一种手性三环酮胺化合物及其合成方法和应用与流程
本发明属于有机化合物合成工艺应用技术领域,具体涉及一种(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物高效便捷合成方法。
背景技术:
Vinblastine和Vincristine是广泛用于抗癌的药物,它们属于双吲哚生物碱。另外一类单萜吲哚类生物碱—白坚木属生物碱也有很好的生物活性,例如Tabersonine对人体癌细胞系SK-BR-3的抑制作用优于顺铂(Cisplatin),Jerantinine-E对人体KB细胞有更强的细胞毒性等等。但是他们在自然界中丰度低,提取较困难,这增加了对其生理学和药理学活性方面的研究难度,阻碍了对该类化合物的进一步研究和应用。
过去的多数路线只能合成单一的非手性天然产物,并不能实现结构多样性的手性合成。“(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物”是合成白坚木属生物碱的高级中间体,利用后期Fischer吲哚化可以对吲哚环进行结构改造与修饰,进而可以对其进一步的生理学和药理学活性方面的研究。因此发明一种“(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物”的不对称合成方法非常重要。
在之前“(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物”的合成中主要是非手性的合成,而手性合成只有3例报道;它们有路线冗长,操作不便,无法大量合成等不足之处。本发明正好解决了这些问题,实现路线短,操作简单,克级规模合成该化合物。
技术实现要素:
本发明提供了一种(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物的合成方法。所述(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物为手性三环酮胺化合物。所述方法能够从廉价易得的原料出发,为后续实现白坚木属生物碱的规模化生产及构效关系研究提供良好的基础。
本发明提供的一种新的制备式(1)(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物的方法,该方法通过3-乙基-5-溴-2-吡喃酮(由DL-苹果酸经3步反应得到)和(S)-2,3-二氢-1H-吡咯-1,2-二甲酸二叔丁酯(由L-焦谷氨酸经3步反应得到)经[4+2]环加成,脱羧,Pd/C催化氢化环化等5个步骤的反应得到。反应路线如路线(1)所示;
具体包括以下步骤:
步骤(a):将式(2)化合物和式(3)化合物溶于有机溶剂中,进行加成反应得到式(4)化合物。
步骤(b):将式(4)化合物溶于三氟乙酸中,进行第一取代反应,得到褐色油状液体5-溴-7-乙基-8-氧代-2,3,3a,4,7,7a-六氢-1H-4,7-(环氧甲烷基)吲哚-2-羧酸;将此油状液体5-溴-7-乙基-8-氧代-2,3,3a,4,7,7a-六氢-1H-4,7-(环氧甲烷基)吲哚-2-羧酸溶于混合溶剂中,加入碳酸氢钠固体和氯甲酸苄酯,进行第二取代反应得到式(5)化合物。
步骤(c):将式(5)化合物、醋酸碘苯和碘溶于有机溶剂中,进行消去反应,得到亚胺阳离子中间体化合物;然后加入三乙基硅氢,进行加成反应得到式(6)化合物。
