苯丙酸苷类化合物及其制备方法和应用与流程
本发明属于医药及保健食品领域,具体涉及一种苯丙酸苷类化合物及其制备方法和应用。
背景技术:
我国已成为糖尿病世界第一大国,目前我国的糖尿病患者超过1亿,而糖尿病前期患者数量超过1.5亿,这为数众多的糖尿病前期患者中每年有10%的人转变为真正的糖尿病,形势十分严峻。糖尿病的大规模爆发和治疗给国家和社会带来了沉重的负担,因此必须采取有效措施来预防和有效治疗。
外周组织对胰岛素的抵抗是糖尿病发生发展的开始阶段,在这一阶段,肌肉、脂肪或肝脏细胞对胰岛素产生抵抗,可发生在受体前、受体或受体后。胰岛素抵抗也贯穿于整个糖尿病期间,直到最后胰岛β细胞衰竭,无法产生胰岛素。在胰岛β细胞还没有彻底衰竭前,如果某种物质能够恢复或促进靶细胞对胰岛素的敏感性,能够促进细胞对葡萄糖的摄入和转化,则该物质可能就具有很好的抗糖尿病作用,也就可能有希望阻止或延缓将处于糖尿病前期的患者向真正的糖尿病转变。
增加胰岛素敏感性的抗糖尿病药物有双胍类和噻唑烷二酮类等,一方面这些药物都有不同程度的副作用,比如噻唑烷二酮类容易引起血液稀释,因引起心血管疾病风险等。另一方面,更重要的是这些药物均会随着用药时间的延长而出现继发性失效,无论是单一疗法还是联合治疗,都没能取得令人满意的结果,我国有三分之二以上二型糖尿病患者的长期血糖控制不达标。因此,仍然需要不断地寻找改善胰岛素敏感性的抗糖尿病药物。
天然产物是寻找新药不可替代的重要源泉,其结构多样性和活性多样性是合成产物无法比拟的。因此从植物或微生物次级代谢产物中寻找具有高效低毒的先导化合物或新药依然是一个重要途径。民族药是我国少数民族人民与疾病长期斗争中积累的宝贵财富,是中华文明的瑰宝。兴安独活(Heracleum dissectum),也称坎库拉,属菊科多年生草本,是东北鄂伦春族药食两用的野生植物,春夏季节其幼嫩的茎叶被当地人用作美味的蔬菜食用,其根作为传统的鄂族药材用于祛风除湿、止痛止泻等。但目前对其化学成分和药理活性的研究报道极少。
技术实现要素:
本发明的目的在于克服现有技术的缺陷,提供一种苯丙酸苷类化合物及其制备方法和应用,能够从兴安独活中提取分离出苯丙酸苷类化合物,该化合物能够促进细胞对葡萄糖的摄入和转化。
本发明是通过以下技术方案来实现:
本发明苯丙酸苷类化合物分子式为C26H36O13。
进一步地,其结构式为:
本发明制备方法,包括以下步骤:
1)取兴安独活的干燥根进行回流提取若干次,将提取液减压浓缩,得到总浸膏或浓缩液;
2)将总浸膏混悬于水中,得到浸膏液,浸膏液或浓缩液经乙酸乙酯萃取若干次后分离出有机层,剩余水层通过正丁醇萃取若干次后合并萃取液,减压除去正丁醇得到正丁醇萃取层;
3)将正丁醇萃取层上样于硅胶柱,以氯仿-甲醇系统作为洗脱液,按体积比(100:0)~(0:100)进行梯度洗脱,对流出液进行检测,将洗脱液体积比为6:1的流份合并,除去溶剂,得到第一次过柱部分;
4)将第一次过柱部分上样于反相硅胶层析柱,以甲醇-水系统作为洗脱液,按体积比(0:100)~(100:0)进行梯度洗脱,将洗脱液体积比为75:30的流份合并,除去溶剂,得到第二次过柱部分;
