在抗蚀剂应用中作为光酸生成剂的磺酸衍生化合物的制作方法
本发明涉及新颖光酸生成剂化合物(“pag”)和包含所述pag化合物的光阻剂组合物。具体地说,本发明pag化合物在有机溶剂中具有极佳溶解度并且在光刻工艺中展现出比常规pag化合物更高的敏感度和比常规pag化合物更好的性能。
背景技术:
光阻剂为将图像转移到衬底的感光性膜。其形成负像或正像。将光阻剂涂布于衬底上后,经由图案化光掩模使涂层曝露于如紫外光的活化能源,在光阻涂层中形成潜像。光掩模具有界定需要转移到底层衬底的图像的对活化辐射不透明和透明的区域。
已证实化学增幅型光阻剂适用于在半导体制造中形成超精细图案的工艺中实现高敏感度。通过将pag与具有酸不稳定结构的聚合物基质掺合来制备这些光阻剂。根据此类光阻剂的反应机制,光酸生成剂在其受到光源照射时生成酸,并且经曝露或照射部分中的聚合物基质的主链或支链在所谓的“曝光后烘烤”(peb)中与所生成的酸反应并且分解或交联,使得聚合物的极性发生改变。这一极性改变造成经照射曝光区域与未曝光区域之间在显影溶液中的溶解度差异,由此在衬底上形成掩模的正像或负像。酸扩散不仅对增加光阻剂敏感度和产出量至关重要,并且也对限制因散粒噪声统计所致的线边缘粗糙度至关重要。
在化学增幅型光阻剂中,成像所需的溶解度转换化学反应并非由曝光直接引起;确切来说,曝光生成在后续peb步骤期间促进溶解度转换化学反应的稳定催化物质。术语“化学增幅”源于以下事实:以光化学方式所生成的每一催化剂分子可促进多个溶解度转换反应事件。转换反应的表观量子效率为催化剂生成的量子效率乘以平均催化链长。原始曝光剂量因后续一系列化学反应事件发生“增幅”。催化剂的催化链长可为极长的(高达数百个反应事件),提供显著曝光增幅。
化学增幅的有利之处在于其可大大改善抗蚀剂敏感度,但其并非没有潜在缺陷。举例来说,由于催化剂分子在数百个反应位点周围移动,因此未对曝露于成像辐射的区域加以必要的限制。抗蚀剂敏感度与成像保真度之间存在潜在取舍。举例来说,增幅型光阻剂经由光掩模曝光,在曝光区域中生成酸催化剂。通过提高peb中的晶片温度,将第一步中生成的潜酸图像转换成可溶和不可溶区域的图像,其允许发生化学反应。一些酸迁移出原先曝光区域而导致“临界尺寸偏差”问题。烘烤后,用溶剂使图像显影。经显影的特征的宽度可能由于酸从曝光区域扩散到未曝光区域中而大于标称掩模尺寸。在增幅型抗蚀剂的历史上,很多情况下这一取舍极少受到关注,因为相对于经印刷的特征大小,催化剂扩散距离并不重要,但当特征大小减小时,扩散距离仍保持大致相同并且催化剂扩散已成为重要问题。
为生成足够的会改变聚合物溶解度的酸,需要一定曝光时间。对于已知pag分子,如n-羟基萘二甲酰亚胺三氟甲磺酸酯(“nit”),这一曝光时间相当长(归因于其在365nm或大于365nm下的低吸收度)。然而,提高所述pag的浓度并不会促使曝光时间变短,因为pag的溶解度为限制因素。另一种可能性为添加敏化剂,其吸收光并且将能量转移到pag,pag随后将释放酸。然而,为了能够将能量转移到极为邻近的pag,必须以相当高的浓度使用所述敏化剂。在所述高浓度下,敏化剂常具有过高的吸收度并且在显影后对抗蚀剂轮廓的形状具有不利影响。
因此,在所属领域中需要展现较好溶解度的pag,其意指将活性较大的分子施加到调配物中,其中相较于由现有技术已知的光阻剂组合物,包含这些化合物的光阻剂组合物对电磁辐射,尤其对波长为200nm到500nm的电磁辐射具有高敏感度,并且同时产生具有较高分辨率的图案化结构。
技术实现要素:
本发明通过提供由式(i)或式(ii)表示的磺酸衍生化合物来满足这一需要:
在一些实施例中,本发明还提供抗蚀剂组合物,其包含成像有效量的一种或多种根据本发明的pag和树脂。
在其它实施例中,本发明提供用于形成本发明光阻剂的浮雕图像的方法,包括用于形成小于四分之一微米尺寸或更小,如小于0.2或小于0.1微米尺寸的高分辨图案化光阻剂图像(例如,具有基本上垂直侧壁的图案化线)的方法。
本发明进一步提供包含衬底的制品,如上面涂布有本发明光阻剂和浮雕图像的微电子晶片或平板显示器衬底。本发明的其它方面揭示于下文中。
附图说明
当结合附图阅读时,由以下详细描述最好地理解本发明。应强调,根据惯例,图式的各种特征不按比例绘制。相反地,为清楚起见,可任意扩大或减小各种特征的尺寸。图式中包括以下诸图:
图1为经uv灯在365nm下照射后化合物t3的uv-vis光谱,其显示强度随能量曝光剂量(mj/cm2)增加而降低;
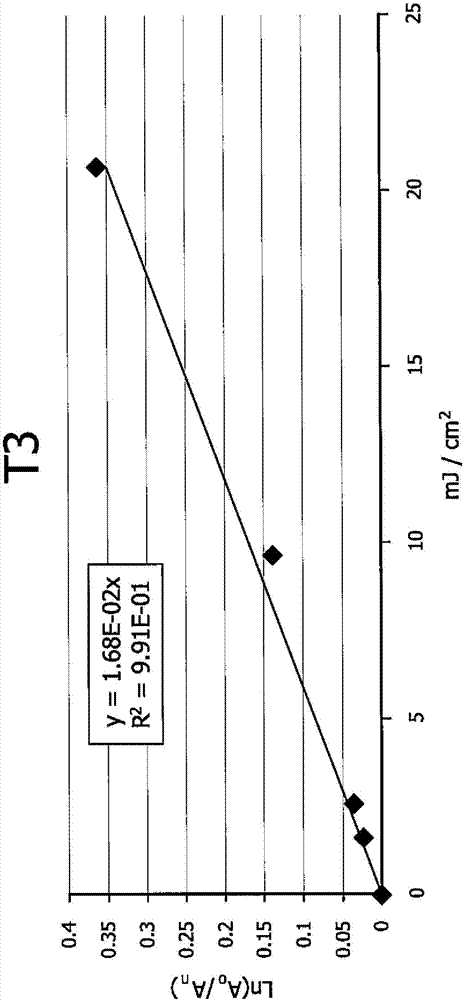
图2为吸光度变化的自然对数与图1的光谱的能量曝光剂量的图,用于得到t3的光反应常数;
图3为具有各种线-间隙(l/s)大小(5、6、7、8、9和10μm)的图案化结构的显微照片,其中采用本发明化合物t2作为pag化合物;和
图4为化合物nit、t1、t2和t5于acn(0.001%w/v)中的uv-vis光谱。
具体实施方式
定义
除非另外说明,否则用于本申请(包括说明书和权利要求书)的以下术语具有下文给出的定义。必须指出,除非上下文另外明确规定,否则如本说明书和所附权利要求书中所使用,单数形式“一(a/an)”和“所述(the)”包括复数个提及物。
所有数值名称,如重量、ph、温度、时间、浓度和分子量,包括范围,为近似值,其可变化10%。应理解,尽管未必总加以明确陈述,但所有数值名称之前均有术语“约”。还应理解,尽管未必总加以明确陈述,但本文所述的试剂仅为示范性的并且此类试剂的等效物在所属领域中为已知的。
参考本发明,除非另外特定定义,否则本文描述中所用的技术和科学术语将具有由所属领域的普通技术人员所通常理解的含义。因此,以下术语预期具有如下含义。
如本文所用,术语“部分”是指分子的特定链段或官能团。化学部分通常公认为嵌入或附接于分子的化学实体。
如本文所用,术语“脂族基”涵盖术语烷基、烯基、炔基,其中的每一个任选地经如以下列举者取代。
如本文所用,“烷基”是指含有1-20(例如,2-18、3-18、1-8、1-6、1-4或1-3)个碳原子的饱和脂族烃基。烷基可为直链、支链、环状或其任何组合。烷基的实例包括(但不限于)甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、正戊基、正庚基或2-乙基己基。烷基可经一个或多个取代基取代(即任选地经取代)或可为如下所阐述的多环。
除非另外特定限制,否则如本文所用,术语“烷基”以及如“烷氧基”和“硫烷基”的衍生术语将直链、支链和环状部分包括在其范围内。
如本文所用,“烯基”是指含有2-20(例如,2-18、2-8、2-6或2-4)个碳原子和至少一个双键的脂族碳基。类似烷基,烯基可为直链、支链或环状或其任何组合。烯基的实例包括(但不限于)烯丙基、异戊二烯基、2-丁烯基和2-己烯基。烯基可任选地经一个或多个如下所阐述的取代基取代。
如本文所用,“炔基”是指含有2-20(例如,2-8、2-6或2-4)个碳原子并且具有至少一个三键的脂族碳基。炔基可为直链、支链或环状或其任何组合。炔基的实例包括(但不限于)炔丙基和丁炔基。炔基可任选地经一个或多个如下所阐述的取代基取代。
“卤素”为周期表第17族的原子,其包括氟、氯、溴和碘。
如本文所用,单独使用或作为如“芳烷基”、“芳烷氧基”或“芳氧基烷基”中较大部分的一部分使用的“芳基”是指单环(例如苯基);双环(例如茚基、萘基、四氢萘基、四氢茚基);和三环(例如芴基、四氢芴基或四氢蒽基、蒽基)环系统,其中单环系统为芳香族,或双环或三环系统中至少一个环为芳香族。双环和三环基团包括苯并稠合2-3元碳环。举例来说,苯并稠合基团包括与两个或更多个c4-8碳环部分稠合的苯基。芳基任选地经一个或多个如下所阐述的取代基取代。