步骤(d):将式(6)化合物溶于有机溶剂中,加入二异丁基氢化铝,进行第一还原反应得到无色油状液体5-溴-7-乙基-8-羟基-2,3,3a,4,7,7a-六氢-1H-4,7-(环氧甲烷基)吲哚-1-羧酸苄基酯。将此无色油状液体5-溴-7-乙基-8-羟基-2,3,3a,4,7,7a-六氢-1H-4,7-(环氧甲烷基)吲哚-1-羧酸苄基酯溶于有机溶剂中,加入Wittig试剂Ph3P=CHCO2Me,进行第一取代反应得到白色固体(3aR,4R,7R,7aS)-5-溴-7-乙基-4-羟基-7-(3-甲氧基-3-氧代丙-1-烯-1-基)-2,3,3a,4,7,7a-六氢-1H-吲哚-1-羧酸苄基酯;将上述白色固体(3aR,4R,7R,7aS)-5-溴-7-乙基-4-羟基-7-(3-甲氧基-3-氧代丙-1-烯-1-基)-2,3,3a,4,7,7a-六氢-1H-吲哚-1-羧酸苄基酯溶于有机溶剂中,依次加入三乙胺和Pd/C,同时进行第二还原反应和第二取代反应得到式(7)化合物。
步骤(e):将式(7)化合物溶于第一有机溶剂中,-78℃~-50℃下加入四氢铝锂,升至25℃~70℃回流进行还原反应,得到淡黄色油状液体(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9-醇;将此淡黄色油状液体溶于第二有机溶剂中,-10℃~0℃下加入戴斯-马丁氧化剂(优选地,0℃下加入戴斯-马丁氧化剂),升温进行氧化反应,得到式(1)(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物。
其中,步骤(a)式(4)化合物的合成中:
所述反应的类型为加成反应。
所述加成反应的温度为90℃~110℃;优选地为,90℃、100℃、110℃;进一步优选地为,110℃。
所述加成反应的时间为24小时~60小时;优选地为,24小时、48小时、60小时;进一步优选地为,60小时。
所述加成反应中,所述有机溶剂选自:甲苯、苯、二氯甲烷;优选地为,甲苯。
所述加成反应中,式(2)化合物和式(3)化合物的摩尔比为1:1~2;优选地为,1:1、1:1.2、1:1.5、1:2;进一步优选地,最佳摩尔比为1:1.5。
其中,步骤(b)式(5)化合物的合成中:
其中,溶于三氟乙酸中反应的类型为第一取代反应。
所述第一取代反应的温度为室温(25℃)。
所述第一取代反应的时间为3小时~10小时;优选地,为5小时。
所述第一取代反应中,三氟乙酸的作用是脱除Boc和t‐Bu,脱除Boc为步骤(d)式(7)化合物的合成做准备,脱除t‐Bu是为步骤(c)式(6)化合物的合成做准备。
所述第一取代反应中,式(4)化合物和三氟乙酸的摩尔比为1:5~15;优选地为,1:5、1:10、1:15;进一步优选地,最佳摩尔比为1:10。
溶于混合溶剂中进行的反应类型为第二取代反应。
所述第二取代反应的温度为室温(25℃)。
所述第二取代反应的时间为15小时~20小时;优选地,为15小时。
所述第二取代反应中,所述混合溶剂选自:四氢呋喃和水的混合溶剂、丙酮和水的混合溶剂;优选地为,四氢呋喃和水的混合溶剂,四氢呋喃和水的最佳体积比为2:1。
所述第二取代反应中,氯甲酸苄酯和碳酸氢钠的摩尔比为1:1~4;优选地,为1:1、1:2、1:4;进一步优选地,最佳摩尔比为1:4。
其中,步骤(c)式(6)化合物的合成中:
溶于有机溶剂后,进行的类型为消去反应。
所述消去反应的温度为25℃~40℃;优选地,为25℃、40℃;进一步优选地,为室温(25℃)。
所述消去反应的时间为0.5~2小时;优选地,为0.5小时、1小时、1.5小时、2小时;进一步优选地为,2小时。
所述消去反应中,所述有机溶剂选自:二氯甲烷、1,2-二氯乙烷;优选地,为二氯甲烷。
优选地,所述消去反应在8W~200W白炽灯的照射下进行;优选地,为8W、25W、50W、100W、150W、200W;进一步优选地,所述消去反应在200W白炽灯的照射下进行。
加入三乙基硅氢后,进行的反应类型为加成反应。
