一种次膦酸酯的制备方法、次膦酸酯及非水电解液与流程
本发明涉及一种次膦酸酯的制备方法、次膦酸酯及非水电解液。
背景技术:
在锂离子二次电池应用领域中,安全性能始终是一个“绊脚石”,如电动汽车领域的起火事件。电动汽车起火事件主要原因在于其使用的锂离子二次电池,而电池起火的原因之一是其使用的可燃的非水电解液。此外,锂离子二次电池的寿命,尤其是高温条件下的容量保持率,也不能满足目前实际应用需求。
目前为止,已有较多文献报道将次膦酸酯用作电解液的成膜添加剂、阻燃添加剂或者不燃溶剂以提升锂/钠离子二次电池的高温特性和安全性能。中国专利cn102916223a公开了一种非水电解液,该非水电解液包含氧化膦、膦酸酯及次膦酸酯等至少一种有机磷化合物。其发明人认为,次膦酸酯可以在正极和/或负极的至少一个电极表面形成一个涂层(sei),该涂层可以改善电池的高温保存特性和高温循环性能。中国专利cn1685556a公开了一种含次膦酸酯的电解液可以防止电解液在高涓流充电或高温储存的过程中分解变质,从而提高二次电池的高温稳定性。日本专利jp1998228928a和jp1999233141a分别公开了通过加入5~100%含量的膦酸酯和/或次膦酸酯以提升电解液不燃性的方法。该方法不仅可以提升二次电池的安全性同时又不损害电池的其他性能。次膦酸酯的应用价值日益受到人们的关注,但对其合成方法的研究却少之又少。
技术实现要素:
为了解决上述问题,本发明提供了一种次膦酸酯的制备方法,包括以下步骤:
(1)通过格氏试剂r1mgx与三卤硫磷x3p=s在溶剂中反应,得到第一中间体
(2)通过卤化剂m’xa与所述第一中间体进行反应,得到第二中间体
(3)通过r2-oh、水与所述第二中间体反应,得到次膦酸酯
其中,r1、r2分别选自烃基、或含有硼、硅、氮、磷、氧、硫、氟、氯、溴及碘中至少一种元素的有机基团;x表示卤素;m’表示金属元素;a表示金属元素m’的化合价。
本发明第一步中,格氏试剂r1mgx在无水条件下与含卤素基团的三卤硫磷x3p=s反应,得到r1基取代的第一中间体
第二步,第一中间体
本发明第二步反应可在溶剂中反应,也可以不使用溶剂。
第三步,r2-oh、水与第二中间体
第三步反应中,醇的羟基氧和水中的氧呈富电子性,亲核进攻磷元素。
在第三步反应中,因为第二中间体极易水解,因此当第二中间体的加入量小于等于2mol时,第三步反应可以在空气氛围下反应得到产物次膦酸酯,即空气中的水分即可满足反应物中对水含量的需求,无需再加水作为反应物添加。这样一方面可以节约制备的成本,更重要的是可以防止由于水的加入量和加入速度没有控制合适而产生的剧烈水解反应引发的危险;同时也可以避免加入过量的水在提纯(如萃取、蒸馏、精馏、干燥)的过程中增加成本。提纯方法包括通过萃取、蒸馏、精馏、干燥等本领域已公开的方法。
在第三步反应当中,当第二中间体用量超过2mol,本发明中水需要作为反应物额外添加。因为当反应投料较大时,空气中的水分含量不足以与第二中间体和醇反应完全。但水含量不能过高,因为商业化的电解液为纯有机溶剂体系,其对水分非常敏感,所以水含量过高会使提纯的成本增加。
同时,第三步反应为无溶剂反应,反应原料绿色环保,因此本反应属于绿色环保节约型反应。同时也大大减少了反应的提纯(如萃取、蒸馏、精馏、干燥)成本,因为本发明的反应产物会被用作电解液添加剂,而电解液对其添加剂的纯度要求非常高。此外,本反应不需要无水无氧操作,这为扩大生产充分地提供了有利条件。