5)将第二次过柱部分上样于高效液相色谱分离柱,用流动相进行等度洗脱,得到苯丙酸苷类化合物。
进一步地,步骤1)的回流提取中采用甲醇、水或体积分数10~95%的乙醇作为提取剂,兴安独活的干燥根与提取剂的质量体积比为1kg:(1~8)L;当提取剂为甲醇或体积分数10~95%的乙醇时,将得到的提取液中溶剂回收得到总浸膏;当提取剂为水时,将提取液的体积浓缩至六分之一到十分之一,得到浓缩液。
进一步地,步骤1)中提取次数为1~6次,每次1~4小时。
进一步地,步骤2)中总浸膏和水的体积比为1:1~1:5。
进一步地,步骤2)中直接经过乙酸乙酯萃取;或者先依次经石油醚和氯仿萃取后,再经过乙酸乙酯萃取;每次萃取均是等体积萃取;每种溶剂分别萃取1~6次。
进一步地,步骤3)中梯度洗脱后每300~800mL收集一个流份;步骤4)中梯度洗脱后每200~500mL收集一个流份;步骤3)和步骤4)中对流出液均是进行TLC检测;步骤5)中等度洗脱的流速为3~6mL/min。
进一步地,步骤5)中流动相是甲醇-0.5%醋酸水系统或乙腈-0.5%醋酸水系统,甲醇-0.5%醋酸水系统中甲醇、水和醋酸的体积比为45:55:0.5;乙腈-0.5%醋酸水系统中乙腈、水和醋酸的体积比为42:58:0.5。
如上所述的苯丙酸苷类化合物在制备抗糖尿病药物和/或保健品中的应用。
与现有技术相比,本发明的有益效果是:
本发明中的苯丙酸苷类化合物对脂肪细胞的甘油三酯的蓄积有促进作用,意即有将细胞外葡萄糖摄入细胞内转化为甘油三酯的促进作用,经试验证明,本发明化合物在3μM时就有显著性促进作用,具有浓度依赖关系,且该化合物具有高效、低毒的特点,有望开发成新的抗糖尿病药物,或者用于制备具有预防和治疗糖尿病作用的保健食品。
本发明中得到的苯丙酸苷类化合物活性较好,但在植物中的含量不高,且极性较大,并且呈现一定的酸性,用常规的色谱方法难以富集并分离得到。本发明中采用特殊的亲水色谱柱,并在流动相中加一定量的醋酸以改善分离,能将其很好地分离纯化得到,具有方法简便有效,得到的化合物纯度高的特点。
【附图说明】
图1为本发明化合物1的1H-NMR图谱;
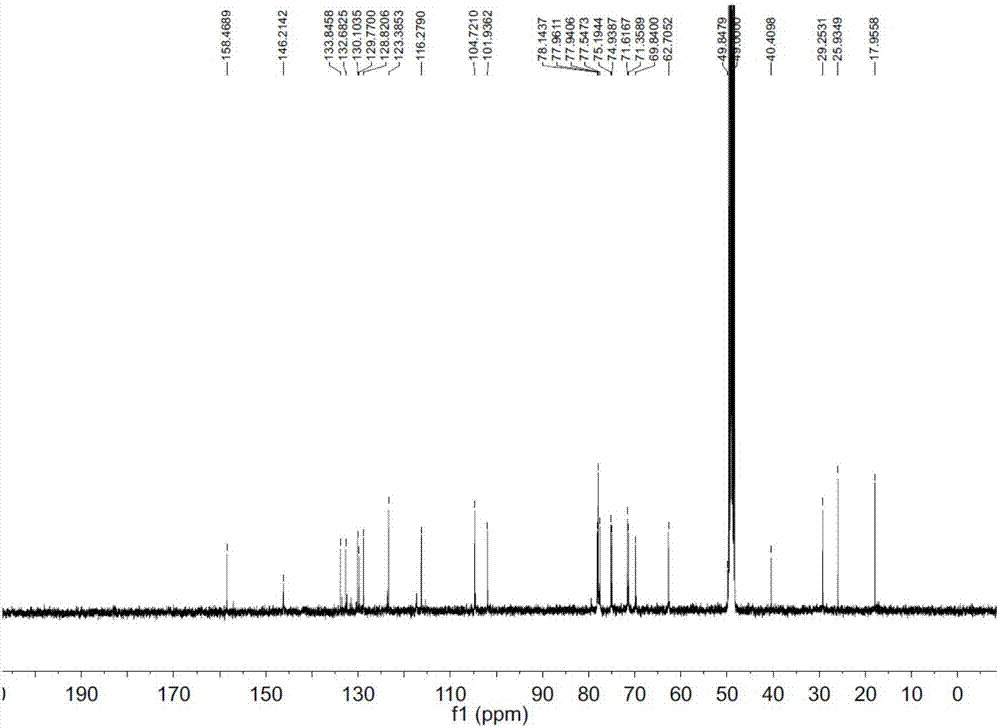
图2为本发明化合物1的13C-NMR图谱;
图3为本发明化合物1的DEPT图谱;
图4为本发明化合物1的1H-1H COSY图谱;
图5为本发明化合物1的HMQC图谱;
图6为本发明化合物1的HMBC图谱;
图7为本发明化合物1的ROESY图谱;
图8为本发明化合物1的HR-ESI-MS图谱。
【具体实施方式】
下面结合具体实施例对本发明作进一步的说明,但并不局限于此。
本发明苯丙酸苷类化合物的制备方法,包括以下步骤:
1)取一定质量(kg)的兴安独活干燥根,用体积为兴安独活干燥根质量1~8倍量的甲醇、体积分数为10~95%的乙醇或水(L)在各自沸点附近加热回流提取1~6次,每次1~4小时,当提取剂为甲醇或体积分数10~95%的乙醇时,合并提取液减压回收除去溶剂,得到总浸膏,当提取剂为水时,合并提取液并将其体积浓缩至六分之一到十分之一,得到浓缩液;