所述加成反应的温度为0~40℃;优选地,为0℃、25℃、40℃;进一步优选地,为室温(25℃)。
所述加成反应的时间为10分钟~1小时;优选地,为10分钟、30分钟、1小时;进一步优选地为,30分钟。
所述加成反应中,所述有机溶剂选自:二氯甲烷、1,2-二氯乙烷;优选地为,二氯甲烷。
式(5)化合物,醋酸碘苯,碘和三乙基硅氢的摩尔比为1:2:0.1~0.5:5;优选地,为1:2:0.1:5、1:2:0.2:5、1:2:0.3:5、1:2:0.4:5、1:2:0.5:5;进一步优选地,最佳摩尔比为1:2:0.2:5。
其中,步骤(d)式(7)化合物的合成中
所述合成无色油状液体的反应的类型为第一还原反应。
所述第一还原反应的温度为–60℃~–40℃;优选地,为–60℃、–50℃、–40℃;进一步优选地,为–40℃。
所述第一还原反应的时间为1小时~5小时;优选地,为1小时、2小时、3小时、4小时、5小时;进一步优选地为,3小时。
所述第一还原反应中,有机溶剂选自:甲苯、四氢呋喃;优选地为,甲苯。
所述第一还原反应中,式(6)化合物与二异丁基氢化铝的摩尔比为1:1.2~2;优选地,为1:1.2、1:1.5、1:2;进一步优选地,最佳摩尔比为1:1.5。
优选地,将式(6)化合物溶于甲苯中,加入二异丁基氢化铝,反应升至–40℃搅拌3小时,得到无色油状液体。将此油状液体溶于甲苯中,加入Ph3P=CHCO2Me,反应升至110℃回流4小时。减压除去甲苯后得到白色固体。将上述白色固体溶于甲醇中,依次加入三乙胺和Pd/C。搅拌8小时,得到式(7)化合物。其中,优选地,所述二异丁基氢化铝在–78℃加入;所述二异丁基氢化铝在–78℃通过自动进样泵历时30分钟加入。
所述合成白色固体的反应类型为取代反应。
所述第一取代反应的温度为25℃~110℃;优选地,为25℃、110℃;进一步优选地为,110℃。
所述第一取代反应的时间为1小时~4小时;优选地,为1小时、2小时、4小时;进一步优选地为,4小时。
所述第一取代反应中,有机溶剂选自:甲苯、四氢呋喃;优选地为,甲苯。
所述第一取代反应中,无色油状液体与Wittig试剂Ph3P=CHCO2Me的摩尔比为1:1.5~2;优选地,为1:1.5、1:2;进一步优选地,最佳摩尔比为1:1.5。
所述第一取代反应中,Wittig试剂Ph3P=CHCO2Me的作用为与半缩醛反应得到α,β-不饱和酯,为式(7)化合物的合成做准备。
所述合成式(7)化合物的反应的类型为还原反应和取代反应。
所述第二还原反应和第二取代反应的温度为25℃。
所述第二还原反应和第二取代反应的时间为5小时~10小时;优选地,为5小时、8小时、10小时;进一步优选地为,8小时。
所述第二还原反应和第二取代反应中,有机溶剂选自:甲醇、乙酸乙酯、乙醇;优选地为,甲醇。
所述第二还原反应和第二取代反应中,白色固体(3aR,4R,7R,7aS)-5-溴-7-乙基-4-羟基-7-(3-甲氧基-3-氧代丙-1-烯-1-基)-2,3,3a,4,7,7a-六氢-1H-吲哚-1-羧酸苄基酯、三乙胺和钯/碳的摩尔比为1:5:0.2~0.5;优选地,为1:5:0.2、1:5:0.5;进一步优选地,最佳摩尔比为1:5:0.2。
其中,步骤(e)式(1)化合物的合成中:
溶于第一有机溶剂后,进行的类型为还原反应。
所述还原反应的温度为25℃~70℃;优选地,为25℃、70℃;进一步优选地为,70℃。
所述还原反应的时间为5小时~15小时;优选地,为5小时、8小时、10小时、15小时;进一步优选地为,10小时。
所述还原反应中,所述第一有机溶剂选自:四氢呋喃、甲苯;优选地,为四氢呋喃。
所述还原反应中,式(7)化合物与四氢铝锂的摩尔比为1:5~10;优选地,为1:5、1:10;进一步优选地,最佳摩尔比为1:5。