作为一种实施方式,所述r1为饱和链状烷基或饱和链状烷氧基。作为优选,r1选自甲基、乙基、丁基、异辛基、2-甲氧基乙基、2-乙氧基乙基或甲氧基甲基。
作为一种实施方式,所述r1为含有烯基、非末端炔基或硝基的不饱和链状基团。作为优选,r1选自乙烯基、1-甲基丙炔基、烯丙基、2-硝基乙基或硝基甲基。
作为一种实施方式,所述r1为饱和环状烷基或含有醚基的饱和环状基团。作为优选,r1选自环己基、环戊基或3-甲氧基环己基。
作为一种实施方式,所述r1为含有烯基、炔基或硝基的不饱和环状基团;或r1为苯基、苯基衍生物基团或杂环基团;作为优选,所述r1选自环戊烯基、3-炔基环己基、3-硝基环己基、苯基、4-甲基苯基、4-甲氧基苯基、二苯基甲基、萘基、呋喃、噻吩或咪唑。
作为一种实施方式,所述r2为碳原子数为1~8的饱和链状烷基。作为优选,所述r2为甲基、乙基、丙基、异丙基、丁基或叔丁基。
作为一种实施方式,所述r2为碳原子数为1~8的氟代烷基。作为优选,所述r2选自2,2,2-三氟乙基、六氟异丙基、七氟异丁基或全氟丁基。
作为一种实施方式,所述r2为含卤素取代基的芳基。作为优选,所述r2为五氟苯基、氟代甲苯基、三氟甲基苯基、三(2,2,2-三氟乙基)苯基或三(2,2,2-三氟乙氧基)苯基。
作为一种实施方式,所述三卤硫磷为三氯硫磷;所述m’选自砷或锑;所述x选自氟、氯、溴及碘中的至少一种。
作为一种实施方式,所述步骤(1)的溶剂选自脂肪族烃类化合物、芳香烃化合物、醚类化合物及酮类化合物中至少一种。作为优选,所述步骤(1)的溶剂选自正己烷、环己烷、苯、甲苯、四氢呋喃及乙醚中的至少一种。
作为一种实施方式,所述步骤(1)中反应温度为-30~60℃;反应时间为2~48小时。作为优选,步骤(1)中反应温度-10~25℃;反应时间为6~24小时。作为一种实施方式,所述步骤(2)中反应温度为25~200℃,反应时间为0.2~10小时。作为优选,步骤(2)中反应温度为50~150℃;反应时间为0.5~5小时。作为一种实施方式,所述步骤(3)中反应温度为0~80℃;反应时间为1~48小时。作为优选,步骤(3)中反应温度为20℃~40℃;反应时间为5~24小时。
作为一种实施方式,步骤(3)中第二中间体与h2o的摩尔比为1:(0.5~1);作为优选,步骤(3)中第二中间体与h2o的摩尔比为1:(0.5~0.8)。步骤(3)中第二中间体与r2oh的摩尔比为1:(1~1.5)。作为优选,步骤(3)中第二中间体与r2oh的摩尔比为1:(1~1.2)。
在步骤(3)中,第二中间体与水反应生成中间产物
本发明提高次膦酸酯
此外,目前商业化的电解液基本为纯有机溶剂体系,对水分非常敏感,所以本发明中控制h2o的量非常的重要。
本发明提高次膦酸酯
此外,过量的醇的提纯(如萃取、蒸馏、精馏、干燥)比较简单且可回收利用。
本发明提高次膦酸酯
本发明的另一个目的是提供一种次膦酸酯,采用本发明上述次膦酸酯的制备方法制备而成。
本发明的另一个目的是提供一种非水电解液,包括本发明上述的次膦酸酯。该电解液可以提升二次电池的高温循环稳定性,同时还具有不燃、阻燃或自熄特性,可以提升二次的电池安全性能。
作为一种实施方式,所述次膦酸酯的质量为非水电解液质量的5~90%。作为优选,所述次膦酸酯的质量为非水电解液质量的10~80%。作为进一步优选,所述次膦酸酯的质量为非水电解液质量的25~60%。
本发明还提供一种非水电解液二次电池,包括正极、负极、隔膜及本发明所述的非水电解液。
