检测人类CYP1A2基因分型的引物及探针、试剂盒、方法与流程
本发明属于基因分型技术领域,具体涉及一种检测人类CYP1A2基因分型的引物及探针、试剂盒、方法。
背景技术:
细胞色素P450超家族中,与药物代谢相关的代谢酶主要是CYP1、CYP2、CYP3家族。CYP1A2是CYP1A亚家族的重要成员,是人肝脏中最重要的CYP酶之一,占肝脏CYP酶总量的13~15%。CYP1A2在肝组织中有特异性表达,芳胺、杂环胺及一些含卤烃化物均是其重要底物。具有遗传多态性,存在个体、种族差异性,导致不同个体、种族的药物代谢能力存在差异。CYP1A2*1F(-163C>A)位于1号内含子,突变会增加CYP1A2的活性。CYP1A2的不同基因型直接影响药物疗效和毒副作用的强弱,因此根据CYP1A2的基因型来指导准确、安全用药具有重要的临床意义。
用于检测基因多态性的方法主要有测序法、PCR-RFLP(聚合酶链反应-限制性片段长度多态性)、基因芯片法、荧光定量PCR法。测序法结果准确,但成本高、耗时长。PCR-RFLP通过酶切电泳的方法检测基因多态性,成本低、结果直观,但操作过程易受污染,导致错误结果。基因芯片法通过核酸杂交检测基因型,优点是通量高,缺点是成本较高,操作繁琐,结果难判读。传统荧光定量PCR法依据其原理不同可分为荧光PCR探针法和荧光PCR熔解曲线法。前者针对目标SNP位点设计两种探针,分别和野生型与突变型匹配,利用扩增的Ct值差异来判读基因型。后者针对目标SNP位点设计一种探针,基于探针与目标核酸之间杂交的准确性(即SNP位点上不同碱基造成熔解温度的差异)来判读基因型。传统的荧光定量PCR法检测的准确度依赖于探针与对应的等位基因之间的亲和程度的差别,即其溶解温度(Tm)的差别,当这种差别并不是很大时,往往导致最终的检测结果存在偏差,准确度不高。随着技术的不断创新,一种基于瓣状核酸内切酶及双荧光标记探针的新型SNP检测技术被开发出来。该方法采用具有瓣状核酸内切酶活性的DNA聚合酶,并利用这种聚合酶在切除瓣状结构中的5’端单链时的切割位置位于瓣状结构的双链部分的第一个碱基和第二个碱基之间这一特性,结合修饰有双重荧光标记及淬灭基团的DNA探针,实现了用一个荧光探针检测目标核酸SNP位点上的分型。
目前针对CYP1A2基因分型的检测方法局限上述基因芯片法、传统荧光定量PCR探针法,其缺点越来越难以满足临床检验的要求。在利用双荧光标记探针的新型SNP检测技术检测CYP1A2基因分型的研究,还没有人报道。
技术实现要素:
本发明目的在于克服现有技术的不足,通过自主设计引物和探针,并结合基于瓣状核酸内切酶及双荧光标记探针的新型SNP检测技术,提供一种检测人类CYP1A2基因分型的引物及探针、试剂盒、方法,以解决目前针对CYP1A2基因分型检测的局限性问题。
为了实现上述发明目的,本发明采用的技术方案如下:
用于检测人类CYP1A2基因分型的引物及探针,包含以下核苷酸序列:
上游引物SEQ ID NO:1:5’-AAACTGAGATGATGTGTGGAGG-3’;
下游引物SEQ ID NO:2:5’-CACGCATCAGTGTTTATCAAA-3’;
探针SEQ ID NO:3:5’-GTGGGCCCAGGACGCATGGTAGATGGA-3’。
本发明提供的引物和探针能准确检测到CYP1A2基因突变位点区域。
本发明还提出一种检测人类CYP1A2基因分型的试剂盒,所述试剂盒包含PCR反应液,所述PCR反应液包括上述引物及探针。
本发明提供的试剂盒成本低,使用简单,能准确检测CYP1A2基因分型。