如本文所用,“芳烷基(aralkyl/arylalkyl)”是指经芳基取代的烷基(例如,c1-4烷基)。上文已定义“烷基”和“芳基”。芳烷基的实例为苯甲基。芳烷基任选地经一个或多个如下所阐述的取代基取代。
如本文所用,“环烷基”是指3-10(例如,5-10)个碳原子的饱和碳环单环或双环(稠合或桥联)。环烷基的实例包括环丙基、环丁基、环戊基、环己基、环庚基、金刚烷基、降冰片基、立方烷基、八氢-茚基、十氢-萘基、双环[3.2.1]辛基、双环[2.2.2]辛基、双环[3.3.1]壬基、双环[3.3.2]癸基、双环[2.2.2]辛基、金刚烷基、氮杂环烷基或((氨基羰基)环烷基)环烷基。
如本文所用,术语“杂芳基”是指具有4到18个环原子的单环、双环或三环系统,其中环原子中的一个或多个为杂原子(例如n、o、s或其组合)并且其中单环系统为芳香族,或双环或三环系统中至少一个环为芳香族。杂芳基包括具有2到3个环的苯并稠合环系统。举例来说,苯并稠合基团包括与一个或两个4到8元杂环脂族部分(例如吲哚嗪基、吲哚基、异吲哚基、3h-吲哚基、吲哚啉基、苯并[b]呋喃基、苯并[b]噻吩基、喹啉基或异喹啉基)稠合的苯并基。杂芳基的一些实例为氮杂环丁烷基(azetidinyl)、吡啶基、1h-吲唑基、呋喃基、吡咯基、噻吩基、噻唑基、恶唑基、咪唑基、四唑基、苯并呋喃基、异喹啉基、苯并噻唑基、二苯并哌喃、噻吨、啡噻嗪、二氢吲哚、苯并[1,3]二氧杂环戊烯、苯并[b]呋喃基、苯并[b]噻吩基、吲唑基、苯并咪唑基、苯并噻唑基、嘌呤基、噌啉基、喹啉基、喹唑啉基、噌啉基、酞嗪基、喹唑啉基、喹喔啉基、异喹啉基、4h-喹嗪基、苯并-1,2,5-噻二唑基或1,8-萘啶基。
单环杂芳基包括(但不限于)呋喃基、噻吩基、2h-吡咯基、吡咯基、恶唑基、噻唑基、咪唑基、吡唑基、异恶唑基、异噻唑基、1,3,4-噻二唑基、2h-哌喃基、4-h-哌喃基、吡啶基、哒嗪基、嘧啶基、吡唑基、吡嗪基或1,3,5-三嗪基。单环杂芳基根据标准化学命名法编号。
双环杂芳基包括(但不限于)吲哚嗪基、吲哚基、异吲哚基、3h-吲哚基、吲哚啉基、苯并[b]呋喃基、苯并[b]噻吩基、喹啉基、异喹啉基、吲哚嗪基、异吲哚基、吲哚基、苯并[b]呋喃基、苯并[b]噻吩基、吲唑基、苯并咪唑基、苯并噻唑基、嘌呤基、4h-喹嗪基、喹啉基、异喹啉基、噌啉基、酞嗪基、喹唑啉基、喹喔啉基、1,8-萘啶基或喋啶基。双环杂芳基根据标准化学命名法编号。
杂芳基任选地经一个或多个如下所阐述的取代基取代。
如本文所用,“杂芳基烷基”是指经杂芳基取代的烷基(例如,c1-4烷基)。上文已定义“烷基”和“杂芳基”。杂芳基烷基任选地经一个或多个如下所阐述的取代基取代。
如本文所用,“酰基”是指甲酰基或rx--c(o)--(如-烷基-c(o)--,也称为“烷基羰基”),其中先前已定义“烷基”。
如本文所用,术语“酰氧基”包括直链酰氧基、支链酰氧基、环酰氧基、环状酰氧基、未经杂原子取代的酰氧基、经杂原子取代的酰氧基、未经杂原子取代的cn-酰氧基、经杂原子取代的cn-酰氧基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基和羧酸酯基。
如本文所用,“烷氧基”是指烷基-o--基团,其中先前已定义“烷基”。
如本文所用,当用作端基时,“羧基”是指--cooh、--coorx、--oc(o)h、--oc(o)rx;或当用作内部基团时,其是指--oc(o)--或--c(o)o--。
如本文所用,“烷氧羰基”意指--coor,其中r为如上文所定义的烷基,例如甲氧羰基、乙氧羰基等。
如本文所用,当在末端使用时,“磺酰基”是指--s(o)2--rx并且当在内部使用时其指--s(o)2--。
术语“烷硫基”包括直链烷硫基、支链烷硫基、环烷硫基、环状烷硫基、未经杂原子取代的烷硫基、经杂原子取代的烷硫基、未经杂原子取代的cn-烷硫基和经杂原子取代的cn-烷硫基。在某些实施例中,涵盖低碳烷硫基。
如本文所用,术语“胺”或“氨基”包括氮原子共价键结到至少一个碳或杂原子的化合物。术语“胺”或“氨基”还包括--nh2并且还包括经取代的部分。所述术语包括“烷氨基”,其包含氮键结到至少一个其它烷基的基团和化合物。所述术语包括“二烷基氨基”,其中氮原子键结到至少两个其它经独立选择的烷基。所述术语包括“芳氨基”和“二芳氨基”,其中氮分别键结到至少一个或两个经独立选择的芳基。
术语“卤烷基”是指经一个到最大可能数目的卤素原子取代的烷基。术语“卤烷氧基”和“卤硫烷基”是指经一到五个卤素原子取代的烷氧基和硫烷基。
词组“任选地经取代”可与词组“经取代或未经取代”互换使用。如本文所述,本发明化合物可任选地经一个或多个取代基取代,如上文总体上说明或如由本发明的特定类别、子类和种类示范。如本文所述,以上部分或下文介绍的那些部分中的任一个可任选地经本文所述的一个或多个取代基取代。特定基团的每一取代基进一步任选地经一到三个卤基、氰基、侧氧基烷氧基、羟基、氨基、硝基、芳基、卤烷基和烷基取代。举例来说,烷基可经烷巯基(alkylsulfanyl)取代并且烷巯基可任选地经一到三个卤基、氰基、侧氧基烷氧基、羟基、氨基、硝基、芳基、卤烷基和烷基取代。
一般来说,无论之前是否出现术语“任选地”,术语“经取代”均指既定结构中的氢基经指定取代基的基团置换。在上文定义中和下文化合物和其实例的描述中描述特定取代基。除非另外指明,否则任选地经取代的基团可在所述基团的每一可取代位置处具有取代基,并且当任何既定结构中的多于一个位置可经多于一个选自指定基团的取代基取代时,取代基在每个位置处可为相同或不同的。如杂环烷基的环取代基可键结到另一个环,如环烷基,以形成螺双环系统,例如,两个环共享一个共享原子。如所属领域的普通技术人员将认识到,本发明所设想的取代基组合为那些促使形成稳定或化学可行的化合物的组合。
在整个本说明书中所揭示的化合物的改质或衍生物预期适用于本发明的方法和组合物。可通过所属领域的技术人员已知的任何方法制备衍生物并且可针对此类衍生物的所需特性分析其特性。在某些方面中,“衍生物”是指仍保持化合物在化学改质前的所需作用的化学改质化合物。
磺酸衍生光酸生成剂化合物
如下文将更详细地解释,根据本发明的磺酸衍生化合物可用作光酸生成剂。意外地,已发现本发明pag化合物的特征在于极佳溶解度和对电磁辐射的光反应性,尤其对波长在150nm到500nm范围内,优选300到450nm范围内,更优选350nm到440nm范围内,更优选365nm(i线)、405(h线)和436nm(g线)波长下的电磁辐射。
根据本发明的磺酸衍生化合物为由式(i)或式(ii)表示的n-羟基萘二甲酰亚胺磺酸酯衍生物:
其中r0选自由以下组成的群组
氢原子,
具有1到18的碳数的脂族基,其中一个或多个氢原子可经卤素原子取代,
具有2到18的碳数的脂族基,其包含至少一个选自由以下组成的群组的部分:-o-、-s-、-c(=o)-、-c(=o)-o-、-c(=o)-s-、-o-c(=o)-o-、-c(=o)-nh-、-o-c(=o)-nh-、-c(=o)-nra-、-o-c(=o)-nra-和-c(=o)-nrarb,其中ra和rb各自独立地为具有1到10的碳数的脂族基,其可相同或不同并且可连接形成脂环基,并且其中脂族基任选地包含至少一个卤素原子;并且
r选自由以下组成的群组
-ch3、-ch2f、-chf2或-cf3,
具有2到18的碳数的脂族基,其可经一个或多个卤素原子取代,