作为优选,所述的正极材料选自锂镍钴锰复合氧化物、钠镍钴锰复合氧化物、钠镍钴复合氧化物、锂镍钴铝复合氧化物、锂锰镍复合氧化物、橄榄石型锂铁磷氧化物、锂钴氧化物、钠钴氧化物、锂锰氧化物及钠锰氧化物中至少一种。但不限于上述材料。
作为优选,所述的负极材料选自石墨、中间相碳微球、无定型碳、锂钛氧化合物、锂钒氧化合物、硅基材料、锡基材料及过渡金属氧化物中至少一种。所述石墨包括人造石墨及天然石墨;所述非定型碳包括硬碳及软碳。但不限于上述材料。
作为优选,所述隔膜选自聚烯烃熔融拉伸隔膜;或所述隔膜选自pet(聚对苯二甲酸乙二醇酯)、聚偏氟乙烯、芳纶及聚酰胺中至少一种为基材的隔膜;或选自高软化点多孔基体材料上涂布聚烯烃的隔膜。本发明聚烯烃类熔融拉伸隔膜可以是聚丙烯单层隔膜或聚乙烯单层隔膜,或是聚丙烯/聚乙烯/聚丙烯三层复合隔膜等。本发明所述高软化点的多孔基体材料是指软化点高于150℃的多孔基体材料。但不限于上述材料。
本发明所述的非水电解液二次电池,除了使用本发明中所述的正极材料的活性物质、负极材料的活性物质、隔膜以及非水电解液外,对其构造不作限定,对其加工制造工艺也不作具体限定,可以与普通锂离子二次电池相同。如正极、负极、隔膜可采用如下方法制备,电池可以采用如下方法组装:
(a)正极
所述的非水电解液二次电池用正极可以用如下所述的方法制造。
首先,混合粉末状正极活性物质、导电剂和粘接剂,并添加溶剂,制成浆料。正极浆料中各材料的混合比,往往决定锂离子二次电池的电化学性能。一般地,正极浆料中各固体材料成分的总质量作为100质量份,与通常的锂离子二次电池的正极类似,优选将活性正极材料含量设定为80~95质量份、导电材料含量设定为2~15质量份、粘接剂含量设定为1~18质量份。
将所获得的正极浆料涂布于铝箔制集流体的表面,并进行干燥以使溶剂挥发。根据需要,也可以通过辊压法等进行加压,以提高电极密度。由此,可制造片状正极。可根据目标电池,以适当的尺寸裁剪片状正极。正极的制造方法并不局限于所例示的方法,也可以采用其它的方法。在制造正极极片时,作为导电剂,例如可使用碳,可以是无定形碳也可以是结晶碳,包括木炭、焦炭、骨炭、糖炭、活性炭、炭黑、焦炭、石墨化中间相碳微球(mcmb)、软碳、硬碳以及石墨等;从微观结构上来分,所述的碳可以是碳纳米管、石墨薄片、富勒烯、石墨烯等;从微观形貌上来分,所述的碳可以是碳纤维、碳管、碳球等。优选高电子电导率、结构强度好的碳材料。
粘接剂起将正极活性物质粒子连接固定的作用,包括亲水性聚合物即羧甲基纤维素(cmc)、甲基纤维素(mc)、邻苯二甲酸醋酸纤维素(cap)、羟基丙基甲基纤维素(hpmc)、羟丙基甲基纤维素邻苯二甲酸酯(hpmcp)、聚乙烯醇(pva)、聚氧化乙烯(peo)等以及疏水性聚合物材料如聚四氟乙烯(ptfe)、四氟乙烯全氟烷基乙烯醚共聚物(fep)、聚偏氟乙烯(pvdf)、聚乙烯-四氟乙烯共聚物(etfe)等氟系树脂以及醋酸乙烯共聚物、苯乙烯-丁二烯嵌段共聚物(sbr)、丙烯酸改性sbr树脂(sbr系乳胶)、阿拉伯橡胶等橡胶类等中的至少一种。其中,优选使用ptfe、pvdf等氟系树脂。导电子聚合物作为粘结剂具有非常明显的优势,是用于电化学器件中的粘结剂的发展方向。
通过正极活性材料以及上述例示的导电剂和粘接剂等添加于适当的溶剂中,并使其分散或溶解而进行混合,由此制得浆料。
将所调制的浆料涂布于正极集流体上,并使溶剂挥发干燥后,进行辊压。作为代表性的示例,可采用涂布装置(涂布机),以规定的厚度在集流体表面涂布浆料。对涂布厚度并没有特别的限定,可根据正极和电池的形状或用途适当地设定。涂布后,干燥以除去溶剂,在集流体表面形成规定厚度的正极活性物质层,然后根据需要进行辊压处理,获得目标厚度的正极极片。