本发明还提供一种利用上述试剂盒检测人类CYP1A2基因分型的方法,其包括如下步骤:
提取人基因组DNA;
将所述DNA与所述试剂盒配成反应体系,进行荧光定量PCR扩增;
根据所述扩增结果分析所述CYP1A2基因分型。
本发明提供的检测CYP1A2基因分型的方法,用一条荧光探针即可检测目标核酸SNP位点上的多种分型,简化了操作过程,节约了成本,检测结果分辨率高、迅速准确。
另外,本发明还提供利用上述试剂盒在检测CYP1A2基因分型中的用途。该用途可破除现有CYP1A2基因分型检测的局限性,相对现有技术,更易满足临床检验的要求。
附图说明
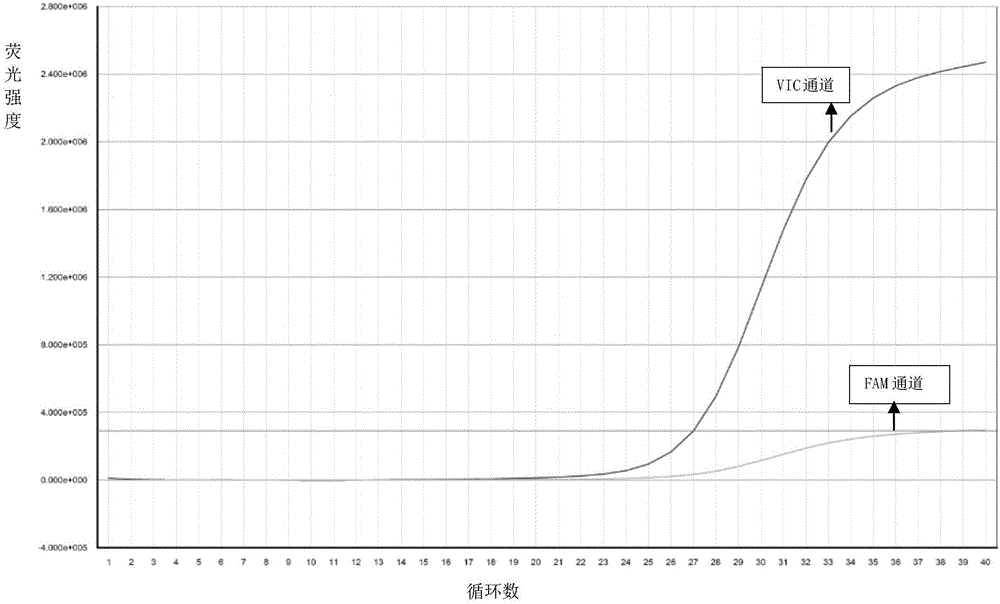
图1是CYP1A2基因-163位点野生型检测结果图;
图2是CYP1A2基因-163位点纯合突变型检测结果图;
图3是CYP1A2基因-163位点杂合型检测结果图;
图4是CYP1A2基因-163位点纯合突变型对比试验检测结果图。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明实施例提供一种用于检测人类CYP1A2基因分型的引物及探针,包含以下核苷酸序列:
上游引物SEQ ID NO:1:5’-AAACTGAGATGATGTGTGGAGG-3’;
下游引物SEQ ID NO:2:5’-CACGCATCAGTGTTTATCAAA-3’;
探针SEQ ID NO:3:5’-GTGGGCCCAGGACGCATGGTAGATGGA-3’。
该探针第7个核苷酸到3’端之间的序列与目标核酸上相应序列互补,该CYP1A2基因的突变为c.-163C>A。本实施例的引物和探针能准确检测CYP1A2基因突变位点区域。
具体地,在探针5’端和3’端分别用两种不同的荧光报告基团标记,优选地,荧光报告基团选自:FAM、HEX、TET、VIC、ROX、CY5、CY3、JOE、ALEX、CAL;在探针的第8位核苷酸位点用荧光淬灭基团标记。在本实施例中,DNA探针的5’端及3’端分别有一个发出绿色荧光(FAM)及红色荧光(VIC)的基团双重标记,而在SNP位点的下一个碱基(沿5’端到3’端的方向地8个碱基)上修饰有一个淬灭基团(IDT公司的ZENTM Internal Quencher),该淬灭基团能够同时淬灭绿色荧光基团及红色荧光基团的荧光。双重荧光标记DNA探针上由SNP位点到其3’端之间的序列与目标核酸上相应序列互补,而由SNP位点到其5’端之间的序列与目标核酸上相应序列不互补,因此形成瓣状结构。
本发明实施例还提供一种检测人类CYP1A2基因分型的试剂盒,该试剂盒包含由上述引物及探针组成的PCR反应液,其中:上游引物浓度为:0.1-0.5μM;下游引物浓度为:0.1-0.5μM;探针浓度为:0.1-0.5μM。具体在本实施例中,上游引物0.5μM;下游引物0.5μM;探针0.5μM。该试剂盒成本低,使用简单,能准确检测CYP1A2基因分型。
具体地,该试剂盒还含有dNTPs,MgCl2。其中dNTPs浓度为:0.2-0.5mM;MgCl2浓度为:0.5-2.5mM;本实施例中,优选dNTPs 0.5mM;MgCl2 2.5mM。具体地,该试剂盒还包含UNG酶和DNA聚合酶,该DNA聚合酶具有瓣状核酸内切酶活性。