具有2到18的碳数的脂族基,其包含至少一个选自由以下组成的群组的部分:-o-、-s-、-c(=o)-、-c(=o)-o-、-c(=o)-s-、-o-c(=o)-o-、-c(=o)-nh-、-o-c(=o)-nh-、-c(=o)-nra-、-o-c(=o)-nra-和-c(=o)-nrarb,其中ra和rb如上文所定义,其中脂族基任选地包含至少一个卤素原子,
具有4到18的碳数的芳基或杂芳基,其中一个或多个氢原子可经卤素原子、脂族基、卤烷基、烷氧基、卤烷氧基、烷硫基、双烷氨基、酰氧基、酰硫基、酰氨基、烷氧羰基、烷基磺酰基、烷基亚磺酰基、脂环基、杂环基、芳基、烷芳基、氰基或硝基取代;和
具有4到18的碳数的芳烷基或杂芳烷基,其中所述芳基或杂芳基中的一个或多个氢原子可经卤素原子、脂族基、卤烷基、烷氧基、卤烷氧基、烷硫基、双烷氨基、酰氧基、酰硫基、酰氨基、烷氧羰基、烷基磺酰基、烷基亚磺酰基、脂环基、杂环基、芳基、烷芳基、氰基或硝基取代,
其限制条件为,如果r为-cf3,那么r0选自由以下组成的群组
氢原子;
具有2到18的碳数的脂族基,其包含至少一个选自由以下组成的群组的部分:-o-、-s-、-c(=o)-、-c(=o)-o-、-c(=o)-s-、-o-c(=o)-o-、-c(=o)-nh-、-o-c(=o)-nh-、-c(=o)-nra-、-o-c(=o)-nra-和-c(=o)-nrarb,其中ra和rb如上文所定义,其中脂族基任选地包含至少一个卤素原子;
-ch2ch(ch3)2、-ch2ch=chch3或-ch2ch2ch=ch2,
由式(a)表示的基团:
-ch2-r11(a),其中r11选自由具有4到18的碳数的脂族基组成的群组,
环丙烯基,和
由式(b)表示的基团:
在一些实施例中,式(i)和(ii)中的r0为具有1到18的碳数的脂族基,其中一个或多个氢原子可经卤素原子取代。优选实例包括甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、正戊基、正己基、2-乙基己基、正庚基、正辛基、正壬基和正癸基。在一个实施例中,式(i)和(ii)中的r0为具有1到18的碳数的脂族基,其中一个或多个氢原子可经卤素原子取代并且r为具有1到18的碳数的脂族基,其可经一个或多个卤素原子取代。优选地,r为具有1到6的碳数的脂族基,并且更优选为具有1到4的碳数的脂族基,其经至少一个氟原子取代。所述pag化合物的实例包括表1中的那些化合物:
表1
在其它实施例中,式(i)和(ii)中的r0为具有2到18的碳数的脂族基,其包含至少一个-c(=o)-o-部分。在一个实施例中,式(i)和(ii)中的r0为具有2到18的碳数的脂族基,其包含至少一个-c(=o)-o-部分,并且r为具有1到18的碳数的脂族基,其可经一个或多个卤素原子取代。优选地,r为具有1到6的碳数的脂族基,并且更优选为具有1到4的碳数的脂族基,其经至少一个氟原子取代。所述pag化合物的实例包括表2中的那些化合物:
表2:
在其它实施例中,式(i)和(ii)中的r0为具有1到18的碳数的脂族基,其中一个或多个氢原子可经卤素原子取代。在另一个实施例中,式(i)和(ii)中的r0为具有1到18的碳数的脂族基,其中一个或多个氢原子可经卤素原子取代,并且r为具有4到18的碳数的芳基或杂芳基,其中一个或多个氢原子可经卤素原子、脂族基或卤烷基取代。所述pag化合物的实例包括表3中的那些化合物:
表3:
在根据本发明的式(i)和(ii)的最优选实施例中,r为-cf3。在所述实施例中,应用上文所述的限制条件。优选实施例包括如下化合物:其中r0为由式(a)表示的基团:-ch2-r11(a),其中r11选自由具有4到18的碳数的脂族基组成的群组。实例包括正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基、2-乙基己基、正庚基、正辛基、正壬基和正癸基。所述优选pag化合物的实例包括表4中的那些化合物:
表4:
在式(i)和(ii)中r为-cf3的其它优选实施例中,r0为具有2到18的碳数的脂族基,其包含至少一个-c(=o)-o-或-o-c(=o)-o-部分,其中所述脂族基任选地包含至少一个卤素原子。所述优选pag化合物的实例包括表5中的那些化合物:
表5:
在式(i)和(ii)中r为-cf3的又其它优选实施例中,r0为具有2到18的碳数的脂族基,其包含至少一个-o-、-s-或-c(=o)-部分,其中所述脂族基任选地包含至少一个卤素原子。所述优选pag化合物的实例包括表6中的那些化合物:
表6:
在式(i)和(ii)中r为-cf3的又其它优选实施例中,r0为具有2到18的碳数的脂族基,其包含至少一个-c(=o)-s-、-c(=o)-nh-、-o-c(=o)-nh-、-c(=o)-nra-、-o-c(=o)-nra-或-c(=o)-nrarb部分,其中ra和rb如上文所定义,并且其中脂族基任选地包含至少一个卤素原子。所述优选pag化合物的实例包括表7中的那些化合物:
表7:
在式(i)和(ii)中r为-cf3的另一个优选实施例中,r0为由式(b)表示的基团:
在本发明的其它实施例中,式(i)和(ii)中的r和r0为具有1到18的碳数的脂族基。所述pag化合物的实例包括化合物t38和t40:
在本发明的其它实施例中,式(i)和(ii)中的r为具有2到18的碳数的脂族基,其包含至少一个-c(=o)-部分并且式(i)和(ii)中的r0为具有1到18的碳数的脂族基。所述pag化合物的实例包括化合物t39:
根据本发明的pag赋予光刻工艺高效率水平并且引起抗蚀剂组合物的曝光区域与未曝光区域之间的对比度和分辨率提高。对pag的量和通过uv照射所供应的能量加以选择以使得其足以达到所需聚缩合。
本发明的pag可适用于正作用型或负作用型化学增幅型光阻剂,即负作用型抗蚀剂组合物,其经历光酸促进的交联反应以使得抗蚀剂涂层的曝光区域相比于未曝光区域较不可溶于显影剂;和正作用型抗蚀剂组合物,其经历一种或多种组合物组分的酸不稳定基团的光酸促进的脱除保护基反应以使得抗蚀剂涂层的曝光区域相比于未曝光区域较可溶于水性显影剂。
用于本发明光阻剂的优选成像波长包括小于300nm的波长,例如248nm;和小于200nm的波长,例如193nm;以及euv,更优选在200nm到500nm范围内,优选在300nm到450nm范围内,甚至更优选在350nm到440nm范围内,最优选在365nm(i线)、405nm(h线)和436nm(g线)的波长下。
式(i)和(ii)化合物的制备(进一步详述于实例中)
对用于制造本发明的n-羟基萘二甲酰亚胺磺酸酯衍生化合物的方法不存在特定限制,并且可使用任何已知合成来制造式(i)和(ii)化合物。对用于制造本发明的n-羟基萘二甲酰亚胺磺酸酯衍生化合物的方法不存在特定限制,并且可使用任何熟知方法来合成这些化合物。流程1中说明我们所采用的合成三氟甲磺酸酯化合物的两种典型途径。可通过以3-溴酸酐为起始物质以类似方式合成在3位具有三键取代基的化合物。起始酸酐(4-溴-1,8-萘二甲酸酐和3-溴-1,8-萘二甲酸酐)可在市面上购得。
如流程1中所示,4-溴-1,8-萘二甲酸酐与末端炔烃之间的薗头偶合(sonagashiracoupling)得到含三键取代基的萘二甲酸酐。这些酸酐经由两种不同方法转化为最终n-羟基萘二甲酰亚胺磺酸酯衍生物。第一种方法涉及使用2.2当量的三氟甲磺酸酐进行一锅反应。第二种方法考虑分离n-羟基酰亚胺中间体并且仅需要1.2当量三氟甲磺酸酐。
流程1.