溶于第二有机溶剂后进行的类型为氧化反应。
所述氧化反应的温度为0℃~25℃;优选地,为0℃、25℃;优选地,为室温(25℃)。
所述氧化反应的时间为30分钟~2小时;优选地,为30分钟、1小时、2小时;进一步优选地为,1小时。
所述氧化反应中,所述第二有机溶剂选自:二氯甲烷、1,2-二氯乙烷;优选地为,二氯甲烷。
所述氧化反应中,淡黄色油状液体和戴斯-马丁氧化剂的摩尔比为1:1.5~5;优选地,为1:1.5、1:2、1:3、1:4、1:5、最佳摩尔比为1:4。
本发明还提出了一类生物碱化合物,其结构如式(4)、式(5)、式(6)、式(7)所示:
在一个具体的实施方式中,合成式(1)化合物的具体步骤包括:
步骤(a)将式(2)化合物和式(3)化合物溶于甲苯中,110℃回流反应60小时。减压除去甲苯后直接柱层析得到白色固体式(4)化合物。
步骤(b)将式(4)化合物溶于三氟乙酸中。室温反应5小时。减压除去三氟乙酸得到褐色油状液体;将此油状液体溶于四氢呋喃和水的混合溶剂中,依次加入碳酸氢钠固体和氯甲酸苄酯,室温下反应15小时。反应液用4N HCl调PH至2-3,减压除掉四氢呋喃。水相用乙酸乙酯萃取5次,无水硫酸钠干燥,过滤,减压除掉溶剂,柱层析分离得到白色固体式(5)化合物。
步骤(c)将式(5)化合物溶于二氯甲烷中,依次加入醋酸碘苯和碘,200W白炽灯的照射下室温反应2小时。然后撤掉白炽灯,加入三乙基硅氢,室温反应30分钟。减压除去甲苯后直接柱层析得到无色油状液体式(6)化合物。
步骤(d)将式(6)化合物溶于甲苯中,–78℃下通过自动进样泵历时30分钟加入二异丁基氢化铝,反应升至–40℃搅拌3小时。饱和酒石酸钾钠溶液淬灭反应,室温下搅拌至反应液澄清。减压除掉甲苯,水相用乙酸乙酯萃取,无水硫酸钠干燥,过滤,减压除掉溶剂得到无色油状液体。将此油状液体溶于甲苯中,加入Ph3P=CHCO2Me,反应升至110℃回流4小时。减压除去甲苯后得到白色固体。将上述白色固体溶于甲醇中,依次加入三乙胺和Pd/C。反应瓶用氢气置换后在氢气氛围(氢气球)下搅拌8小时,过滤,乙酸乙酯洗涤滤饼。减压除掉溶剂,得到淡黄色固体用水从新溶解,乙酸乙酯萃取,无水硫酸钠干燥,过滤,减压除掉溶剂,柱层析分离得到白色固体式(7)化合物。
步骤(e)将式(7)化合物溶于四氢呋喃中,–78℃下缓慢加入四氢铝锂,反应升至70℃回流10小时。反应完毕,冷却至0℃,反应依次用水,15%氢氧化钠水溶液和水淬灭,过滤,乙酸乙酯洗涤滤饼,减压除去甲苯后直接柱层析得到淡黄色油状液体。将此油状液体溶于二氯甲烷中,0℃下加入戴斯-马丁氧化剂,反应升至室温搅拌1小时。反应完毕,依次加入饱和硫代硫酸钠溶液和饱和碳酸氢钠溶液,体系用二氯甲烷萃取无水硫酸钠干燥,过滤,减压除掉溶剂,柱层析分离得到黄色油状式(1)化合物。
在一个具体的实施方式中,合成式(1)化合物的反应路线如路线(1’)所示;
本发明还提出了所述(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉-9(2H)-酮化合物在合成白坚木属生物碱中的应用,实现了白坚木属生物碱家族中(+)-Dehydroaspidospermidine和(+)-Aspidospermidine的合成。
反应如路线(2)所示:
具体包括以下步骤:
步骤i):将式(1)化合物、苯肼溶于第一有机溶剂中,进行取代反应,得到苯腙中间体;将此苯腙中间体溶于第二有机溶剂中,得到式(8)化合物(+)-Dehydroaspidospermidine。
步骤ii):将式(8)化合物(+)-Dehydroaspidospermidine溶于有机溶剂中,加入四氢铝锂,进行还原反应,得到式(9)化合物(+)-Aspidospermidine。
