体依赖性细胞毒性(ADCC)途径消化病原体,或(2)与第一补体成分C1的C1q结合,引发补体依赖性细胞毒性(CDC)途径,从而裂解病原体。对应用于人的治疗而言,当hGH-L-CTP-vFc融合蛋白结合于靶点细胞表面上的hGH受体时,融合蛋白的Fc区域最好不具有不良效应子功能,从而不会溶解或除去这些靶点细胞。因此,hGH-Fc的Fc区域必须是非溶解性的,对结合于FcγRs和Clq从而触发效应子功能方面,Fc区域最好是无活性的。在四种人IgG亚型中,IgG1和IgG3能有效结合FcγRs,IgG4与FcγRs的结合亲和力较低,而IgG2与FcγRs的结合低得难以测定,所以人IgG2几乎没有ADCC效应。此外,人IgG1和IgG3还能有效结合C1q而激活补体级联反应。人IgG2与C1q结合相对弱,而IgG4不与C1q结合(Jefferis R等,Immunol Rev,1998,163:59-76),所以人IgG2CDC效应也较弱。显然,没有一种天然IgG亚型非常适合产生hGH融合蛋白。为了得到不具效应子功能的非裂解性Fc,最有效方法是对Fc片段上补体、受体结合域突变改造,调节Fc与相关受体的结合亲和力,降低或消除ADCC和CDC效应,只保留Fc的长循环半衰期特性,而不产生细胞毒性。更多的非裂解性Fc变体所包含突变位点可以参见Shields RL等,J Biol Chem,2001,276(9):6591-604或中国发明专利CN 201280031137.2。
药代动力学改善的Fc变体
IgG的血浆半寿期取决于它与FcRn的结合,一般在pH 6.0时结合,在pH 7.4(血浆pH)时解离。通过对两者结合位点的研究,改造IgG上与FcRn结合的位点,使之在pH 6.0时结合能力增加。已经证明对于结合FcRn重要的人Fcγ结构域的一些残基的突变可增加血清半衰期。尤其,在人Fcγ1中,当三个残基被其他19个常规氨基酸替代时,一些点突变显示增加的FcRn结合亲和力(Hinton等,J Biol Chem,279(8):6213-6216,2004)。Hinton等发现T250Q和M428L 2个突变体分别使与FcRn的结合增加3和7倍。同时突变2个位点,则结合增加28倍。在恒河猴体内,M428L或T250QM428L突变体显示血浆半寿期增加2倍(Paul R.Hinton等,J Immunol,2006,176:346-356)。此外,有研究对五种人源化抗体的Fc段进行T250Q/M428L突变不仅改善了Fc与FcRn的相互作用,且在随后的体内药代动力学试验中,发现以皮下注射给药,Fc突变抗体与野生型抗体相比药代动力学参数改善,体内暴露量增加,清除率降低,皮下生物利用度得到了改善(Datta-Mannan A等.MAbs.Taylor&Francis,2012,4(2):267-273.)。
羧基末端肽(CTP)
CTP是一段来自人绒毛膜促性腺激素(hCG)的β-亚基羧基末端的短肽。四种与生殖相关的多肽类激素促卵泡激素(FSH)、黄体生成素(LH)、促甲状腺素(TSH)和绒毛膜促性腺激素(hCG)含有相同的α-亚基和各自特异的β-亚基。与其它三种激素相比,hCG体内半衰期明显延长,这主要来源于其β-亚基上特有的羧基末端肽(CTP)(Fares FA等,Proc Natl Acad Sci USA,1992,89:4304-4308)。CTP含有37个氨基酸残基,它具有4个O-糖基化位点,终端是唾液酸残基。带负电、高度唾液酸化的CTP能够抵抗肾脏对它的清除作用,从而延长蛋白在体内的半衰期。因而,发明人创造性地在适当长度的柔性连接肽之后增加至少一个CTP多肽,使得融合蛋白的半衰期进一步延长,生物利用度提高。