(b)负极
本发明中所述的负极极片,由本发明所述的活性物质材料与导电剂、粘合剂、溶剂按一定比例混合制成浆料后均匀涂覆于铜箔上,再经干燥和滚压制成。
上述对电池极片的制造的描述是基于当前常规的大规模制造工艺,但并不排除以后有望实现的等离子喷涂技术、3d打印技术等应用于锂离子二次电池极片的制造。
(c)隔膜
隔膜是电池的关键组成部分之一,位于电池的正、负电极之间,用来隔离正、负电极,避免电池内部短路,同时又保证离子在充放电时能够顺利通过。用于电池的隔膜是一种多孔结构的电子绝缘薄膜,具有高的离子传导性能和良好的机械强度,能够在电解液中长期稳定存在,不发生化学反应。在二次电池中,隔膜性能的优劣直接影响着电池的内阻、容量、充放电电流密度、循环寿命和安全等关键性能。
本发明所述的电池对隔膜的材料、结构没有特殊限定。可以选用聚烯烃类熔融拉伸隔膜,主要为聚丙烯,聚乙烯单层隔膜,或是聚丙烯/聚乙烯/聚丙烯三层复合隔膜;也可以选用以pet(聚对苯二甲酸乙二醇酯)无纺布为隔膜。
(d)电池的形状、结构
本发明中所述的一种长寿命可快充非水电解质电池,由上述的正极、负极、隔膜以及非水系电解液构成,可为圆柱型、方型等各种形状,外包装可以是金属壳也可以是铝塑膜,可以根据实际应用需要设计。
附图说明
图1为本发明实施例1和对比例1中电池的充放电曲线;
图2为本发明实施例1和对比例1中电池的容量循环曲线;
图3为本发明实施例1和对比例1的能量效率循环曲线;
图4为本发明实施例2的充放电曲线;
图5为本发明实施例2的容量循环曲线;
图6为本发明实施例2的能量效率循环曲线。
具体实施方式
以下的具体实施例对本发明进行了详细的描述,然而本发明并不限制于以下实施例。
实施例1:
第一步反应:向反应容器中加入170.0g的三氯硫磷和1.5l溶剂四氢呋喃,并在室温下搅拌均匀,然后加入含191.0g正丁基溴化镁的四氢呋喃溶液继续搅拌18h得到反应液。将反应液倒入500ml10wt%的硫酸溶液中,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到74.0g产物
第二步反应:将70.0g上述产物转移至单口圆底烧瓶中,加入85.9g三氟化锑后开始搅拌,并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至165℃反应1h得到无色液体40.2g。将无色液体再次减压蒸馏纯化得到34.4g无色液体产物
第三步反应:将30.0g上述无色液体产物转移至两口圆底烧瓶中,保持反应容器与空气接通,以便利用空气中的水分,27℃下缓慢搅拌0.5小时之后,在0.5小时内缓慢搅拌加入14.9g三氟乙醇,并继续反应10小时结束,通过减压蒸馏得到34.5g(收率约89%)产物
非水电解液配制:配制碳酸乙烯酯(ec)、碳酸甲乙酯(emc)以及碳酸二甲酯(dmc)的非水混合溶剂,ec:emc:dmc的体积比为30:50:20,然后向其中加入
电池制作:11.5ah层叠铝塑膜软包装电池,正极材料采用lini0.6co0.2mn0.2o2(ncm622),负极材料使用硬碳,常温1c放电时,能量密度约150wh/kg。
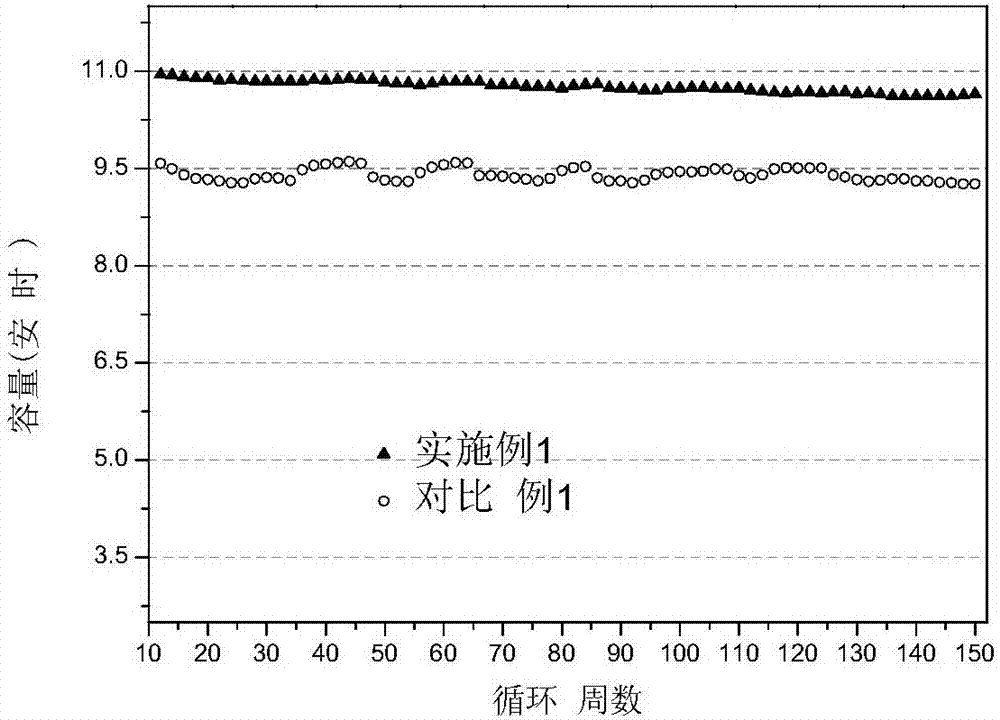