2)将总浸膏混悬于水中,总浸膏和水的体积比为1:1~1:5,得到浸膏液,浸膏液或浓缩液分别依次用等体积的有机溶剂萃取,其中,第一次萃取时用石油醚对浸膏液或浓缩液进行等体积萃取,之后每次萃取均是分离出上一次萃取后的有机层,将剩余的水层用有机溶剂等体积进行下一次萃取;每种溶剂各萃取1~6次,合并萃取液,常压或减压蒸馏除去有机溶剂,分别得到各萃取层和水层。有机溶剂包括石油醚、氯仿、乙酸乙酯和正丁醇等,且萃取次序为先用极性小的溶剂,再用极性大的有机溶剂,石油醚和氯仿可以省去。
3)取正丁醇萃取层,通过采用柱层析纯化等分离方法,得到本发明的苯丙酸苷类化合物。
柱层析包括以下三个阶段:
第一阶段:将正丁醇萃取层上样于硅胶柱,以氯仿-甲醇系统作为洗脱液,按体积比(100:0)~(0:100)进行梯度洗脱,每300~800mL收集一个流份,对流出液进行TLC检测,合并相同流份,共得到30个流份,分别命名为FrB1-FrB30,常压或减压蒸干溶剂,取其中的FrB26流份,即洗脱液体积比为6:1的流份,作为第一次过柱部分;
第二阶段:将第一次过柱部分,上样于反相硅胶层析柱,以甲醇-水系统作为洗脱液,按体积比(0:100)~(100:0)进行梯度洗脱,每200~500mL收集一个流份,对流出液进行TLC检测,合并相同流份,减压除去溶剂,得到3个亚流份,分别命名为FrB26.1-FrB26.3,将FrB26.2流份,即洗脱液体积比为75:30的流份,作为第二次过柱部分;
第三阶段:将第二次过柱部分,上样于高效液相色谱分离柱,分离得到苯丙酸苷类化合物。
其中,高效液相色谱分离柱为江苏汉邦的Megres C18柱,高效液相色谱分离是用示差折光检测器,以甲醇-0.5%醋酸水系统或乙腈-0.5%醋酸水系统作为流动相,按3~6mL/min进行等度洗脱。甲醇-0.5%醋酸水系统中甲醇、水和醋酸的体积比为45:55:0.5;乙腈-0.5%醋酸水系统中乙腈、水和醋酸的体积比为42:58:0.5。
本发明中得到的苯丙酸苷类化合物,其结构式如下:
本发明中的苯丙酸苷类化合物在制备抗糖尿病药物和/或保健品中的应用:本发明在对兴安独活进行化学成分和药理活性的研究中发现,从中分离得到的苯丙酸苷类化合物具有很好的改善脂肪细胞胰岛素敏感性的作用,能够显著促进脂肪细胞摄入葡萄糖并转化为甘油三酯,因此,有望将其开发成具有改善胰岛素抵抗的抗糖尿病药物和/或保健品。
实施例1
1、苯丙酸苷类化合物1的提取和分离
1)取坎库拉干燥根6kg,用体积量为坎库拉干燥根质量5倍的甲醇加热回流提取3次,每次2小时,合并提取液减压回收溶剂,得总浸膏;
2)将总浸膏混悬于4倍量水中,用石油醚等体积萃取4次,然后经氯仿,乙酸乙酯和正丁醇等体积萃取4次,萃取层减压除去溶剂后分别得到石油醚层,氯仿层,乙酸乙酯层,正丁醇层。
3)取正丁醇萃取层100g,首先通过采用硅胶柱色谱,氯仿/甲醇按体积比(v/v)为100:0~0:100进行梯度洗脱,每500mL收集一个流份,经TLC检识合并相同流份后共得到30个流份(FrB1-FrB30)。
4)其中第26个流份FrB26(7.0g)经反相硅胶柱色谱分离,用MeOH/H2O按体积比(v/v)为100:0~0:100进行梯度洗脱,每200mL收集一个流份,经TLC检识合并相同流份后得到3个流份(FrB26.1-FrB26.3)。