此外,通过在hGH与Fc变体间增加CTP肽,相当于增加了一段刚性连接肽。这一方面保证了N-端融合的hGH不会影响Fc变体与FcRn的结合位点,从而影响半衰期;另外Fc的Protein A结合位点对于制备工艺中纯化步骤很重要,连接CTP保证N-端融合的hGH也不会“罩住”它与protein A的结合位点。另一方面,CTP的添加也使得约25KD大小的Fc片段不会干扰N-端融合的hGH的正确折叠,从而使其生物学活性/功能的下降或丧失。具有多个糖基侧链的刚性CTP多肽,相对于(GGGGS)n这类柔性连接肽的无规则卷曲,它可以形成稳定的立体构象,这种“阻隔”作用促使hGH和Fc段独立折叠形成正确的三维构象而互不影响各自的生物活性。
融合蛋白及其生产方法
本发明融合蛋白通常由生物合成的方法制备。根据本发明所述的核苷酸序列,本技术领域人员可方便地用各种已知方法制得本发明的编码核酸。这些方法例如但不限于:PCR,DNA人工合成等,具体的方法可参见J.萨姆布鲁克,《分子克隆实验指南》。作为本发明的一种实施方式,可通过分段合成核苷酸序列再进行重叠延伸PCR的方法来构建本发明的编码核酸序列。
本发明还提供了一种表达载体,包含编码本发明的融合蛋白的序列以及与之操作性相连的表达调控序列。所述的“操作性相连”或“可操作地连于”指这样一种状况,即线性DNA序列的某些部分能够调节或控制同一线性DNA序列其它部分的活性。例如,如果启动子控制序列的转录,那么它就是可操作地连于编码序列。
表达载体可采用市售的例如但不限于:pcDNA3、pIRES、pDR,pUC18等可用于真核细胞系统表达的载体。本领域技术人员可以根据宿主细胞来选择合适的表达载体。
根据已知空载表达载体的酶切图谱,本领域技术人员可按照常规方法通过限制性酶剪切与拼接,将本发明的融合蛋白的编码序列插入合适的限制性位点,制得本发明的重组表达载体。
本发明还提供了表达本发明融合蛋白的宿主细胞,其中含有本发明的融合蛋白的编码序列。所述的宿主细胞优选的是真核细胞,例如但不限于CHO,COS细胞,293细胞,RSF细胞等。作为本发明的优选方式,所述的细胞是CHO细胞,其可良好地表达本发明的融合蛋白,可获得结合活性良好,稳定性良好的融合蛋白。
本发明还提供一种用重组DNA制备本发明融合蛋白的方法,其步骤包括:
1)提供编码融合蛋白的核酸序列(如SEQ ID NO:3序列);
2)将1)的核酸序列插入到合适的表达载体,获得重组表达载体;
3)将2)的重组表达载体导入合适的宿主细胞;
4)在适合表达的条件下培养转化宿主细胞;
5)收集上清液,并纯化融合蛋白产物。
将所述编码序列导入宿主细胞可采用本领域的多种已知技术,例如但不限于:磷酸钙沉淀,原生质体融合,脂质体转染,电穿孔,微注射,反转录法,噬菌体转导法,碱金属离子法。
有关宿主细胞的培养和表达可参见Olander RM Dev Biol Stand 1996;86:338。可通过离心去除悬浮液中的细胞和残渣,收集清液。可通过琼脂糖凝胶电泳技术进行鉴定。
可将上述制备获得的融合蛋白纯化为基本均一的性质,例如在SDS-PAGE电泳上呈单一条带。例如,当重组蛋白为分泌表达时,可以采用商品化的超滤膜来分离所述蛋白,例如Millipore、Pellicon等公司产品,首先将表达上清浓缩。浓缩液可采用凝胶层析的方法进一步加以纯化,或采用离子交换层析的方法纯化。例如阴离子交换层析(DEAE等)或阳离子交换层析。凝胶基质可为琼脂糖、葡聚糖、聚酰胺等常用于蛋白纯化的基质。Q-或SP-基团是较为理想的离子交换基团。最后,还可用羟基磷灰石吸附层析,金属螯合层析,疏水相互作用层析和反相高效液相色谱(RP-HPLC)等方法对上述纯化产物进一步精制纯化。上述所有纯化步骤可利用不同的组合,最终使蛋白纯度达到基本均一。