其中,步骤i)式(8)化合物(+)-Dehydroaspidospermidine的合成中:
溶于第一有机溶剂后,进行的类型为取代反应。
所述取代反应的温度为25℃~80℃;优选地,为25℃、50℃、80℃;进一步优选地为,80℃。
所述取代反应的时间为1小时~4小时;优选地为,1小时、2小时、3小时、4小时;进一步优选地为,3小时。
所述取代反应中,所述第一有机溶剂选自:苯、甲苯;优选地为,苯。
所述取代反应中,式(1)化合物与苯肼的摩尔比为1:1.2~2;优选地,为1:1.2、1:1.5、1:2;进一步优选地,最佳摩尔比为1:1.5。
溶于第二有机溶剂进行的反应类型为重排反应。
所述重排反应的温度为90℃~120℃;优选地为,90℃、100℃、110℃、120℃;进一步优选地为,120℃。
所述重排反应的时间为1小时~5小时;优选地为,1小时、2小时、3小时、4小时、5小时;进一步优选地为,4小时。
所述重排反应中,所述第二有机溶剂选自:甲苯和乙酸、乙酸;优选地为,乙酸。
其中,步骤ii)式(9)化合物(+)-Aspidospermidine的合成中:
溶于有机溶剂后,进行的类型为还原反应。
所述还原反应的温度为25℃~70℃;优选地,为25℃、50℃、70℃;进一步优选地为,70℃。
所述还原反应的时间为5小时~15小时;优选地,为5小时、10小时、13小时、15小时;进一步优选地,为13小时。
所述还原反应中,所述有机溶剂选自:四氢呋喃、甲苯;优选地,为四氢呋喃。
所述还原反应中,式(1)化合物与苯肼的摩尔比为1:5~10;优选地,1:5、1:10;进一步优选地,最佳摩尔比为1:10。
在一个具体的实施方式中,合成式(9)化合物的反应路线如路线(2’)所示:
本发明的有益效果在于,所述合成(31S,6aR,9aR)-6a-乙基十氢-1H-吡咯并[3,2,1-ij]喹啉
-9(2H)-酮化合物的方法从廉价易得的原料出发,合成路线短,无需高功率反应器、损耗低,条件简单,操作方便,收率为10%~15%,制备的式(1)化合物为光学纯化合物。为后续实现该类天然产物的规模化生产及构效关系研究提供良好的技术支持。
附图说明
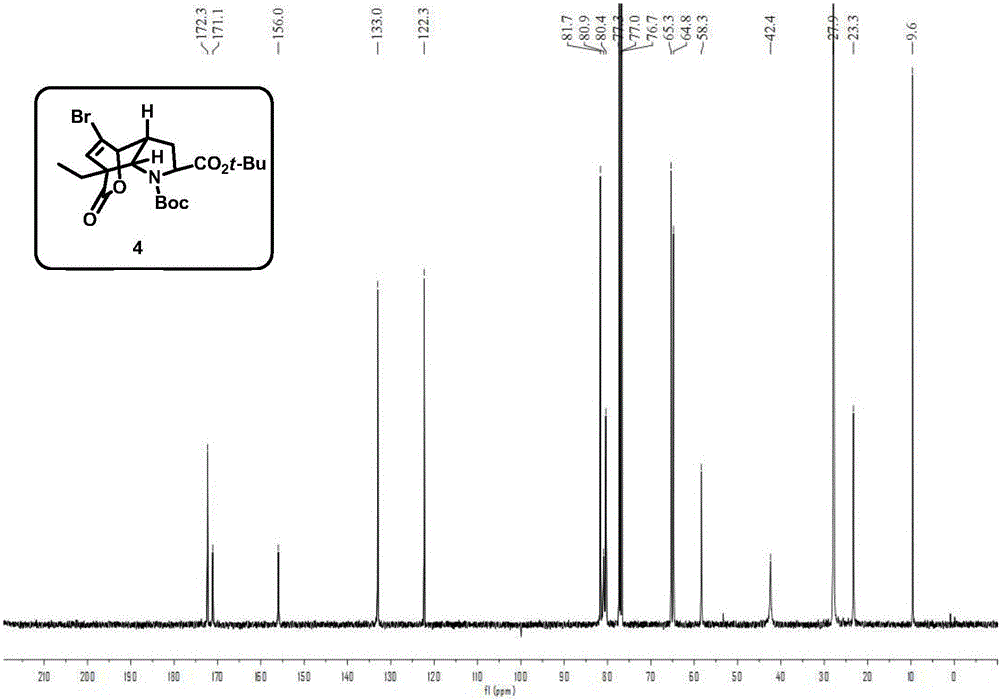
图1为式(4)化合物的氢谱。
图2为式(4)化合物的碳谱。
图3为式(5)化合物的氢谱。
图4为式(5)化合物的碳谱。
图5为式(6)化合物的氢谱。
图6为式(6)化合物的碳谱。
图7为式(7)化合物的氢谱。
图8为式(7)化合物的碳谱。
图9为式(1)化合物的氢谱。
图10为式(1)化合物的碳谱。
图11为式(8)化合物(+)-Dehydoaspidospermidine的氢谱。
图12为式(8)化合物(+)-Dehydroaspidospermidine的碳谱。
图13为式(9)化合物(+)-Aspidospermidine的氢谱。
图14为式(9)化合物(+)-Aspidospermidine的碳谱。
具体实施方式
结合以下具体实施例和附图,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
实施例1
式(4)化合物的合成
将式(2)化合物(8.36g,41.18mmol,1.0equiv.)和式(化合物3)(16.64g,61.76mmol,1.5equiv.)溶于甲苯(206mL)中,110℃回流反应60小时。减压除去甲苯后直接柱层析(石油醚:乙酸乙酯=20:1)得到白色固体式(4)(10.40g,54%yield),另外回收原料式(2)化合物(2.56g).