5)然后FrB26.2用半制备高效液相色谱纯化,采用Megres C18柱,流动相是MeOH/0.5%HAc水溶液(45:55,v/v),流速为3.0mL/min,得到化合物1(tR 17分钟)。
本发明通过理化常数和现代波谱学技术手段(HR-ESI-MS,1D-NMR,2D-NMR)鉴定化合物的结构,化合物1为drupanin-4-O-β-D-glucopyranosyl-(1→6)-β-D-glucopyranoside。化合物1的结构鉴定过程如下所述。
2、苯丙酸苷类化合物的结构鉴定
1)化合物1为淡黄色粉末;其图8的HR-ESI-MS m/z 579.2070[M+Na]+(calcd579.2048),确定分子式为C26H36O13。在图1的1H-NMR中,化学位移δH 7.41(1H,dd,J=8.5,1.9Hz),7.31(1H,d,J=1.9Hz)和7.20(1H,d,J=8.5Hz)的质子信号显示该化合物中含有一个1,2,4-三取代苯环结构,质子δH 7.56(1H,d,J=15.9Hz)和6.29(1H,d,J=15.9Hz)表明分子中存在一个反式双键。在图2的13C NMR谱和图3的DEPT谱中,共有26个碳,包括1个羧基碳,2个芳香季碳,1个连氧芳香季碳,6个SP2杂化次甲基碳,1个SP2杂化季碳,8个连氧次甲基碳,2个连氧亚甲基碳,2个芳香次甲基碳,1个亚甲基碳和2个甲基碳。在图2的13CNMR谱和图6的HMBC谱中,羧基碳δC 170.1和两个双键质子δH 7.56(1H,d,J=15.9Hz)及6.29(1H,d,J=15.9Hz)相关,表明分子中存在一个桂皮酸的结构。另外,季碳δC 133.8和δC 123.4(相应的氢信号是δH 5.32,1H,t,J=7.3Hz)相关,说明分子中还存在一个三取代双键的结构片段。在1H NMR谱和图4的1H-1H COSY谱中,质子δH 3.38(2H,m)和SP2杂化的次甲基δH5.32(1H,t,J=7.3Hz)相关,HMBC谱中两个甲基质子δH 1.71(3H,s)and 1.73(3H,s)和季碳δC133.8相关,以上信息揭示分子中存在一个异戊烯基片段,且该片段连接在桂皮酸的C-3位,因为HMBC谱中可观察到异戊烯基片段中的亚甲基质子δH 3.38(2H,m)和双键质子5.32(1H,t,J=7.4Hz)与苯环的C-3位碳δC 132.6相关。化合物1的核磁数据与化合物drupanin的十分相似。
在1H和13C NMR谱中,两个端基质子δH 4.94(1H,d,J=7.3Hz)及4.35(1H,d,J=7.7Hz)和他们相应的碳信号δC 101.9,104.7表明分子中存在两个糖单位。酸水解后用GC分析表明这两分子糖为葡萄糖,且构型为β型,这从端基质子的偶合常数也能判定,分别为7.3和7.7Hz。外侧葡萄糖的1位连在内侧葡萄糖的6位,这可从HMBC谱中得到验证,外侧葡萄糖的端基质子δH 4.35(1H,d,J=7.7Hz,H-1″)与内侧葡萄糖的6位亚甲基碳δC 69.8(C-6′)相关。