可利用含有所述融合蛋白的特异性抗体、受体或配体的亲和层析柱对表达的融合蛋白进行纯化。根据所使用的亲和柱的特性,可利用常规的方法,如高盐缓冲液、改变pH等方法洗脱结合在亲和柱上的融合性多肽。可选择地,所述的融合蛋白的氨基端或羧基端还可含有一个或多个多肽片段,作为蛋白标签。任何合适的标签都可以用于本发明。例如,所述的标签可以是FLAG,HA,HAl,c-Myc,6-His或8-His等。这些标签可用于对融合蛋白进行纯化。
药物组合物
本发明还提供了一种药物组合物,它含有有效量(如0.000001-90wt%;较佳的0.1-50wt%;更佳的,5-40wt%)的本发明的融合蛋白,以及药学上可接受的载体。通常,可将有效量的本发明融合蛋白配制于无毒的、惰性的和药学上可接受的水性载体介质中,其中pH通常约为5-8,较佳地,pH约为6-8。术语“有效量”或“有效剂量”是指可对人和/或动物产生功能或活性的且可被人和/或动物所接受的量。“药学上可接受的”的成分是适用于人和/或哺乳动物而无过度不良副反应(如毒性、刺激和变态反应)的,即具有合理的效益/风险比的物质。术语“药学上可接受的载体”指用于治疗剂给药的载体,包括各种赋形剂和稀释剂。
药学上可接受的载体包括(但并不限于):盐水、缓冲液、葡萄糖、水、甘油、乙醇、及其组合。通常药物制剂应与给药方式相匹配,本发明的药物组合物可以被制成针剂形式,例如用生理盐水或含有葡萄糖和其他辅剂的水溶液通过常规方法进行制备。所述的药物组合物宜在无菌条件下制造。活性成分的给药量是治疗有效量。本发明的药物制剂还可制成缓释制剂。
本发明所述的融合蛋白的有效量可随给药的模式和待治疗的疾病的严重程度等而变化。优选的有效量的选择可以由本领域普通技术人员根据各种因素来确定(例如通过临床试验)。所述的因素包括但不限于:所述的融合蛋白的药代动力学参数例如生物利用度、代谢、半衰期等;患者所要治疗的疾病的严重程度、患者的体重、患者的免疫状况、给药的途径等。通常,当本发明的融合蛋白每天以约0.00001mg-50mg/kg动物体重(较佳的0.0001mg-10mg/kg动物体重)的剂量给予,能得到令人满意的效果。例如,由治疗状况的迫切要求,可每天给予若干次分开的剂量,或将剂量按比例地减少。
附图说明
图1、显示了在pAPG-3表达载体内Spe I-EcoR I片段的融合蛋白的核苷酸序列及推导的氨基酸序列。成熟蛋白中的肽接头位置用下划虚线标出。在Fc区域中,粗体的核苷酸和相应的氨基酸变体用下划线标出。
图2、显示了hGH融合蛋白刺激Nb2-11细胞增殖的能力。
图3、显示了hGH融合蛋白的药时曲线。
图4、显示了hGH融合蛋白给药后各组的生长曲线。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如Sambrook等人,分子克隆:实验室手册(New York:Cold Spring Harbor Laboratory Press,1989)中所述的条件,或按照制造厂商所建议的条件。
实施例1、构建编码hGH融合蛋白的表达质粒
编码hGH前导肽和成熟hGH的基因序列以及不同长度柔性肽接头、不同长度CTP刚性肽和不同IgG Fc变体的基因序列都是人工优化的CHO细胞偏爱密码子,经化学合成方法获得。为了便于将目的片段插入表达载体的特定位点,在所合成片段5’和3’端各有一个限制性酶内切位点,分别为SpeI和EcoRI。测序验证后的融合基因用SpeI和EcoRI酶切,然后插入到以PCDNA3.1为模板并改造后的表达质粒PXY1A1的相应酶切位点间,得到融合基因表达质粒pAPG。PXY1A1质粒包含但不限于以下重要表达元器件:1)人巨细胞病毒早期启动子和哺乳动物细胞高外源表达所需增强子;2)双重筛选标记物,在细菌中具有卡那霉素抗性,在哺乳动物细胞中具有G418抗性;3)鼠二氢叶酸还原酶(DHFR)基因表达框,当宿主细胞为DHFR基因缺陷型时,氨甲蝶呤(MTX)能共扩增融合基因和DHFR基因(参见美国专利US 4,399,216)。