由于DNA聚合酶具有瓣状核酸内切酶活性,当正向引物被延伸至荧光探针处时,荧光探针上不与目标核酸互补的序列会被聚合酶切除。切割位置在瓣状结构的双链部分的第一个碱基和第二个碱基之间。当目标核酸上SNP位点的碱基与荧光探针上的对应碱基互补时,则在SNP位点和下一个碱基之间切除。因此,绿色荧光基团与淬灭基团分离,从而产生绿色荧光。随着聚合酶继续切割荧光探针上与目标核酸互补的序列,淬灭基团也与另一端的红色荧光基团分离,从而产生红色荧光。因此,在绿色荧光基团与红色荧光基团的发光效率大致上相同的情况下,若目标核酸上的SNP位点的碱基与荧光探针上的对应碱基互补,测得的绿色荧光的强度与红色荧光的强度大致上相等。当目标核酸的SNP位点上的碱基与荧光探针上相应位置的碱基不互补时,聚合酶在荧光探针上进行切割的位置在修饰有淬灭基团的碱基之后。因此,在荧光探针上不与目标核酸互补的序列被切除下来之后,绿色荧光基团与淬灭基团仍保留在该被切除下来的序列上,因此绿色荧光基团的荧光仍旧被淬灭基团淬灭,从而不产生绿色荧光,但淬灭基团与红色荧光基团分离,从而产生红色荧光,此时测得的绿色荧光的强度与红色荧光的强度相比可忽略不计。当核酸样本中同时含有一半的SNP位点的碱基与荧光探针上的对应碱基互补的目标核酸和另一半的SNP位点的碱基不与荧光探针上的对应碱基互补的目标核酸,在这种情况下,SNP位点的碱基与荧光探针上的对应碱基互补的目标核酸同时产生绿色荧光和红色荧光,而SNP位点的碱基不与荧光探针上的对应碱基互补的目标核酸只产生红色荧光。由于两种目标核酸的含量相等,在绿色荧光基团与红色荧光基团的发光效率大致上相同的情况下,测得的所述绿色荧光的强度约为红色荧光的强度的一半。
本实施例的试剂盒检测SNP的特异性来源于具有瓣状核酸内切酶活性的DNA聚合酶切除瓣状结构时的切割位置的准确性,而不是基于DNA探针与目标核酸之间的杂交的准确性(即SNP位点上不同碱基造成熔解温度的差异),因此,该试剂盒检测SNP的准确性和可靠性显著地高于现有技术。
本发明实施例的试剂盒反应体系采用UNG-dUTP防污染系统,其中,所属UNG酶可以选择性水解断裂含有dU的双链或单链DNA中的尿嘧啶糖苷键,形成的有缺失碱基的DNA链,在碱性介质以及高温下会进一步水解断裂,因此在反应体系中加入适量dUTP,在PCR扩增前,利用UNG酶作用能有效防止实验室内扩增产物导致的污染,从而对整个检测体系包括样品的浓度、干扰因素进行质量控制。且在95℃时UNG酶可被灭活,不影响后续PCR扩增效果。
更具体地,本发明实施例试剂盒设置有阳性对照品和阴性对照品,阳性对照品包含两种质粒,阴性对照品为TE缓冲液。该两种质粒分别包含CYP1A2基因的-163C野生型核酸序列和-163A突变型核酸序列。两种质粒按摩尔浓度比1:1的比例组成。通过阳性、阴性对照,使本实施例技术方法更加准确真实。
本发明实施例还提供一种检测人类CYP1A2基因分型的方法,其包括如下步骤:
提取人基因组DNA;
将DNA与上述试剂盒配成反应体系,进行荧光定量PCR扩增;
根据扩增结果分析CYP1A2基因分型。
具体地,该反应体系为40ul。其中,DNA质量:10-500ng,优选100ng;PCR反应液34.5ul,混合酶0.5ul。荧光定量PCR扩增的反应程序为:50℃2min;95℃预变性2min;95℃变性15s;62℃延伸45s;35个循环。
本方法利用瓣状核酸内切酶结合双重荧光标记DNA探针进行荧光PCR反应实现了用一条荧光探针检测目标核酸SNP位点上的多种分型,简化了操作、节约了成本。基于该方法,准确检测CYP1A2基因分型。
最后,根据本发明的实施例,本发明人能理所当然想到上述试剂盒在检测CYP1A2基因分型中的用途。该用途可破除现有CYP1A2基因分型检测的局限性,相对现有技术,更易满足临床检验的要求。