同时,内侧葡萄糖的端基质子δH 4.94(1H,d,J=7.3Hz,H-1′)与苯环上的碳δC 158.4(C-4)相关,表明糖链连在苯环的4位。其它位置的连接均通过综合解析2D-NMR包括图5的HMQC,图6的HMBC,图4的1H-1H COSY和图7的ROESY得以确定。因此,化合物1鉴定为drupanin-4-O-β-D-glucopyranosyl-(1→6)-β-D-glucopyranoside,即3-异戊烯基桂皮酸-4-O-β-D-吡喃葡萄糖基-(1→6)-β-D-吡喃葡萄糖苷。是一个未见文献报道的新化合物。其结构式如下:
其核磁数据见表1。
表1本发明中化合物1的1H NMR和13C NMR数据
下面对本发明中所分离鉴定的化合物1进一步做药理活性检测。
3、脂肪细胞甘油三酯脂肪蓄积实验
实验方法:
3T3-L1细胞(5.0×104cells/well)接种于48孔板中,24小时后,加入分化培养基(含有10%FBS的DMEM(high glucose:4500mg/L,1μM的地塞米松,0.5mM的IBMX和5μg/mL的胰岛素)培养3天,然后培养基换成维持培养基(含有10%FBS的DMEM(high glucose:4500mg/L和5μg/mL的胰岛素)再培养2天后,更换新鲜维持培养基继续培养2天,在第8天,吸去培养基,每孔加入200μL蒸馏水,超声破碎,用甘油三酯液体试剂盒测定细胞破碎液中的TG含量。样品溶于DMSO,在每次更换培养基时加入,其中DMSO的终浓度为0.1%,Troglitazone用作阳性对照化合物。数值表示为与对照组相比的TG含量增加值MEAN±SEM(n=4).*p<0.05,**p<0.01(与对照组相比)。
实验结果见表4。
表4.化合物1对3T3-L1脂肪细胞的TG蓄积作用
实验结果:表4中的数据表明,本发明中的化合物1具有很好的促进脂肪细胞蓄积甘油三酯的作用,化合物1在3μM时就有显著性促进作用(p<0.01),且随着浓度的增加,促进作用也随之增强,意即具有浓度依赖关系。
结合以上实验及其实验结果,表明本发明中的苯丙酸苷类化合物具有极强的改善脂肪细胞对胰岛素的敏感性,促进对葡萄糖的摄入和转化,因此有望开发为新的抗糖尿病药物;或者用于制备预防和治疗糖尿病的保健食品。
实施例2
1)取坎库拉干燥根6kg,用体积量为坎库拉干燥根质量3倍的95%乙醇加热回流提取5次,每次1小时,合并提取液减压回收溶剂,得总浸膏;
2)将总浸膏混悬于2倍量水中,用乙酸乙酯等体积萃取1次,然后经正丁醇等体积萃取6次,萃取层减压除去溶剂后分别得到乙酸乙酯层,正丁醇层。
3)取正丁醇萃取层100g,首先通过采用硅胶柱色谱,氯仿/甲醇按体积比(v/v)为100:0~0:100进行梯度洗脱,每500mL收集一个流份,经TLC检识合并相同流份后共得到30个流份(FrB1-FrB30)。
4)其中第26个流份FrB26(7.0g)经反相硅胶柱色谱分离,用MeOH/H2O按体积比(v/v)为100:0~0:100进行梯度洗脱,每200mL收集一个流份,经TLC检识合并相同流份后得到3个流份(FrB26.1-FrB26.3)。