如表1所示,本发明构建了一系列hGH-Fc融合蛋白,它们具有不同长度的柔性肽接头、CTP组成及也位置不同,以及几种不同亚型的IgG Fc变体(vFc)元件组成。其中APG-3的核苷酸序列及翻译的氨基酸序列如图1所示。
表1、构建的几种hGH融合蛋白组成
实施例2、瞬时表达不同融合蛋白和体外活性测定
实施例1中得到的一系列表达质粒,在30ml的摇瓶里使用DNAFect LT试剂(ATGCell)转染3×107CHO细胞,经转染的细胞在无血清生长培养基中生长5天,根据对应的ELISA方法测定上清液中的融合蛋白浓度,并用实施例5中描述的方法测定其体外生物学活性。ELISA结果显示几种质粒在该条件下的瞬时表达量相差不多,但是它们的活性却显示出较大的差异(表2)。其中,我们将APG-1的摩尔比活性定义为100%。APG-2的活性为APG-1的80.9%,表明仅靠延长肽接头并不能有效改善融合蛋白的活性,即彼此的空间位阻效应并不能随着肽接头的延伸而削弱。甚至,过长的肽接头不仅不能提高融合蛋白的活性,反而会使蛋白错误折叠,以无活性的多聚体形式分泌。我们推测原因,过长的柔性肽接头使融合蛋白的灵活度更高,使其能够自由转动,这可能使hGH的立体结构更加靠近Fc区,而在二者之间加入CTP,一方面相当增加了一端刚性肽接头,使彼此远离,更重要的是CTP含多个糖基侧链,相对于柔性肽接头的无规则卷曲形态,CTP可以形成固定的空间构象,能够有效的分开融合蛋白的不同功能区,这更有利于两部分独立折叠成正确的三维构象,保持了较高的活性。通过APG-1、APG-2和APG-3的活性比较验证了这种推测的正确性。而CTP置于Fc C端的APG-4的活性仅为CTP置于Fc N端的APG-3的61.3%,APG-4的活性与不含CTP的APG-1相似。而APG-3的活性比APG-4高,APG-5、APG-6和APG-7的活性也要远高于其他融合蛋白。以上结果,证实CTP对融合蛋白的活性极为关键,并且CTP置于Fc的N端更有利于提高融合蛋白的活性。
表2、瞬时表达的各种融合蛋白的体外活性EC50值比较
实施例3、融合蛋白在转染细胞系中的表达
将重组的表达质粒转染入哺乳动物宿主细胞系,以表达hGH-L-CTP-vFc融合蛋白。为了稳定高水平的表达,优选的宿主细胞系是DHFR酶缺陷型CHO-细胞(参见美国专利US 4,818,679)。一种优选的转染方法是电穿孔,也可以使用其它方法,包括磷酸钙共沉降、脂转染和原生质融合。在电穿孔中,用设置为300V电场和1050μFd电容的Gene Pulser Electroporator(Bio-Rad Laboratories,Hercules,CA),在比色杯内放置5×107个细胞,随后加入50μg用BspCI线性化的质粒进行电穿孔。在转染两天后,将培养基改成含0.6mg/mL G418的生长培养基。转染约2周后,对G418药物具有抗性的转染子可长成肉眼可见的细胞群落。用抗人IgG Fc的ELISA分析方法,筛选对选择用药具有抗性的转染子。也可用抗hGH的ELISA进行融合蛋白表达量的定量分析。通过极限稀释96孔培养板,亚克隆产生高水平Fc融合蛋白的孔。