电池性能测试:在45℃环境温度下,将上述软包装电池在2.00~4.20v电压范围内充放电,即恒流(充电倍率为2c)充电至4.20v,接着4.20v恒压充电(倍率为0.1c),然后恒流(放电倍率2c)放电至2.00v。电池充电的恒流比在94~98%间。充放电的效率基本稳定在100%左右。首次放电容量可以达到10.85ah,循环150周可以放电容量为10.64ah,容量保持率98.06%。电芯首次比能量可以达到142.10wh/kg,循环150周可以达到139.50wh/kg。电池的能量转换效率基本稳定在95%左右。能量转换效率为放电能量与充电能量的比值。具体测试结果请见图1、图2和图3。
实施例2:
第一步和第二步反应同实施例1,不同的是,第三步反应为将36g无色液体产物
非水电解液配制:配制碳酸乙烯酯(ec)、碳酸甲乙酯(emc)以及碳酸二甲酯(dmc)的非水混合溶剂,ec:emc:dmc的体积比为30:50:20,然后向其中加入
电池制作:10ah层叠铝塑膜软包装电池,正极材料采用lini0.6co0.2mn0.2o2(ncm622),负极材料使用硬碳,电池能量密度较高,常温1c放电时,能量密度约130wh/kg。
电池性能测试性能:在45℃环境温度下,将上述软包装电池在2.00~4.20v电压范围内充放电,即恒流(充电倍率为2c)充电至4.20v,接着4.20v恒压充电(倍率为0.1c),然后恒流(放电倍率2c)放电至2.00v。电池充电的恒流比在94~98%间。充放电的效率基本稳定在100%左右。首次放电容量可以达到10.98ah,循环150周可以放电容量为10.88ah,容量保持率99.09%。电芯首次比能量可以达到138.20wh/kg,循环150周可以达到135.20wh/kg。电池的能量转换效率在94~95%之间。能量转换效率为放电能量与充电能量的比值。具体测试结果请见图4、图5和图6。
实施例3:
第一步反应:向反应容器中加入170.0g三氯硫磷和1.5l溶剂四氢呋喃,并在室温(25℃)下搅拌均匀。然后加入399.1g乙基溴化镁的四氢呋喃溶液(399.1g指乙基溴化镁的质量)继续在25℃搅拌至反应48小时得到反应液。将反应液倒入500ml10wt%的硫酸溶液,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到69.0g产物
第二步反应:将69.0g上述产物转移至单口圆底烧瓶中,加入108.0g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至144℃反应2小时得到无色液体57.0g。将无色液体再次减压蒸馏纯化得到52.5g无色液体产物
第三步反应:将50.0g上述无色液体产物转移至两口圆底烧瓶中,保持反应容器与空气接通,以便利用空气中的水分,0℃下缓慢搅拌0.5小时之后,在0.5小时内缓慢加入25.2g正丁醇,并继续0℃下反应48小时结束,通过减压蒸馏得到52.4g(收率约86%)产物
实施例4:
第一步反应:向反应容器中加入500.0g三氯硫磷和1.6l溶剂四氢呋喃,并在室温下搅拌均匀。然后加入779.2g正丁二基溴化镁的四氢呋喃溶液(779.2g指丁二基溴化镁的质量)继续在60℃下搅拌至反应2小时得到反应液,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到182.0g(收率约52%)产物