光阻剂组合物
本发明光阻剂组合物包含(i)至少一种选自式(i)和(ii)的光酸生成剂;(ii)至少一种可为碱可溶或不可溶的光阻聚合物或共聚物;(iii)有机溶剂;和任选的(iv)添加剂。
根据本发明的包含式(i)和(ii)的光酸生成剂的光阻剂组合物适用作多种应用中的光阻剂,尤其用于制造电子装置,包括平板显示器(在这一情况下,光阻剂可为经涂布的玻璃衬底或氧化铟锡层)和半导体装置(在这一情况下光阻剂可涂布于硅晶片衬底上)。可使用多种曝光辐射,包括用波长为200nm到500nm,优选在300nm到450nm范围内,更优选在350nm到440nm范围内,甚至更优选在365nm(i线)、436nm(g线)或405nm(h线)下的电磁辐射曝光,其中波长为365nm的电磁辐射尤其优选。
根据本发明的光阻剂组合物包含一种或多种光阻聚合物或共聚物作为组分(ii),其可以可溶于或不可溶于显影剂溶液中。根据本发明的光阻剂组合物可用于正型或负型组合物。在正型组合物的情况下,组分(ii)的溶解度在与从根据本发明的化合物释放的酸反应后提高。在这一情况下,具有酸不稳定基团的光阻聚合物或共聚物用作组分(ii),其不可溶于碱水溶液,但其在酸存在下以催化方式脱除保护基以使得其变得可溶于溶液中。在负型组合物的情况下,组分(ii)的溶解度在与从根据本发明的化合物释放的酸反应后降低。在这一情况下,光阻聚合物或共聚物用作组分(ii),其可溶于显影剂溶液中,但在酸存在下发生交联以使得其变得不可溶于碱水溶液中。因此,光阻聚合物或共聚物能够在酸存在下被赋予在显影剂溶液中的经改变的溶解度。显影剂溶液优选为水溶液,更优选为碱水溶液。
可用作正型组合物中的组分(ii)的光阻聚合物的实例包括(但不限于)芳香族聚合物,如用酸不稳定基团保护的羟基苯乙烯的均聚物或共聚物;丙烯酸酯,如聚(甲基)丙烯酸酯,其具有至少一个含有脂环侧基的单元,并且具有与聚合物主链和/或与脂环基、环烯聚合物、环烯顺丁烯二酸酐共聚物、环烯乙烯醚共聚物、硅氧烷侧接的酸不稳定基团;倍半硅氧烷、碳硅烷;和寡聚物,包括多面寡聚倍半硅氧烷、碳水化合物和其它笼型化合物。上述聚合物或寡聚物按需要用碱水溶液可溶性基团、酸不稳定基团、极性官能团和含硅基团适当官能化。
可用作本发明的正型组合物中的组分(ii)的共聚物的实例包括(但不限于)聚(对羟基苯乙烯)-甲基丙烯酸甲基金刚烷基酯(phs-madma)、聚(对羟基苯乙烯)-甲基丙烯酸2-乙基-2-金刚烷基酯(phs-eadma)、聚(对羟基苯乙烯)-甲基丙烯酸2-乙基-2-环戊酯(phs-ecpma)、聚(对羟基-苯乙烯)-甲基丙烯酸2-甲基-2-环戊酯(phs-mcpma)或phs-eve。
正型组合物中的至少一种组分(ii)优选为聚(羟基苯乙烯)-树脂,其中至少一部分羟基经保护基取代。优选保护基选自由叔丁氧基羰氧基、叔丁氧基、叔戊氧基羰氧基和缩醛基组成的群组。另外,适合作为组分ii)者为在ep1586570a1的段落[0068]到[0114]中描述为“含酸可解离基团的树脂”的所有聚合物和共聚物。ep1586570a1中关于这些树脂的揭示内容以引用的方式并入本文中,并且形成本发明揭示内容的一部分。
优选的负型组合物包含在曝露于酸后将固化、交联或硬化的材料的混合物。优选的负作用型组合物包含聚合物粘合剂(如酚类或非芳香族聚合物)作为组分(ii)、交联剂组分作为添加剂(iv)和根据本发明的光酸生成剂组分作为组分(i)。适用于所述负型光阻剂组合物的聚合物粘合剂和交联剂以及其用途已揭示于ep-a-0164248和us5,128,232中。用作组分(ii)的优选酚类聚合物包括酚醛清漆和聚(乙烯基苯酚)。酚醛清漆树脂为苯酚与醛的热塑性缩合产物。适合与醛(尤其甲醛)缩合形成酚醛清漆树脂的苯酚的实例包括苯酚、间甲酚、邻甲酚、对甲酚、2,4-二甲苯酚、2,5-二甲苯酚、3,4-二甲苯酚、3,5-二甲苯酚和百里香酚(thymol)。酸催化的缩合反应导致形成适合的酚醛清漆树脂,其分子量可从约500道尔顿(dalton)变化到100,000道尔顿。聚乙烯苯酚树脂为可通过相应单体在阳离子催化剂存在下嵌段聚合、乳液聚合或溶液聚合所形成的热塑性聚合物。适用于制造聚乙烯基苯酚树脂的乙烯基苯酚可例如通过水解市售香豆素(coumarin)或经取代的香豆素,随后使所得羟基肉桂酸脱羧基来制备。适用的乙烯苯酚还可通过相应羟基烷基苯酚的脱水或通过由经取代或未经取代的羟基苯甲醛与丙二酸反应所产生的羟基肉桂酸的脱羧基来制备。由所述乙烯基苯酚制备的优选聚乙烯基苯酚树脂的分子量范围为约2,000道尔顿到约60,000道尔顿。用作组分(iv)的优选交联剂包括基于胺的材料(包括三聚氰胺)、甘脲、基于苯并胍胺的材料和基于脲的材料。三聚氰胺-甲醛聚合物通常尤其适合。所述交联剂可在市面上购得,例如三聚氰胺聚合物、甘脲聚合物、基于脲的聚合物和苯并胍胺聚合物,如由氰特(cytec)以商标名cymeltm301、303、1170、1171、1172、1123和1125以及beetletm60、65和80所出售的那些聚合物。
根据本发明的组合物包含至少一种有机溶剂作为组分(iii)。有机溶剂可为任何能够溶解组分(ii)和组分(i)以产生均匀溶液的溶剂,并且可使用一种或多种选自已知用作常规化学增幅型抗蚀剂的溶剂的材料的溶剂。有机溶剂的特定实例包括酮,如丙酮、甲基乙基酮、环己酮、甲基异戊基酮和2-庚酮;多元醇和其衍生物,如乙二醇、乙二醇单乙酸酯、二乙二醇、二乙二醇单乙酸酯、丙二醇、丙二醇单乙酸酯、二丙二醇,或二丙二醇单乙酸酯的单甲醚、单乙醚、单丙醚、单丁醚或单苯醚;环醚,如二恶烷;和酯,如乳酸甲酯、乳酸乙酯(el)、乙酸甲酯、乙酸乙酯、乙酸丁酯、丙酮酸甲酯、丙酮酸乙酯、甲氧基丙酸甲酯和乙氧基丙酸乙酯。这些有机溶剂可单独使用,或作为含有两种或更多种不同溶剂的混合溶剂使用。尤其优选的有机溶剂(iii)选自由酮、醚和酯组成的群组。
另外,根据本发明的组合物还可任选地包含至少一种不同于组分(i)、(ii)和(iii)的添加剂。举例来说,其它任选的添加剂包括光化和对比染料、抗条纹剂、塑化剂、速度增进剂、敏化剂等。所述任选的添加剂通常将在光阻剂组合物中呈较小浓度,但其中填充剂和染料可呈相对较大浓度,如,呈抗蚀剂的干组分的总重量的5到30重量%的量。
根据本发明的光阻剂组合物中通常所采用的一种添加剂为碱性淬灭剂。碱性淬灭剂的目的为中和底层光阻层的表面区域中由达到预期作为光阻层的未曝光(暗)区域处的杂散光所生成的酸。这使得可通过控制未曝光区域中不合需要的脱除保护基反应来改善散焦区域中的聚焦深度和曝光宽容度。因此,可最小化或避免所形成抗蚀剂图案中的轮廓不规则性,例如颈缩和t型尖顶。
为实现碱性淬灭剂与底层光阻层的暗区中所生成的酸之间的有效相互作用,碱性淬灭剂应为非表面活性剂型。即,碱性淬灭剂不应为因例如相对于外涂层组合物的其它组分来说较低的表面自由能而迁移到外涂层的顶面的类型。在这一情况下,碱性淬灭剂明显不应在光阻层介面处与所生成的酸相互作用,从而防止酸脱除保护基。因此,碱性淬灭剂应为存在于外涂层/光阻层介面处的类型,无论其均匀分布于外涂层中或在介面处形成分级或分离层。此类分离层可通过选择具有相对于外涂层组合物的其它组分来说较高的表面自由能的碱性淬灭剂来实现。
适合碱性淬灭剂包括例如:直链和环状酰胺以及其衍生物,如n,n-双(2-羟基乙基)棕榈酰胺、n,n-二乙基乙酰胺、n1,n1,n3,n3-四丁基丙二酰胺、1-甲基氮杂环庚-2-酮、1-烯丙基氮杂环庚-2-酮和1,3-二羟基-2-(羟基甲基)丙-2-基氨基甲酸叔丁酯;芳香族胺,如吡啶和二-叔丁基吡啶;脂族胺,如三异丙醇胺、正叔丁基二乙醇胺、三(2-乙酰氧基-乙基)胺、2,2',2",2'"-(乙烷-1,2-二基双(氮烷三基))四乙醇和2-(二丁氨基)乙醇、2,2',2"-氮基三乙醇;环脂族胺,如1-(叔丁氧基羰基)-4-羟基哌啶、1-吡咯烷甲酸叔丁酯、2-乙基-1h-咪唑-1-甲酸叔丁酯、哌嗪-1,4-二甲酸二-叔丁酯和n(2-乙酰氧基-乙基)吗啉。在这些碱性淬灭剂中,1-(叔丁氧基羰基)-4-羟基哌啶和三异丙醇胺为优选的。尽管碱性淬灭剂的含量将取决于例如底层光阻层中光酸生成剂的含量,但以外涂层组合物的总固体计,其通常以0.1wt%到5wt%、优选0.5wt%到3wt%、更优选1wt%到3wt%的量存在。