为了实现融合蛋白较高水平的表达,宜用受MTX药物抑制的DHFR基因进行共扩增。在含有递增浓度MTX的生长培养基中,用DHFR基因共扩增转染的融合蛋白基因。用极限稀释的亚克隆能在高达6μM MTX培养基中生长的转染子。测定分泌率对亚克隆的细胞系进行进一步的分析。分泌率水平超过约30(较佳地约50)μg/106(即百万)个细胞/24小时的细胞系适应使用无血清生长培养基的悬浮培养。然后再用条件培养基纯化融合蛋白。
实施例4、融合蛋白的纯化与定性
用1N NaOH将含有融合蛋白的条件培养基滴定到pH 7~8,然后用0.45微米的硝酸纤维素过滤器过滤。将滤液加样到磷酸盐缓冲液盐水(PBS)平衡的Protein A柱上。待融合蛋白结合于Protein A柱后,弃去流出的组分。用PBS洗涤该柱,直到280nm处的OD值低于0.01。然后用0.1M pH为3.75的柠檬酸缓冲液洗脱结合的融合蛋白。洗脱液用0.4体积的1M K2HPO4中和,合并含有纯化蛋白的组分,并用PBS透析。然后用0.22微米的硝酸纤维素过滤器过滤,并存储在4℃。在非还原条件下,由SDS-PAGE对蛋白产物进行鉴定和分析。用BSA作为标准,通过BCA蛋白质分析定量该融合蛋白。
实施例5、融合蛋白的体外生物活性分析
利用Nb2-11细胞增殖来检测重组hGH-L-CTP-Fc融合蛋白的体外生物学活性。
用含有10%胎牛血清的Fischer's培养基正常培养小鼠淋巴瘤细胞Nb2-11(美国ATCC细胞库)。使用无血清培养基,1000ng/ml起始3倍梯度稀释融合蛋白,获得8个不同浓度的样本,每孔100μl,添加至96孔板,培养基为阴性对照。取处于对数期生长期的细胞,用无血清培养基洗涤细胞一次,调整密度为每毫升3×106个细胞,每孔100μl加入到上述96孔板中。在37℃、5%CO2培养箱中培养48小时,使用CCK-8试剂盒(Cell Counting Kit,购自上海翊圣生物科技有限公司)检测细胞增殖情况。用酶标仪测定在450nm处的吸光度,将OD读数相对于融合蛋白的浓度作图,所得剂量反应曲线计算融合蛋白的生物活性。
图2显示了hGH融合蛋白刺激Nb2细胞增殖的能力。表3为不同融合蛋白的半数有效浓度值(half maximal effective concentration,EC50)。由于生长激素C端的氨基酸与其功能密切相关,Fc直接与hGH的C段连接会影响其生物活性。在hGH与Fc中加入连接肽后,hGH融合蛋白的活性提高。由结果可以看出,APG-3的活性与APG-1相比,活性提高将近一倍。这可能是由于CTP一方面发挥自身的功能,另一方面作为Fc和hGH之间的刚性连接肽,CTP与柔性肽偶联,这种结构使融合蛋白折叠成更有利的三维结构,保证了hGH的生物活性。
表3、hGH融合蛋白的EC50值
实施例6、融合蛋白的药代动力学测定
雄性SPF级SD大鼠(购自上海必凯实验动物有限公司),预饲一周后每组选用体重294g左右的的大鼠3只,单次0.176mg/kg给予静脉注射APG-1和APG-3融合蛋白,考察血药浓度和时间的变化规律。对照组和给药组分别在给药后0、0.5、1、2、3、4、5、8、10、24、48、72h经眼眶采血,血液在室温放置30min后5000r/min离心10min,分离出血清,-20℃保存。用针对hGH特异的ELISA方法测定各时间点血清中hGH含量。通过软件PKSOLVER,计算各组主要药代动力学参数。各组药代动力学参数结果如表4。
表4、hGH融合蛋白的药代动力学参数