式(4)化合物的检测数据如下:1H NMR(400MHz,CDCl3)δ6.39(s,1H),4.74(s,1H),4.30(t,J=10.8Hz,2H),3.08(d,J=7.2Hz,1H),2.40(dd,J=22.9,9.7Hz,1H),2.00–1.85(m,3H),1.44(s,9H),1.39(s,9H),1.02(t,J=7.2Hz,3H).13C NMR(100MHz,CDCl3)δ172.3,171.1,156.0,133.0,122.3,81.7,80.9,80.4,65.3,64.8,58.3,42.4,27.9,23.3,9.6.IR(neat,cm-1)2925,1763,1736,1460,1227,1152,1044.HRMS(ESI)[M+NH4]+Calcd for C21H34BrN2O6489.1595,Found 489.1595.[α]D25-115.4(c 1.0,CHCl3)
实施例2
式(5)化合物的合成
将式(4)化合物(10.40g,22.02mmol,1.00equiv.)溶于三氟乙酸(20mL)中。室温反应5小时。减压除去三氟乙酸得到褐色油状液体(无需柱层析分离纯化);
将此油状液体溶于四氢呋喃(140mL)和水(70mL)的混合溶剂中,依次加入碳酸氢钠固体(37.34g,440.33mmol,20.00equiv.)和氯甲酸苄酯(18.77g,110.08mmol,5.00equiv.),室温下反应15小时。反应液用4N HCl调PH至2-3,减压除掉四氢呋喃。水相用乙酸乙酯萃取5次,无水硫酸钠干燥,过滤,减压除掉溶剂,柱层析分离(石油醚:乙酸乙酯=2:1)得到白色固体式(5)化合物(6.48g,66%yield for two steps)。
式(5)化合物的检测数据如下:1H NMR(400MHz,CDCl3)δ7.22(d,J=18.3Hz,4H),6.34(s,1H),5.94(s,1H),4.99(s,2H),4.73(s,1H),4.49(d,J=7.4Hz,1H),4.30(d,J=9.4Hz,1H),3.07(dd,J=17.5,8.5Hz,1H),2.43(dt,J=13.8,9.1Hz,1H),2.14–1.98(m,1H),1.90(s,1H),0.96(s,3H).13C NMR(100MHz,CDCl3)δ177.3,170.9,156.4,135.3,132.7,128.5,128.3,128.1,122.6,80.6,68.0,64.7,63.9,58.0,42.8,28.4,23.2,9.5.IR(neat,cm-1)2925,1753,1394,1257,1185,1087,1009.HRMS(ESI)[M+NH4]+Calcd for C20H24BrN2O6467.0812,Found467.0812.[α]D25-109.9(c 0.5,CHCl3).
实施例3
式(6)化合物的合成
将式(5)化合物(6.48g,14.43mmol,1.00equiv.)溶于新蒸的二氯甲烷(140mL)中,依次加入醋酸碘苯(9.30g,28.86mmol,2.00equiv.)和碘(0.73g,2.89mmol,0.20equiv.),200W白炽灯的照射下室温反应2小时。然后撤掉白炽灯,加入三乙基硅氢(8.37g,72.15mmol,5.00equiv.),室温反应30分钟。减压除去甲苯后直接柱层析(石油醚:乙酸乙酯=20:1)得到无色油状液体式(6化合物)(5.38g,92%yield)。
式(6)化合物的检测数据如下:1H NMR(400MHz,CDCl3)δ7.38–7.29(m,5H),6.38(s,1H),5.10(dd,J=29.0,12.2Hz,2H),4.94(t,J=1.9Hz,1H),4.21(d,J=211.6Hz,2H),3.23(td,J=11.6,7.3Hz,1H),2.93(t,J=9.6Hz,1H),2.16–1.87(m,3H),1.81(s,1H),1.02(m,3H).13C NMR(100MHz,CDCl3)δ171.8,155.6,136.0,131.6,128.4,128.1,127.8,122.6,83.8,67.5,61.5,56.2,47.0,43.1,28.2,22.2,8.8.IR(neat,cm-1)2926,1759,1702,1407,1259,1196,1095,1011.HRMS(ESI)[M+NH4]+Calcd for C19H24BrN2O4423.0914,Found 423.0912.[α]D25-98.2(c 1.0,CHCl3).