5)然后FrB26.2用半制备高效液相色谱纯化,采用Megres C18柱,流动相是MeOH/0.5%HAc水溶液(45:55,v/v),流速为3.0mL/min,得到化合物1(tR 17分钟)。
实施例3
1)取坎库拉干燥根6kg,用体积量为坎库拉干燥根质量8倍的水加热回流提取2次,每次3小时,合并提取液减压回收溶剂,得总浸膏;
2)将总浸膏混悬于1倍量水中,用乙酸乙酯等体积萃取2次,然后经正丁醇等体积萃取3次,萃取层减压除去溶剂后分别得到乙酸乙酯层,正丁醇层。
3)取正丁醇萃取层100g,首先通过采用硅胶柱色谱,氯仿/甲醇按体积比(v/v)为100:0~0:100进行梯度洗脱,每800mL收集一个流份,经TLC检识合并相同流份后共得到30个流份(FrB1-FrB30)。
4)其中第26个流份FrB26(7.0g)经反相硅胶柱色谱分离,用MeOH/H2O按体积比(v/v)为100:0~0:100进行梯度洗脱,每500mL收集一个流份,经TLC检识合并相同流份后得到3个流份(FrB26.1-FrB26.3)。
5)然后FrB26.2用半制备高效液相色谱纯化,采用Megres C18柱,流动相是乙腈-0.5%醋酸水系统(42:58,v/v),流速为3.0mL/min,得到化合物1(tR 18.6分钟)。
实施例4
1)取坎库拉干燥根6kg,用体积量为坎库拉干燥根质量4倍的10%乙醇加热回流提取4次,每次2小时,合并提取液减压回收溶剂,得总浸膏;
2)将总浸膏混悬于5倍量水中,用石油醚等体积萃取2次,然后经氯仿,乙酸乙酯和正丁醇等体积萃取2次,萃取层减压除去溶剂后分别得到石油醚层,氯仿层,乙酸乙酯层,正丁醇层。
3)取正丁醇萃取层100g,首先通过采用硅胶柱色谱,氯仿/甲醇按体积比(v/v)为100:0~0:100进行梯度洗脱,每300mL收集一个流份,经TLC检识合并相同流份后共得到30个流份(FrB1-FrB30)。
4)其中第26个流份FrB26(7.0g)经反相硅胶柱色谱分离,用MeOH/H2O按体积比(v/v)为100:0~0:100进行梯度洗脱,每400mL收集一个流份,经TLC检识合并相同流份后得到3个流份(FrB26.1-FrB26.3)。
5)然后FrB26.2用半制备高效液相色谱纯化,采用Megres C18柱,流动相是乙腈-0.5%醋酸水系统(42:58,v/v),流速为3.0mL/min,得到化合物1(tR 18.6分钟)。
对比例1
将步骤5)中的流动相换成其它流动相或不同比例的相同流动相(或者色谱柱替换成一般的反相色谱柱),其它步骤和条件与实施例1相同。发现无法分离出苯丙酸苷类化合物。
本发明公开了一种苯丙酸苷类化合物及其应用。所述的苯丙酸苷类化合物是从鄂伦春族药食两用的植物坎库拉中提取分离得到,经过实验研究发现该类化合物对3T3-L1脂肪细胞的甘油三酯蓄积有促进作用,意即对细胞摄入葡萄糖有促进作用,且该类化合物从可食用野生植物中提取分离得到,具有高效低毒的特点,有望开发成具有抗糖尿病作用的药物和/或保健品。上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!