由结果看出,APG-3和APG-1融合蛋白的体内半衰期分别为2.6和2.4小时,半衰期基本一致,这一点也可以从MRT参数中看出来,二者在体内停留的时间也接近。从图3的药时曲线上发现APG-3的血药浓度一直比APG-1高,推测可能是APG-3结构的改变导致了其药代动力学的性质发生了变化;APG-3的总体清除率只有APG-1的一半,表明APG-3的体内清除相对慢,而APG-3的表观分布容积是APG-1的两倍,说明APG-1进入体内后迅速的分布到组织中,导致了其血药浓度低于APG-3。表观分布容积和总体清除率发生了改变,二者共同作用的结果是APG-3的血药浓度比APG-1要高,同时药物的体内暴露量(AUC)也高。
APG-1经过Fc位点的突变后,推测其比未突变的相比暴露量增加、清除率降低。APG-3在APG-1的基础上增加了CTP的结构,进一步降低清除率和增加AUC,在达到了上述两个目的之后,由于引入了CTP刚性结构又近一步降低了表观分布容积,使其在组织中分布更少,血液中药物含量更高,导致体内暴露量更高,所以APG-3的结构组合优势不仅体现在优越的药代参数上,而且其药效可能也优于APG-1,因为APG-3经过改造后血药浓度更高,意味着游离的药物在体内多于APG-1,所以APG-3的药效好于APG-1,生物利用度更高,可以预期其临床给药剂量也将降低。由此可见,APG-3在生物学活性和药代动力学方面表现出优异的性能。
实施例7、融合蛋白的体内生物学活性测定
选取4周龄SPF级雄性SD大鼠,体重60-80g,由中国食品药品检定研究院实验动物中心提供。
试验前2周清洁条件下手术摘除垂体,去垂体手术后2周为恢复期,给药前选择大鼠体重变化小于手术前体重±10%的合格健康动物,去垂体大鼠按体重均匀随机分为6组,5只/组。以短效rhGH(商品名norditropin,Novo Nordisk A/S生产)作为本实验的阳性参照药1(Y1),比活3IU/mg;以PEG-rhGH(长春金赛药业有限责任公司)作为阳性参照药2(Y2),比活6IU/mg;APG-3低、中、高剂量设定分别为5mg/kg/14d、15mg/kg/14d和45mg/kg/14d。根据分子量大小和摩尔数,APG-3的中剂量15mg/kg/14d与阳性参照药Y1和Y2相当。给药方式为颈部皮下注射,不同剂量APG-3融合蛋白和Y2,每鼠于第1天和第8天各给药1次;Y1每天给药1次,连续14天。
给药后每只大鼠每天称重,第15天用二氧化碳窒息处死全部大鼠,称重。每只动物第i天(di)体重(bwi)与给药前即第1天(d1)体重(bw1)之差即增加的体重(必要时,实验结束后可进行尸检,切开蝶鞍区,肉眼检查有无垂体残留,剔除有垂体残存的动物)。体重增加计算公式:Δbw=bwi–bw1,其中:Δbw为体重增重;bw1为给药前d1体重;bwi为给药后第i天体重。计量资料以均数标准差表示,结果见表5。图4显示了给药后各组的生长曲线。
表5、hGH融合蛋白给药前后大鼠体重变化(n=7)
从结果可以看出,与模型组相比,在给药后第8天各给药组Δbw均显著增加,但各给药组间Δbw值差异不显著;而在给药后第15天,APG-3对大鼠的体重增长具有非常显著的促进作用,高剂量APG-3诱导垂体切除大鼠体重增加(Δbw值)是rhGH(Y1)和PEG-rhGH(Y2)组的约1.5倍;而中剂量APG-3诱导的体重增加略优于Y1,与Y2基本一致。我们发现,APG-3高剂量组在第2次给药后对大鼠体重增加,与PEG-rhGH组相比表现出更显著的促进作用,而在第1次给药后一周内,两组的Δbw值却不具有统计学差异。从这种差异性结果分析推测出APG-3相比PEG-rhGH应具有更长的体内活性半衰期,因此重复给药后,这种体内蓄积效应使得APG-3组在第2次给药后生长曲线的上升趋势更陡峭。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!