实施例4
式(7)化合物的合成
将式(6)化合物(5.38g,11.98mmol,1.00equiv.)溶于甲苯(120mL)中,–78℃下通过自动进样泵历时30分钟加入二异丁基氢化铝(12mL,17.98mmol,1.50equiv.,1.5M in toluene)反应升至–40℃搅拌3小时。饱和酒石酸钾钠溶液(20mL)淬灭反应,室温下搅拌至反应液澄清。减压除掉甲苯,水相用乙酸乙酯(3x 50mL)萃取,无水硫酸钠干燥,过滤,减压除掉溶剂得到无色油状液体(无需柱层析分离纯化)。
将此油状液体溶于甲苯(120mL)中,加入Ph3P=CHCO2Me(6.01g,17.98mmol,1.50equiv.),反应升至110℃回流4小时。减压除去甲苯后直接柱层析(石油醚:乙酸乙酯=3:1)得到白色固体(4.12g,74%yield for two steps)。
将上述白色固体溶于甲醇(887mL)中,依次加入三乙胺(4.48g,44.35mmol,5.00equiv.)和Pd/C(10wt%,1.88g,1.77mmol,0.20equiv.)。反应瓶用氢气置换后在氢气氛围(氢气球)下搅拌8小时,过滤,乙酸乙酯(3x 50mL)洗涤滤饼。减压除掉溶剂,得到淡黄色固体用水(10mL)从新溶解,乙酸乙酯(5×50mL)萃取,无水硫酸钠干燥,过滤,减压除掉溶剂,柱层析分离(二氯甲烷:甲醇=20:1)得到白色固体式(7)化合物(1.43g,72%yield for two steps)。
式(7)化合物的检测数据如下:1H NMR(400MHz,CDCl3)δ3.90–3.77(m,2H),3.29(t,J=11.1Hz,1H),3.12(d,J=4.6Hz,1H),2.41–2.30(m,2H),2.23–2.16(m,1H),2.08–1.73(m,6H),1.71–1.66(m,1H),1.56–1.47(m,1H),1.26–1.15(m,1H),1.11(d,J=13.4Hz,1H),0.86(t,J=7.5Hz,3H).13C NMR(100MHz,CDCl3)δ167.9,66.8,64.0,45.6,40,1,32.9,30.7,29.7,27.8,27.6,25.1,17.5,7.1.IR(neat,cm-1)3375,2923,2854,1736,1615,1462,1288,1251.HRMS(EI)[M]+Calcd for C13H21NO2223.1572,Found 223.1570.[α]D25-87.7(c 0.5,CHCl3).
实施例5
式(1)化合物的合成
将式(7)化合物(1.43g,6.40mmol,1.00equiv.)溶于四氢呋喃(64mL)中,–78℃下缓慢加入四氢铝锂(1.22g,32.02mmol,5.00equiv.),反应升至70℃回流10小时。反应完毕,冷却至0℃,反应依次用H2O(1.2mL),15%NaOH(1.2mL)和H2O(3.6mL)淬灭,过滤,乙酸乙酯洗涤滤饼,减压除去甲苯后直接柱层析得到淡黄色油状液体(无需柱层析分离纯化)。
将此油状液体溶于二氯甲烷中(130mL)中,0℃下加入戴斯-马丁氧化剂(10.86g,25.61mmol,4.00equiv.),反应升至室温搅拌1小时。反应完毕,依次加入饱和硫代硫酸钠溶液(50mL)和饱和碳酸氢钠溶液(50mL),体系用二氯甲烷(3x 50mL)萃取无水硫酸钠干燥,过滤,减压除掉溶剂,柱层析分离(石油醚:乙酸乙酯=2:1)得到黄色油状式(1)化合物(1.08g,77%yield for two steps)。
式(1)化合物的检测数据如下:1HNMR(400MHz,CDCl3)δ2.97(td,J=8.7,3.0Hz,2H),2.63(dd,J=7.4,5.5Hz,1H),2.44–2.31(m,2H),2.29–2.17(m,2H),1.94–1.83(m,3H),1.76(td,J=11.2,4.0Hz,1H),1.73–1.64(m,1H),1.63–1.52(m,2H),1.48–1.42(m,2H),1.34–1.25(m,1H),1.07(td,J=13.3,4.6Hz,1H),0.90(t,J=7.6Hz,3H).13C NMR(100MHz,CDCl3)δ211.2,73.4,53.1,52.8,48.1,36.7,34.7,32.8,30.0,26.0,21.2,21.2,7.0.IR(neat,cm-1)2926,1730,1460,1261,1075,1023.HRMS(EI)[M]+Calcd for C13H21NO 207.1623,Found 207.1626.[α]D25-25.6(c 0.5,CHCl3).