第二步反应:将180.0g上述产物转移至单口圆底烧瓶中,加入328.3g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至167℃反应3.6小时得到无色液体163.6g。将无色液体再次减压蒸馏纯化得到154.6g无色液体产物
第三步反应:将300.0g上述无色液体产物转移至两口圆底烧瓶中,27℃下在30min内加入19.0g去离子水并继续搅拌0.5小时之后,0.7小时内缓慢搅拌加入104.0g乙醇,并在60℃继续反应6小时结束,通过减压蒸馏得到281.0g(收率约91%)产物
实施例5:
第一步反应:向反应容器中加入500.0g三氯硫磷和3.0l溶剂四氢呋喃,并在室温下搅拌均匀。然后加入1.6kg苯基溴化镁的四氢呋喃溶液(1.6kg指苯基溴化镁的质量)继续在45℃下搅拌至反应36小时得到反应液。将反应液倒入1.5l10wt%的硫酸溶液,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到274.4g产物
第二步反应:将160.0g上述产物转移至单口圆底烧瓶中,加入291.8g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至167℃反应3.6小时得到无色液体132.9g。将无色液体再次减压蒸馏纯化得到126.7g无色液体产物
第三步反应:将120.0g上述无色液体产物转移至两口圆底烧瓶中,0.7小时内缓慢搅拌加入34.1g乙醇,并在60℃下继续反应8小时结束,通过减压蒸馏得到111.0g(收率约91%)产物
实施例6:
第一步反应:向反应容器中加入500.0g三氯硫磷和3.0l溶剂四氢呋喃,并在室温下搅拌均匀。然后加入1.6kg烯丙基溴化镁的四氢呋喃溶液(1.6kg指烯丙基溴化镁的质量)继续在45℃下搅拌至反应36h得到反应液。将反应液倒入1.5l10wt%的硫酸溶液,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到274.4产物
第二步反应:将160.0g该产物转移至单口圆底烧瓶中,加入291.8g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至167℃反应3.6小时得到无色液体137.8g。将无色液体再次减压蒸馏纯化得到126.7g无色液体产物
第三步反应:将120.0g该无色液体产物转移至两口圆底烧瓶中,0.7小时内缓慢搅拌加入41.6g乙醇,并在60℃下继续反应8小时结束,通过减压蒸馏得到111.0g(收率约91%)产物
实施例7:
第一步反应:向反应容器中加入500.0g三氯硫磷和3.0l溶剂四氢呋喃,并在室温下搅拌均匀。然后加入1.6kg
第二步反应:将160.0g上述产物转移至单口圆底烧瓶中,加入291.8g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至167℃反应3.6h得到无色液体133.3g。将无色液体再次减压蒸馏纯化得到126.7g无色液体产物
第三步反应:将100g上述第二步产物
实施例8:
第一步反应:向反应容器中加入500.0g三氯硫磷和3.0l溶剂四氢呋喃,并在室温下搅拌均匀。然后加入1.6kg对氟苯基溴化镁的四氢呋喃溶液(1.6kg指苯二基溴化镁的质量)继续在45℃下搅拌至反应36小时得到反应液。将反应液倒入事先准备的1.5l10wt%的硫酸溶液,用乙醚萃取并分离得到有机层,用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到274.4g产物