另一种概念为使碱性部分连接到pag分子。在这一情况下,淬灭剂为pag的一部分并且极接近照射后所形成的酸。这些化合物对电磁辐射,尤其对波长在200nm到500nm范围内的电磁辐射,更尤其对波长为365nm(i线)的电磁辐射具有高敏感度,并且同时允许产生具有比由现有技术已知的含有淬灭剂作为添加剂的光阻剂组合物要高的分辨率的图案化结构。遵循这一概念的化合物为例如t26、t30和t31。
本发明抗蚀剂的树脂粘合剂组分通常以足以使得抗蚀剂的曝光涂层可用如碱性水溶液显影的量使用。更具体地说,树脂粘合剂宜占抗蚀剂总固体的50重量%到约90重量%。光敏性组分应以足以在抗蚀剂涂层中生成潜像的量存在。更具体地说,光敏性组分宜以抗蚀剂总固体的约1重量%到40重量%的量存在。通常,较少量的光敏性组分将适合化学增幅型抗蚀剂。
根据一优选实施例,根据本发明的组合物包含:
(i)0.05wt.%到15wt.%,优选0.1wt.%到12.5wt.%并且最优选1wt.%到10wt.%的至少一种式(i)或(ii)光酸生成剂化合物;
(ii)5wt.%到50wt.%,优选7.5wt.%到45wt.%并且最优选10wt.%到40wt.%的至少一种可为碱可溶或不可溶的光阻聚合物或共聚物;和
(iv)0wt.%到10wt.%、优选0.01wt.%到7.5wt.%并且最优选0.1wt.%到5wt.%的其它添加剂,其中组合物中的剩余部分为有机溶剂(iii)。
由于在本发明化合物中,充当在曝露于电磁辐射后所释放的酸基的淬灭剂的碱性官能团为光酸生成剂化合物的一部分,因此无需添加单独的碱性组分作为淬灭剂(在由现有技术已知的光阻剂组合物中其为必需的)。根据本发明组合物的一优选实施例,这一组合物优选包含小于5wt.%、更优选小于1wt.%、甚至更优选小于0.1wt.%,并且最优选0wt.%的不同于组分(i)到(iv)的碱性化合物,如氢氧化物、羧酸酯、胺、亚胺和酰胺。
本发明的光阻剂一般遵循已知程序制备,但改用本发明pag取代调配所述光阻剂时所用的先前光敏性化合物。举例来说,本发明抗蚀剂可通过将光阻剂组分溶解于以下适合溶剂中而制备为涂层组合物:例如二醇醚,如2-甲氧乙基醚(二乙二醇二甲醚)、乙二醇单甲醚、丙二醇单甲醚;乳酸酯,如乳酸乙酯或乳酸甲酯,其中乳酸乙酯为优选的;丙酸酯,尤其丙酸甲酯和丙酸乙酯;赛路苏酯(cellosolveester),如赛路苏乙酸甲酯;芳香族烃,如甲苯或二甲苯;或酮,如甲基乙基酮、环己酮和2-庚酮。光阻剂的固体含量通常在光阻剂组合物总重量的5重量%与35重量%之间变化。
可根据已知程序使用本发明光阻剂。尽管本发明光阻剂可以干膜涂覆,但其优选以液体涂层组合物的形式涂覆于衬底上,通过加热干燥以去除溶剂,优选直到涂层无粘性,经由光掩模曝光以激活辐射,任选地进行曝光后烘烤以产生或增大抗蚀剂涂层的曝光区域与非曝光区域之间的溶解度差异,并且随后优选用碱性显影剂水溶液显影以形成浮雕图像。上面经本发明抗蚀剂涂覆并且经适当加工的衬底可为任何用于涉及光阻剂的工艺中的衬底,如微电子晶片。举例来说,衬底可为硅、二氧化硅或铝-氧化铝微电子晶片。还可采用砷化镓、陶瓷、石英或铜衬底。还宜采用用于液晶显示器和其它平板显示器应用的衬底,例如玻璃衬底、经氧化铟锡涂布的衬底等。液体涂层抗蚀剂组合物可通过任何标准手段涂覆,如旋转、浸渍或滚涂。曝光能量应足以有效激活辐射敏感性系统的光敏性组分以在抗蚀剂涂层中产生图案化图像。适合的曝光能量通常在约1mj/cm2到300mj/cm2范围内。如上所述,优选的曝光波长包括小于200nm,如193nm。适合的曝光后烘烤温度为约50℃或大于50℃,更具体地说,约50℃到140℃。对于酸硬化的负作用型抗蚀剂,视需要可采用在约100℃到150℃的温度下显影后烘烤数分钟或更长时间以进一步固化显影后所形成的浮雕图像。显影和任何显影后固化之后,通过显影裸露的衬底表面随后可进行选择性加工,例如根据所属领域中已知的程序化学蚀刻或镀覆光阻剂的裸露的衬底区域。适合的蚀刻剂包括氢氟酸蚀刻溶液和等离子气体蚀刻,如氧气等离子蚀刻。
复合物
本发明提供一种用于制造复合物的工艺,所述复合物包含衬底和涂覆于所述衬底上而呈图案化结构的涂层,所述工艺包含以下步骤:
(a)在衬底表面上涂覆一层根据本发明的组合物并且至少部分去除有机溶剂(iii);
(b)将所述层的所选区域曝露于电磁辐射,由此于曝露于电磁辐射的区域中从化合物(i)释放出酸;
(c)任选地对所述层进行加热以在其中释放有酸的区域中赋予化合物(ii)于水溶液中的经改变的溶解度;和
(d)至少部分去除所述层。
在工艺步骤(a)中,在衬底表面上涂覆一层根据本发明的组合物,随后至少部分去除有机溶剂(iii)。
衬底可为任何尺寸和形状,并且优选为适用于光刻的那些衬底,如硅、二氧化硅、绝缘体上硅(silicon-on-insulator,soi)、应变硅(strainedsilicon)、砷化镓;经涂布的衬底,包括涂有氮化硅、氮氧化硅、氮化钛、氮化钽的那些衬底;超薄栅氧化物,如氧化铪;金属或经金属涂布的衬底,包括涂有钛、钽、铜、铝、钨、其合金的那些衬底;和其组合。优选地,本文中的衬底表面包括待图案化的临界尺寸层,包括例如一个或多个门级层或衬底上用于半导体制造的其它临界尺寸层。所述衬底优选地可包括硅、soi、应变硅和其它此类衬底材料,其形成为具有如20cm、30cm或更大直径的尺寸或适用于晶片制造生产的其它尺寸的圆形晶片。
将根据本发明的组合物涂覆于衬底上可通过任何适合方法完成,包括旋涂、喷涂、浸涂、刀片刮涂或其类似方法。涂覆所述层光阻剂优选通过使用涂布道旋涂光阻剂来完成,其中将光阻剂分配于旋转晶片上。在旋涂工艺期间,可以高达4,000rpm,优选500rpm到3,000rpm,并且更优选1,000rpm到2,500rpm的速度旋转晶片。旋转经涂布的晶片以去除有机溶剂(iii),并且在加热板上烘烤以从膜去除残留溶剂和自由体积,使其均匀致密。
在工艺步骤(b)中,使所选的层区域曝露于电磁辐射,由此于曝露于电磁辐射的区域中从化合物(i)释放出酸。如上所述,可使用各种曝光辐射,包括用波长为365nm(i线)、436nm(g线)或405nm(h线)的电磁辐射曝光,其中波长为365nm的电磁辐射为尤其优选的。
此类图案逐次曝光可使用如步进器的曝光工具进行,其中膜经由图案掩模受到照射并且因此图案逐次曝光。所述方法优选使用在能够有高分辨率的波长下产生活化辐射,包括极紫外线(euv)或电子束辐射的先进曝光工具。应了解,使用活化辐射进行曝光可分解包含于光阻层中的曝光区域中的本发明组分并且生成酸和分解副产物,并且所述酸随后实现聚合物化合物(ii)的化学变化(使酸敏性基团去阻断以生成碱可溶性基团,或者催化曝光区域中的交联反应)。所述曝光工具的分辨率可小于30nm。
在工艺步骤(c)中,可任选地对层进行加热以在其中释放有酸的区域中赋予化合物(ii)于水溶液中的经改变的溶解度。在这一所谓的“曝光后烘烤”中,产生或增大涂层的曝光区域与未曝光区域之间的溶解度差异。曝光后烘烤条件通常包括约50℃或大于50℃的温度,更具体地说在约50℃到约160℃范围内的温度,持续10秒到30分钟,优选30秒到200秒。根据本发明工艺的特定实施例,在工艺步骤(b)之后和(d)之前不进行热处理。
在工艺步骤(d)中,用水溶液,优选碱水溶液至少部分去除层。这一去除操作可通过用适合显影剂处理经曝光的光阻层来完成,所述适合显影剂能够选择性地去除膜的曝光部分(其中光阻剂为正型)或去除膜的未曝光部分(其中光阻剂为负型)。优选地,光阻剂为基于具有酸敏性(可脱除保护)基团的聚合物的正型光阻剂,并且显影剂优选为不含金属离子的四烷基氢氧化铵溶液。
根据本发明所制得的复合物的特征在于,其包含衬底和涂覆于衬底表面上而呈图案化结构的涂层,其中所述涂层包含根据本发明的化合物。