本发明先后进行过多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
实施例1检测人类CYP1A2基因分型的试剂盒
引物和探针设计
发明人根据NCBI人CYP1A2基因序列,设计能检测-163位点基因多态性的引物和探针核苷酸序列如下:
上游引物SEQ ID NO:1:5’-AAACTGAGATGATGTGTGGAGG-3’;
下游引物SEQ ID NO:2:5’-CACGCATCAGTGTTTATCAAA-3’;
探针SEQ ID NO:3:5’-GTGGGCCCAGGACGCATGGTAGATGGA-3’。
其中,探针第7个核苷酸到3’端之间的序列与目标核酸上相应序列互补,探针用双荧光报告基团标记,其5’端用FAM标记,3’端用VIC标记,探针第8位T标记淬灭基团ZENTMInternal Quencher。
CYP1A2荧光定量PCR反应液的配制
本实施例PCR反应液浓度配置如下:缓冲液,dNTPs(含dUTP)0.5mM;MgCl2 2.5mM;上游引物0.5μM;下游引物0.5μM;探针0.5μM。
混合酶
试剂盒中,所用混合酶由尿嘧啶糖基化酶(UNG酶)和DNA聚合酶混合而成,其中,DNA聚合酶为具有瓣状核酸内切酶活性的DNA聚合酶,选自New England Biolabs公司的Taq DNA聚合酶。该组成的混合酶活性浓度为2-4U。
对照品
根据NCBI数据库CYP1A2基因-163位点基因多态性设计阳性对照品序列,合成序列,构建DNA重组质粒。CYP1A2阳性对照品包括2种质粒分别为-163C野生型、-163A突变型。具体是由-163C野生型、-163A突变型重组质粒按1:1混合而成。且阴性对照品为TE缓冲液。
根据上述的PCR反应液、混合酶、对照品即组成本实施例提供的用于检测人类CYP1A2基因分型的试剂盒。
需要说明的是:PCR反应液中各组分的含量不限于上述浓度,只要满足如下都可用于后续试验,即:dNTPs浓度0.2-0.5mM;MgCl2浓度0.5-2.5mM;上游引物浓度0.1-0.5μM;下游引物浓度0.1-0.5μM;探针浓度0.1-0.5μM。
实施例2CYP1A2基因分型的荧光定量PCR检测及分析
(一)人血液样本基因组DNA的提取
本实施例利用深圳华因康基因科技有限公司宝安分公司生产的血液基因组提取纯化试剂盒提取样本DNA,具体操作如下:
(1)从提取试剂盒中取出1.5ml离心管若干个,每个管加入抗凝血200μl。
(2)加入500μl缓冲液CLG溶液到抗凝血中,盖紧离心管盖,漩涡震荡20s以上,充分颠倒混匀,必须充分混匀或漩涡震荡以确保释放基因组DNA。
(3)加入100μl缓冲液PP溶液,漩涡震荡或来回颠倒10s。
(4)12,000g离心5min。
(5)从提取试剂盒中取出DNA纯化柱,将步骤(4)中的上清液转移至纯化柱,8000g离心1min。为提高DNA提取量,离心后,滤液可再回倒离心一次。
(6)倒弃过柱后滤液,将DNA纯化柱放入同一收集管中,往纯化柱中加入750μl缓冲液WⅠ,室温放置2min后,8000g离心1min(确认缓冲液WⅠ已加入无水乙醇)。
(7)倒弃过柱后滤液,往纯化柱中加入750μl Buffer缓冲液WⅡ,8000g离心1min(确认缓冲液WⅡ已加入无水乙醇)。
(8)倒弃过柱后滤液,将纯化柱12,000g离心2min。