第二步反应:将160.0g上述产物转移至单口圆底烧瓶中,加入291.8g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至167℃反应3.6小时得到无色液体130.7g。将无色液体再次减压蒸馏纯化得到126.7g无色液体产物
第三步反应:将600g上述第二步产物
实施例9:
第一步反应:向反应容器中加入170.0g三氯硫磷和1.5l溶剂四氢呋喃,并在-30℃下搅拌均匀。然后加入399.1g乙基溴化镁的四氢呋喃溶液(399.1g指乙基溴化镁的质量)继续在-30℃搅拌至反应48小时得到反应液。将反应液倒入500ml10wt%的硫酸溶液,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到53.1g产物
第二步反应:将50.0g上述产物转移至单口圆底烧瓶中,加入78.3g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至25℃反应10小时得到无色液体27.8g。将无色液体再次减压蒸馏纯化得到22.4g无色液体产物
第三步反应:将438.0g上述无色液体产物转移至两口圆底烧瓶中,27℃下在30min内加入48.6g去离子水并继续搅拌0.5小时之后,,在0.5小时内缓慢加入222.0g正丁醇,并继续0℃下反应48小时结束,通过减压蒸馏得到422.7g(收率约79%)产物
实施例10:
同实施例2,不同的是步骤(3)的反应温度为80℃,反应时间为2小时,得到产物
实施例11:
第一步反应:向反应容器中加入250.0g三氯硫磷和1.6l溶剂四氢呋喃,并在室温下搅拌均匀。然后加入389.6g正丁二基溴化镁的四氢呋喃溶液(389.6g指丁二基溴化镁的质量)继续在60℃下搅拌至反应2小时得到反应液。将反应液倒入540ml10wt%的硫酸溶液,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到91.0g(收率约52%)产物
第二步反应:将80.0g上述产物转移至单口圆底烧瓶中,加入145.9g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至200℃反应0.2小时得到无色液体49.5g。将无色液体再次减压蒸馏纯化得到43.8g无色液体产物
第三步反应:将30.0g上述无色液体产物转移至两口圆底烧瓶中,27℃下在30min内加入2.5g去离子水并继续搅拌0.5小时之后,0.7小时内缓慢搅拌加入10.4g乙醇,并在60℃继续反应6小时结束,通过减压蒸馏得到27.4g(收率约89%)产物
对比例1:
非水电解液配制:配制碳酸乙烯酯(ec)、碳酸甲乙酯(emc)以及碳酸二甲酯(dmc)的非水混合溶剂,ec:emc:dmc的体积比为30:50:20,然后向其中加入成膜添加剂碳酸亚乙烯酯(vc,含量为非水电解液体重质量的2.0wt%)。缓慢加入电解质盐lipf6并加以冷却,形成浓度为12.5wt%的非水电解液。
电池制作:11.5ah层叠铝塑膜软包装电池,正极材料采用lini0.6co0.2mn0.2o2(ncm622),负极材料使用硬碳,常温1c放电时,能量密度约130wh/kg。