将式(i)和(ii)的光酸生成剂化合物用于官能团的光诱导聚合、光诱导交联、光诱导降解和光诱导转化也在本发明的范围内。本发明化合物尤其适用于保护性涂层、智能卡、3d快速原型设计或添加剂制造、牺牲性涂层、胶粘剂、抗反射涂层、全息图、检流和电镀掩模、离子植入掩模、抗蚀剂、化学增幅型抗蚀剂、光感测应用、印刷电路板(pcb)图案化、mems制造、平板显示器上的tft层图案化、柔性显示器上的tft层图案化、用于显示器的像素图案化、用于lcd的滤色片或黑色基质,或封装工艺中的半导体图案化,和半导体制造保护性涂层、智能卡、3d快速原型设计或添加剂制造、牺牲性涂层、胶粘剂、抗反射涂层、全息图、检流和电镀掩模、离子植入掩模、抗蚀剂、化学增幅型抗蚀剂、光感测应用上或滤色片中的tsv相关图案化。
以下实例预期说明以上发明内容,并且不应解释为使其范围变窄。所属领域的技术人员应易于认识到,所述实例表明许多其它的可以实践本发明的方式。应理解,可进行诸多变化和修改,同时仍在本发明的范围内。
实例
溶解度
溶解度为pag评估中的重要因素。高溶解度不仅使pag纯化变得容易并且还使pag能够在光阻剂和不同溶剂系统中以广泛范围的浓度使用。为测试pag的溶解度,缓慢添加溶剂直到pag完全溶解并且在澄清溶液中观察不到浑浊。表8列举一些代表性n-羟基萘二甲酰亚胺磺酸酯衍生物对比nit在20℃下在各种有机溶剂中的溶解度(w/w%)。所有本发明化合物在pgmea和环己酮中均展现出显著高于比较化合物a和b以及商用基准物(nit)的溶解度。应注意,化合物t2和t5在测试溶剂中展现出极高溶解度。这些结果表明,本发明中的n-羟基萘二甲酰亚胺磺酸酯衍生物可以高浓度用于感光性组合物中。由于pag的溶解度随温度改变而显著变化,因此高溶解度可改善感光性组合物的溶液稳定性,使得可允许组合物用于广泛范围的操作温度,而无需担心pag从组合物再结晶。
表8
*丙二醇单甲醚乙酸酯
**γ-丁内酯
光反应性
光阻剂组合物通常包含pag、聚合物、添加剂和溶剂。光阻剂组合物的性能主要取决于pag和聚合物组分的特性。为调配高性能光阻剂组合物,通常选择感光性较强的pag。pag的感光性通常与所生成的酸的强度和pag的光反应性直接相关。对于一系列产生相同潜酸的pag来说,其感光性仅与其光反应性相关。因此,评估pag的感光性可通过研究其光反应性来实现。光反应性越高,感光性越高。可通过在低曝光强度(以避免不生成所需酸的副反应)下使pag在其稀释溶液中光解来研究光反应性。照射后,pag浓度的变化可通过在最大吸收波长下测量pag的吸光度来测定。
在室温下在空气中于乙腈中进行pag的光解。酸指示剂染料四溴酚蓝的钠盐(tbpbna)(其在618nm下具有最大吸收度)购自奥德里奇(aldrich)(指示剂级别)并且按原样使用。在365nm下使用cole-parmer(科尔帕默)uv15w基准灯(ew-97605-50)对pag溶液(3×10-5m)进行照射。使用来自eit公司的uvpowerpuckii辐射计测量光强度。在赛默科技(thermoscientific)evolution201紫外可见分光亮度计上运作uv-vis光谱。
在乙腈中检查nit、比较化合物a和b、t1、t2、t3以及t5的光解。参看图1,在365nm下经uv灯照射后,t3的uv-vis光谱变化显示强度随能量曝光剂量(mj/cm2)增加而降低。照射后,在362nm(λmax)下的吸收带逐渐减小,表明光反应随能量曝光剂量增加而有所进展。假设光反应为一级,吸光度变化的自然对数与能量曝光剂量的图提供t3的光反应常数(即线性趋势线的斜率)(图2)。在相同照射条件下以类似方式测定其它化合物的光反应常数。将a、b、t1、t2、t3和t5的常数与标准化为1的nit常数进行比较,得到相对光反应性(表9)。根据本发明的pag(t1、t2和t3)的光反应性为nit的光反应性的8到10倍并且为比较化合物a的光反应性的3到4倍。含九氟丁磺酸基的pag(t1和t3)的光反应性比含三氟甲磺酸基的那些pag的光反应性强。通过观察酸指示剂tbpbna在618nm下的光谱变化来确认照射后酸的形成。
表9:溶解度与光反应性的对比
*=丙二醇单甲醚乙酸酯
**=n-羟基萘二甲酰亚胺三氟甲磺酸酯(nit)
抗蚀剂评估
参看表10,通过遵循这一通用程序制备四种不同的本发明光阻剂组合物:将50gphs-eve聚合物溶液(pgmea中的约30wt%聚合物含量;约35%经eve封端的oh基,mw=32,000,mw/mn=1.88)和50gpgmea预混合。向这一混合物中添加1.3mmolpag,并且使用0.0263g(20mol%pag)三乙胺作为淬灭剂。搅拌混合物直到固体完全溶解。随后将组合物储存于黑暗中以用于通过光刻的后续图案研究。
表10:组合物概述和曝光
图案化结构的制备
遵循这一通用程序通过光刻使用组合物1-4制备图案化结构。通过旋涂器(1000rpm,40s,型号ace-200)将各组合物涂布于经声波处理清洗的裸玻璃晶片上。在110℃下在加热板(lklab韩国(lklabkorea),型号pdlp-250,含sus盖板)上软烘烤涂层60秒并且随后通过经各种线-间隙(l/s)大小(5、6、7、8、9和10μm)图案化的光掩模,使用由材成工程公司(jaesungengineeringco.)(韩国)制造的含i线(365nm)led光源的cooluv-100掩模校准器,以不同曝光时间曝露于约100-280mj/cm2的i线照射。1-2分钟等待时间之后,通过在23℃下将晶片浸入2.38wt%氢氧化四甲铵(tmah)水溶液中1分钟来去除曝露于辐射的区域中的涂层,产生图案化结构。通过浸入去离子水中洗涤20秒并且通过温和地吹氮气进行干燥。
通过高分辨率显微镜仔细分析组合物1-4的所得图案化结构(图3)。通过逐步改变曝光时间并且在显影和干燥之后使用显微镜(来自艾姆斯科普公司(amscopeco.)的型号mu500,放大率:×500并且最小分辨率10μm)检查图案,来确定适当的曝光时间。当图案显示出与掩模相同的大小时,达到适当的曝光时间。在过度曝光的情况下,各线之间的间隙宽度宽于掩模的相应特征,并且在曝光不足的情况下,各线之间的间隙宽度小于掩模的相应特征。通过在显影和干燥之后检查起离或缺失特征来确定图案质量。++=无起离无缺失特征,+=无起离,一些小特征缺失,o=起离或所有小特征缺失。因此,根据本发明的化合物独特地展现出高溶解度和高敏感度(短曝光时间)。
uv-vis光谱
如图4中所示,本发明pag化合物在有机溶剂中具有极佳溶解度并且在汞灯的i线下具有强吸收。化合物t1和t2在362nm下展现吸收最大值,并且化合物t5在353nm下显示吸收最大值。其在汞灯的i线下的吸光度远远大于例如nit的吸光度,其为现有技术的商用pag性能基准。因此,相对于现有技术,本发明化合物展现出作为光刻中的pag的较高敏感度和较好性能。
pag化合物的制备
实例2、3、4、5、6、7、8和9描述根据本发明的磺酸衍生物的合成实例。
实例1:比较化合物b的合成
将4-溴-1,8-萘二甲酸酐(263g,0.9492mol)、pph3(19.92g,75.94mmol)、et3n(201.7g,1.99mol)和2lthf装入5l烧瓶内。在氮气下搅拌混合物1h。在氮气下向这一混合物中添加cui(5.42g,28.5mmol)和pd(pph3)2cl2(6.66g,9.492mmol)。将混合物加热到回流,并且在2.5h内逐滴添加含1-己炔(101.8g,1.0mol)的0.5lthf。添加后,将混合物保持回流14.5h。反应的tlc监测显示出少量未反应的酸酐。在0.5h内添加额外量的1-己炔(14.5g,142mmol),并且将混合物再保持回流2h。使混合物缓慢冷却到室温。添加20ml去离子水。过滤得到黄色固体,并且将溶剂从滤液中完全去除,得到深棕色固体。将两种固体合并并且随后溶解于1lch2cl2中。用1.5l去离子水洗涤溶液。分离并且旋转蒸发ch2cl2溶液,得到208g粗固体。从320mlacn再结晶,得到200g(产率:70%)呈淡黄色晶体的b-i1。应注意,b-i1不经进一步纯化即用于后续反应。mp:153-4℃。1hnmr(300mhz,cdcl3)δ:8.50(d,2h),8.25(d,1h),7.65(t,1h),7.58(d,1h),2.53(t,2h),1.62(p,2h),1.45(六重峰,2h),0.91(t,3h)。13cnmr(75.5mhz,cdcl3)δ:13.6,19.6,22.2,30.5,103.5,117.0,118.8,127.5,129.8,130.2,130.6,131.7,132.4,133.4,133.8,160.0,160.4。