(9)将纯化柱转移至新的1.5ml离心管中,室温放置5min充分挥发乙醇。加70~100μl收集液EB(收集液EB预热到65℃有利于提高DNA的洗脱效率),室温放置1min,12,000离心2min洗脱基因组DNA。可将过柱后的洗脱液吸出再加到纯化柱膜上,室温放置1min,12,000g离心1min可以提高DNA的洗脱量。
最后,基因组DNA的浓度和质量通过微量紫外分光光度计测定OD260和OD280吸光值(德国Implen的NanoPhotometerTMP-Clas微量分光光度计、北京凯奥科技发展有限公司的K2800核酸分析仪),DNA浓度在2ng/μl~100ng/μl;OD260/OD280值在1.5~2.0之间,满足后续试验要求。
纯化的DNA样本在4℃保存不超过7天;未能及时检测的DNA样本于-20℃保存,保存时间不超过3个月;纯化DNA样本反复冻融不超过5次。
(二)试剂准备
从试剂盒中取出PCR反应液,在冰上解冻,计算每个反应体系所需要的PCR反应管数,管数为N(N=n样本数+1阴性对照+1阳性对照,N≥3)。按下表1分别取计算量的PCR反应液和混合酶到合适的离心管中,颠倒混匀10次,2000g离心10秒,标记不同的反应体系。
表1
(三)加样
阳性对照和阴性对照取出,在冰上解冻,在涡旋振荡器混匀,2000g离心10秒。向所设定的PCR反应管中分别加入样本、阳性对照、阴性对照各5ul,盖紧管盖,并做好记录。混匀各PCR反应管中的PCR反应体系和所加的样本。将PCR反应管2000g离心10秒。转移至检测区,置于PCR仪上,记录样本摆放顺序。
需要说明的是,上述样本、阳性对照DNA质量浓度在2ng/μl~100ng/μl之间,即每个反应体系为10-500ng都可以满足试验要求。
(四)PCR扩增
ABI7500荧光定量PCR仪,选择FAM(Reporter:FAM;Quencher:none)检测通道;VIC(Reporter:VIC;Quencher:none)检测通道。参比荧光(Passive Reference)设置为None,阈值线设为50000。
PCR扩增程序:50℃2min;95℃预变性2min;95℃变性15s;62℃延长45s;35个循环,在62℃45s时收集荧光,即荧光收集设置在“步骤3:62℃,45s”。
(五)检测结果分析
如反应体系检测结果在VIC检测通道无Ct或Ct大于37.5,则判断样本的基因组DNA浓度低于检测限。当反应体系检测结果在VIC检测通道Ct小于或等于37.5的情况下,按以下表2判断(即比较样本FAM通道和VIC通道的荧光强度),CYP1A2基因分型如下表3所示。
表2
表3
在本实施例检测结果中,CYP1A2基因-163位点的检测结果有3种。如图1所示:FAM荧光强度和VIC通道荧光强度相近,即-163位点为CC野生型;如图2所示:FAM通道荧光强度相比VIC通道荧光强度可忽略不计,即-163位点为AA纯合突变型;如图3所示:FAM荧光强度为VIC通道荧光强度一半,即-163位点为CA杂合型。
本实施例设计了普通TaqMan探针进行对比试验,针对野生(FAM标记)及突变位点(VIC标记)分别设计普通TaqMan探针。检测纯合突变型样本的结果如图4所示,FAM标记的野生型匹配探针有明显扩增信号(理论上此时应该没有扩增信号或可忽略不计),其结果相对本发明所设计的双标记探针准确性明显降低。
由此可知,该CYP1A2基因分型检测方法,具有操作简单、迅速、分辨率高、结果更准确的特点。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!