电池性能测试:在45℃环境温度下,将上述软包装电池在2.00v~4.20v电压范围内充放电,即恒流(充电倍率为2c)充电至4.20v,接着4.20v恒压充电(倍率为0.1c),然后恒流(放电倍率2c)放电至2.00v。电池充电的恒流比在循环初期在93.00%左右,后衰减至86.00%左右。充放电的效率基本稳定在100%左右。首次放电容量为9.57ah,循环150周可以放电容量为9.26ah,容量保持率为96.76%。电芯首次比能量可以达到115.80wh/kg,循环150周为112.30wh/kg。具体测试结果请见图1、图2和图3。
对比例2:
同实施例8,不同的是第三步反应中,将600g第二步产物
对比例3:
同实施例8,不同的是第三步反应中加入去离子水的量为58.05g,得到反应53.3g(收率约53%)产物
对比例4:
第一步反应:向反应容器中加入250.0g三氯硫磷和1.6l溶剂四氢呋喃,并在室温下搅拌均匀。然后加入389.6g正丁二基溴化镁的四氢呋喃溶液(389.6g指丁二基溴化镁的质量)继续在70℃下搅拌至反应1小时得到反应液。将反应液倒入540ml10wt%的硫酸溶液,用乙醚萃取并分离得到有机层,有机层用无水硫酸镁干燥后减压蒸馏除去溶剂得到固体混合物。将固体混合物用甲苯和乙醇重结晶得到白色固体,真空干燥后得到36.8g(收率约21%)产物
第二步反应:将35.0g上述产物转移至单口圆底烧瓶中,加入63.8g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至200℃反应0.2小时得到无色液体27.2g。将无色液体再次减压蒸馏纯化得到21.6g无色液体产物
第三步反应:将20.0g上述无色液体产物转移至两口圆底烧瓶中,0.5小时内缓慢搅拌加入6.9g乙醇,并在60℃继续反应6小时结束,通过减压蒸馏得到20.3g(收率约97.1%)产物
对比例5:
第一步反应:同实施例9,得到产物
第二步反应:将50.0g上述产物转移至单口圆底烧瓶中,加入78.3g三氟化锑后开始搅拌。并且将单口圆底烧瓶连接减压蒸馏装置,减压下加热至10℃反应12小时得到无色液体9.6g。将无色液体再次减压蒸馏纯化得到7.1g无色液体产物
第三步反应:将7.0g上述无色液体产物转移至两口圆底烧瓶中,在0.5小时内缓慢加入3.5g正丁醇,并继续0℃下反应48小时结束,通过减压蒸馏得到5.9g(收率约69%)产物
本发明所有实施例及对比例的收率计算方法为:(实际得到的产物质量/理论产物质量)×100%。
从实施例1和对比例1中可知:对比例1的容量保持率不仅比实施例1低且对比例1的放电容量随测试环境的变化波动较大;此外,对比例1的容量发挥较低,仅为实施例1的88.20%。最后,对比例1的电池的能量转换效率在88%-91%间浮动变化,远低于实施例1。
从实施例8和对比例2中可知:在第三步反应中其他反应条件相同时,实施例8(先加水和第二中间体,再加醇的反应物加入顺序)得到的产物收率明显高于对比例2(先加醇和第二中间体,再加水的反应物加入顺序)。
从实施例8和对比例3可知:第三步反应中,当加入水的量与加入第二中间体的量的摩尔比大于1.5:1时,次膦酸酯的产率会明显降低。
从实施例8和对比例4可知:第一步反应中反应时间为1小时,反应温度为70℃,即步骤(1)中的反应时间和反应温度都不在本发明要求的范围内时,得到的第一步产物产率非常低。
从实施例9和对比例5可知:当第二步反应中反应温度为10℃,反应时间为12小时,即步骤(2)中的反应时间和反应温度都不在本发明要求的范围内时,得到的第二步产物产率非常低。
- 还没有人留言评论。精彩留言会获得点赞!