将b-i1(100g,0.3593mol)、h2noh·hcl(25.49g,0.3593mol)和吡啶(369.49g,4.671mol)装入1l烧瓶内。将混合物加热到回流,持续4h。tlc监测反应显示b-i1消失。使用冰盐浴将反应混合物冷却到-13℃。向这一混合物中逐滴添加三氟甲磺酸酐(222.85g,0.7905mol),并且在添加期间将温度保持在10℃以下。在4h内完成添加。添加2l去离子水,并且在室温下搅拌所得混合物1h。过滤得到139g黄色固体。从100mlacn和1.2lmeoh再结晶,得到115g(产率:75%)呈淡黄色粉末的化合物b。mp:112-4℃。1hnmr(300mhz,cdcl3)δ:8.60(d,1h),8.55(d,1h),8.42(d,1h),7.72(m,2h),2.55(t,2h),1.65(p,2h),1.45(六重峰,2h),0.95(t,3h)。13cnmr(75.5mhz,cdcl3)δ:13.6,19.7,22.2,30.5,77.5,103.6,120.0,121.7,127.4,127.6,130.9,131.0,132.1,132.2,133.1,134.5,158.7,159.0。
实例2:化合物t1的合成
将b-i1(54.3g,0.195mol)、150mldmf和h2noh·hcl(13.84g,0.215mol)装入1l烧瓶内。向浆料混合物中逐滴添加48%koh溶液(12.04g,0.215mol),并且在添加期间将温度保持在25℃下。添加后,在室温下搅拌反应混合物4h。添加500ml去离子水。在室温下搅拌混合物2h。过滤并且用去离子水洗涤,得到黄色固体。在真空中在60℃下干燥固体隔夜,得到57g(产率:99%)羟基亚胺b-i2。应注意,b-i2不经进一步纯化即用于后续反应。mp:163-6℃。1hnmr(300mhz,dmso)δ:10.75(s,1h),8.42(m,2h),8.32(d,1h),7.82(t,1h),7.70(d,1h),2.58(t,2h),1.60(p,2h),1.45(六重峰,2h),0.95(t,3h)。13cnmr(75.5mhz,dmso)δ:13.4,18.8,21.5,30.0,77.4,101.5,121.4,122.7,126.0,127.5,127.8,130.0,130.5,130.97,131.00,131.8,160.2,160.5。
通过羟基亚胺b-i2与c4f9so2f反应,以30%产率合成九氟丁磺酸酯t1。mp:125-8℃。1hnmr(300mhz,cdcl3)δ:8.55(d,1h),8.50(d,1h),8.45(d,1h),7.73(m,2h),2.55(t,2h),1.65(p,2h),1.45(六重峰,2h),0.95(t,3h)。
实例3:化合物t2的合成
通过遵循与b-i1相同的程序以68%产率以类似方式合成酸酐中间体t2-i1,其中用1-辛炔替换1-己炔。应注意,t2-i1不经进一步纯化即用于后续反应。mp:100-1℃。1hnmr(300mhz,cdcl3)δ:8.65(d,1h),8.60(d,1h),8.45(d,1h),7.78(m,2h),2.54(t,2h),1.66(p,2h),1.46(p,2h),1.29(m,4h),0.85(t,3h)。
将t2-i1(36g,0.1626mol)、75mldmf和h2noh·hcl(13.58g,0.1952mol)装入1l烧瓶内。向浆料混合物中逐滴添加48%koh溶液(10.95g,0.1952mol),并且在添加期间将温度保持在25℃下。添加后,在室温下搅拌反应混合物4h。添加250ml去离子水。在室温下搅拌混合物2h。过滤并且用去离子水洗涤,得到黄色固体。在真空中在60℃下干燥固体隔夜,得到35g(产率:93%)羟基亚胺t2-i2。应注意,t2-i2不经进一步纯化即用于后续反应。mp:142-6℃。1hnmr(300mhz,cdcl3)δ:8.80(brs,1h),8.54(m,2h),8.42(d,1h),7.70(m,2h),2.54(t,2h),1.67(p,2h),1.48(p,2h),1.29(m,4h),0.85(t,3h)。
将t2-i2(25g,77.8mmol)、乙腈(100ml)和吡啶(9.23g,116.7mmol)装入500ml烧瓶内。将混合物冷却到0℃,并且随后在添加期间在5℃以下逐滴添加三氟甲磺酸酐(27.4g,197.2mmol)。添加后,将反应混合物升温到室温并且在室温下搅拌5h。将300ml去离子水添加到混合物中。过滤得到黄色固体。将固体溶解于100gch2cl2中,并且使溶液通过短硅胶垫。去除ch2cl2并且从异丙醇(200g)与乙腈(20g)的混合物再结晶,得到黄色固体,将其在真空下在50℃下干燥隔夜,得到15.9g(产率:45%)t2。mp:66-8℃。1hnmr(300mhz,cdcl3)δ:8.60(m,2h),8.44(d,1h),7.74(t,1h),2.55(d,2h),1.67(p,2h),1.48(p,2h),1.28(m,4h),0.83(t,3h)。13cnmr(75.5mhz,cdcl3)δ:14.0,20.0,22.6,28.4,28.7,31.3,77.5,103.7,120.0,121.7,127.3,127.5,130.8,130.9,132.1,132.2,133.1,134.5,158.7,159.0。
实例4:化合物t3的合成
通过羟基亚胺t2-i2与c4f9so2f反应,以42%产率合成九氟丁磺酸酯t3。mp:115-6℃。1hnmr(300mhz,cdcl3)δ:8.60(m,2h),8.42(d,1h),7.74(m,2h),2.52(t,2h),1.65(p,2h),1.45(m,2h),1.28(m,2h),0.95(t,3h)。13cnmr(75.5mhz,cdcl3)δ:14.0,20.0,22.5,28.4,28.7,31.3,77.5,103.6,120.0,121.7,127.3,127.5,130.8,130.9,132.0,132.2,133.1,134.4,158.7,158.9。
实例5:化合物t5的合成
通过遵循与b-i1相同的程序以28%产率以类似方式合成酸酐中间体t5-i1,其中用丁酸炔丙酯(易于在三乙胺存在下通过炔丙醇与丁酰氯的反应制备)替换1-己炔。应注意,t5-i1不经进一步纯化即用于后续反应。mp:135-7℃。1hnmr(300mhz,cdcl3)δ:8.63(m,2h),8.49(d,1h),7.84(m,2h),5.02(s,2h),2.36(t,2h),1.67(六重峰,2h),0.94(t,3h)。
将t5-i1(4.9g,15.2mmol)、10mldmf和h2noh·hcl(1.078g,16.72mmol)装入烧瓶内。向浆料混合物中逐滴添加48%koh溶液(0.938g,16.72mmol),并且在添加期间将温度保持在25℃下。添加后,将反应混合物在室温下搅拌隔夜。添加30ml去离子水,并且在室温下搅拌混合物1h。过滤并且用去离子水洗涤,得到黄色固体。固体从ch2cl2与ea的混合物再结晶,得到3.5g(产率:68%)羟基亚胺t5-i2。mp:180-2℃。1hnmr(300mhz,cdcl3)δ:8.60(m,2h),8.50(d,1h),7.82(m,2h),5.02(s,2h),2.36(t,2h),1.67(六重峰,2h),0.94(t,3h)。