实施例6
式(8)化合物(+)-Dehydoaspidospermidine的合成
将式(1)(207.3mg,1.0mmol,1.0equiv.)和苯肼(162.2mg,1.5mmol,1.5equiv.)溶于苯(10mL)中,反应升至80℃回流3小时。反应完毕,冷却至室温,减压除去苯后直接得到淡黄色油状液体(无需柱层析分离纯化)。
将此油状液体溶于乙酸(10mL)中,反应升至120℃回流4小时。反应完毕,冷却至室温,用2M的氢氧化钠溶液调PH至8-9,二氯甲烷(3x 10mL)萃取,无水硫酸钠干燥,过滤,减压除掉溶剂,柱层析分离(石油醚:乙酸乙酯:三乙胺=100:10:0.5)得到黄色油状液体式(8)化合物(+)-Dehydroaspidospermidine(178.6mg,64%yield for two steps)。
式(8)化合物(+)-Dehydroaspidospermidine的检测数据如下:1H NMR(500MHz,CDCl3)δ7.50(d,J=7.6Hz,1H),7.32(d,J=7.2Hz,1H),7.28(td,J=7.6,1.2Hz,1H),7.15(td,J=7.4,0.9Hz,1H),3.20–3.16(m,2H),3.10(ddd,J=14.2,12.0,5.2Hz,1H),2.75(ddd,J=14.1,10.4, 3.5Hz,1H),2.58(ddd,J=11.5,8.5,5.7Hz,1H),2.45(td,J=12.6,3.1Hz,1H),2.40(s,1H),2.22–2.13(m,2H),1.92–1.79(m,1H),1.64(dd,J=12.4,5.6Hz,1H),1.61–1.53(m,2H),1.46(d,J=13.5Hz,1H),1.00(td,J=13.6,4.9Hz,1H),0.70–0.58(m,2H),0.49(t,J=7.4Hz,3H).13C NMR(125MHz,CDCl3)δ192.4,154.4,147.1,127.4,125.1,121.0,120.1,79.0,61.3,54.5,52.0,36.5,35.1,33.2,29.7,27.2,23.7,22.0,7.3.IR(neat,cm-1)2931,1709,1660,1459,1257,1012.HRMS(EI)[M]+Calcd for C19H24N2280.1939,Found 280.1937.[α]D25198.0(c 0.5,CHCl3).
实施例7
式(9)化合物(+)-Aspidospermidine的合成
将式(8)化合物(+)-Dehydroaspidospermidine(140.0mg,0.50mmol,1.0equiv.)溶于四氢呋喃(5mL)中,–78℃下分三批等量加入四氢铝锂(190.0mg,5.0mmol,10.0equiv.)反应升至70℃回流13小时。反应完毕,冷却至室温,向反应体系中分别用水(190μL),15%氢氧化钠溶液(190μL)and水(570μL).搅拌30分钟后过滤除掉固体不容物。减压除去溶剂后直接柱层析(石油醚:乙酸乙酯:三乙胺=100:10:1)得到白色固体式(9)化合物(+)-Aspidospermidine(117.8mg,83%yield)。
式(8)化合物(+)-Aspidospermidine的检测数据如下:1H NMR(400MHz,CDCl3)δ7.08(d,J=7.4Hz,1H),7.01(td,J=7.6,1.2Hz,1H),6.72(td,J=7.4,0.9Hz,1H),6.62(d,J=7.7Hz,1H),3.51(dd,J=11.0,6.2Hz,1H),3.12(dd,J=10.1,7.5Hz,1H),3.05(d,J=10.8Hz,1H),2.36–2.17(m,3H),2.03–1.88(m,2H),1.82–1.68(m,1H),1.67–1.59(m,2H),1.54–1.33(m,4H),1.12(td,J=27.0,13.5Hz,1H),1.05(dt,J=13.5,3.0Hz,1H),0.93–0.82(m,1H),0.64(t,J=7.5Hz,3H).13C NMR(100MHz,CDCl3)δ149.4,135.6,127.0,122.7,118.9,110.2,71.2,65.6,53.8,53.3,52.9,38.8,35.6,34.4,29.9,28.0,23.0,21.7,6.7.IR(neat,cm-1)2931,2778,1606,1480,1462,1256,1178,1008.HRMS(EI)[M]+Calcd for C19H26N2282.2096,Found 282.2099.[α]D2520.8(c0.5,EtOH).
本发明的保护内容不局限于以上实施例。在不背离发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。
- 还没有人留言评论。精彩留言会获得点赞!