将t5-i2(3.0g,8.89mmol)、乙腈(15ml)和吡啶(1.06g,13.34mmol)装入烧瓶内。将混合物冷却到0℃,并且随后在添加期间在5℃以下逐滴添加三氟甲磺酸酐(3.13g,11.11mmol)。添加后,使反应混合物升温到室温并且搅拌2h。将100ml去离子水添加到混合物中。过滤得到黄色固体。将固体溶解于10gch2cl2中,并且使溶液通过短硅胶垫。去除ch2cl2并且从异丙醇(20g)与去离子水(2g)的混合物再结晶,得到黄色固体,将其在真空下在50℃下干燥隔夜,得到3.2g(产率:76%)t5。mp:87-9℃。1hnmr(300mhz,cdcl3)δ:8.61(m,2h),8.50(d,1h),7.83(m,2h),5.01(s,2h),2.36(t,2h),1.67(六重峰,2h),0.93(t,3h)。13cnmr(75.5mhz,cdcl3)δ:13.6,18.4,35.9,52.2,82.3,94.7,120.8,121.3,121.8,127.2,128.1,131.5,131.8,132.1,133.3,134.2,158.5,158.8,172.9。
实例6:化合物t35的合成
通过遵循与b-i1相同的程序以64%产率以类似方式合成酸酐中间体t35-i1,其中用丁酸炔丙酯(易于在三乙胺存在下通过炔丙醇与乙酰氯的反应制备)替换1-己炔。应注意,t35-i1不经进一步纯化即用于后续反应。mp:173-6℃。1hnmr(300mhz,cdcl3)δ:8.60(m,2h),8.46(d,1h),7.82(m,2h),5.01(s,2h),2.13(s,3h)。
将t35-i1(10g,34.0mmol)、15mldmf和h2noh·hcl(2.60g,37.4mmol)装入烧瓶内。向浆料混合物中逐滴添加48%koh溶液(2.10g,37.4mmol),并且在添加期间将温度保持在25℃下。添加后,将反应混合物在室温下搅拌隔夜。添加30ml去离子水。在室温下搅拌混合物1h。过滤并且用去离子水洗涤,得到固体,将其在真空中在60℃下干燥,得到9.0g(产率:86%)羟基亚胺t35-i2。应注意,t35-i1不经进一步纯化即用于后续反应。
将t35-i2(8.7g,28.1mmol)、乙腈(50ml)和吡啶(3.33g,42.1mmol)装入烧瓶内。将混合物冷却到0℃,并且随后在添加期间在5℃以下逐滴添加三氟甲磺酸酐(9.9g,35.1mmol)。添加后,将反应混合物升温到室温并且在室温下搅拌2h。将100ml去离子水添加到混合物中。过滤得到黄色固体。将固体溶解于10gch2cl2中,并且使溶液通过短硅胶垫。去除ch2cl2并且从异丙醇(50ml)与acn(25ml)的混合物再结晶,得到黄色固体,将其在真空下在50℃下干燥隔夜,得到8g(产率:64%)t35。mp:157-9℃。1hnmr(300mhz,cdcl3)δ:8.61(m,2h),8.50(d,1h),7.83(m,2h),5.01(s,2h),2.13(s,3h)。13cnmr(75.5mhz,cdcl3)δ:20.7,52.4,82.4,94.4,121.4,121.8,127.3,128.1,128.5,131.6,131.8,132.1,133.4,134.2,158.5,158.8,172.9。
实例7:化合物t38的合成
将b-i2(15g,51.1mmol)、60mlacn和et3n(5.7g,56.2mmol)装入1l烧瓶内。将混合物冷却到0℃,并且逐滴添加ch3so2cl(6.44g,56.2mmol)。添加后,将反应混合物升温到室温并且在室温下搅拌3h。添加200ml去离子水,并且在室温下搅拌混合物1h。过滤并且用去离子水洗涤,得到18.6g黄色固体。固体从ea再结晶,得到16.7g(产率:88%)t38。mp:121-2℃。1hnmr(300mhz,cdcl3)δ:8.60(m,2h),8.45(d,1h),7.72(m,2h),3.52(s,3h),2.55(t,2h),1.62(p,2h),1.50(m,2h),0.95(t,3h)。
实例8:化合物t40的合成
将b-i2(20g,68.2mmol)、80mlacn和et3n(7.6g,75mmol)装入1l烧瓶内。将混合物冷却到0℃,并且逐滴添加buso2cl(11.74g,75mmol)。添加后,将反应混合物升温到室温并且在室温下搅拌3h。添加200ml去离子水,并且在室温下搅拌混合物1h。过滤并且用去离子水洗涤,得到黄色固体。固体从acn再结晶,得到25.6g(产率:91%)t40。mp:110-1℃。1hnmr(300mhz,cdcl3)δ:8.45(m,2h),8.35(d,1h),7.62(m,2h),3.62(t,2h),2.52(t,2h),2.02(m,2h),1.62(m,2h),1.45(m,4h),0.95(m,6h)。
实例9:化合物t39的合成
将b-i2(15g,51.1mmol)、80mlacn和et3n(5.7g,56.2mmol)装入1l烧瓶内。将混合物冷却到0℃,并且逐滴添加樟脑磺酰氯(14.1g,56.2mmol)。添加后,将反应混合物升温到室温并且在室温下搅拌3h。添加200ml去离子水,并且在室温下搅拌混合物1h。过滤并且用去离子水洗涤,得到黄色固体。固体从ea再结晶,得到18g(产率:70%)t39。mp:124-6℃。1hnmr(300mhz,cdcl3)δ:8.60(m,2h),8.50(d,1h),7.65(m,2h),4.15(d,1h),3.85(d,1h),2.55(t,2h),2.40(m,2h),1.30-2.10(brm,9h),1.12(s,3h),0.95(m,6h)。
实例10:比较化合物a的合成
通过遵循与b-i1相同的程序以75%产率以类似方式合成酸酐中间体a-i1,其中用苯乙炔替换1-己炔。应注意,a-i1不经进一步纯化即用于后续反应。
将a-i1(81g,271.5mmol)、250mldmf和h2noh·hcl(18.4g,285.1mmol)装入1l烧瓶内。向浆料混合物中逐滴添加48%koh溶液(16.0g,285.1mmol),并且在添加期间将温度保持在25℃下。添加后,在室温下搅拌反应混合物4h。添加250ml去离子水。在室温下搅拌混合物2h。过滤并且用去离子水洗涤,得到黄色固体。在真空中在60℃下干燥固体隔夜,得到80g(产率:94%)羟基亚胺a-i2。应注意,a-i2不经进一步纯化即用于后续反应。mp:194-9℃。
将a-i2(55g,175.5mmol)、乙腈(200ml)和吡啶(23.6g,298.4mmol)装入500ml烧瓶内。将混合物冷却到0℃,并且随后在添加期间在5℃以下逐滴添加三氟甲磺酸酐(74.3g,263.3mmol)。添加后,将反应混合物升温到室温并且在室温下搅拌隔夜。将混合物加热到回流持续30分钟并且冷却到室温。将200ml去离子水添加到混合物中并且在室温下搅拌10分钟。过滤得到黄色固体,将其溶解于1lch2cl2中,并且使溶液通过短硅胶垫。对溶液进行旋转蒸发直到剩余100gch2cl2。过滤得到黄色固体,将其在真空下在50℃下干燥隔夜,得到46g(产率:59%)a。mp:193-5℃。1hnmr(300mhz,dmso)δ:8.94(d,1h),8.70(d,1h),8.63(d,1h),8.18(d,1h),8.10(dd,1h)7.82(m,2h),7.52(m,3h)。
尽管上文已参考某些特定实施例和实例加以说明和描述,然而本发明并不打算限于所展示的细节。确切来说,可在权利要求书的等效物的范畴与范围内并且在不偏离本发明精神的情况下在细节方面作出各种修改。举例来说,明确预期这一文件中所有宽泛地叙述的范围在其范围内包括所有属于较宽范围内的较窄范围。另外,一个实施例的特征可并入另一个实施例中。
- 还没有人留言评论。精彩留言会获得点赞!