形成抗蚀剂下层膜的组合物及使用该组合物的抗蚀剂图案的形成方法与流程
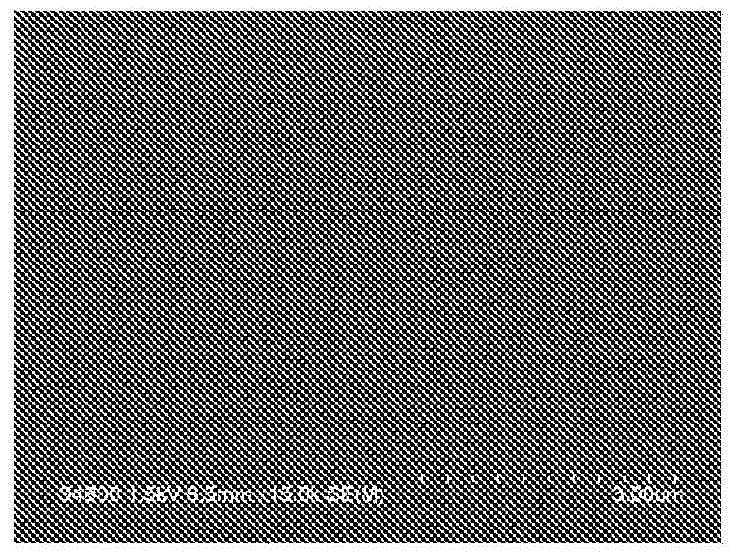
本发明涉及即使在形成薄的膜厚(例如20nm以下)的抗蚀剂下层膜的情况下,也可以形成没有缺陷的均匀的膜的形成光刻用抗蚀剂下层膜的组合物,以及使用该形成抗蚀剂下层膜的组合物的抗蚀剂图案形成方法。
背景技术:
一直以来,在半导体装置的制造中,利用使用抗蚀剂组合物的光刻进行微细加工。上述微细加工为在硅晶片等半导体基板上形成光致抗蚀剂组合物的薄膜,在其上隔着描绘有器件的图案的掩模图案来照射紫外线等活性光线,进行显影,将所得的光致抗蚀剂图案作为保护膜对基板进行蚀刻处理,从而在基板表面形成对应于上述图案的微细凹凸的加工法。近年来,半导体器件的高集成度化进展,所使用的活性光线也从i射线(波长365nm)、KrF准分子激光(波长248nm)向ArF准分子激光(波长193nm)的短波长化。与此相伴,活性光线从半导体基板的漫反射、驻波的影响成为大问题。因此为了解决该问题,广泛研究了在抗蚀剂与半导体基板之间设置防反射膜(BottomAnti-ReflectiveCoating:BARC)的方法。该防反射膜也被称为抗蚀剂下层膜。作为这样的防反射膜,从其使用的容易性等出发,对由具有吸光部位的聚合物等形成的有机防反射膜进行了大量的研究。专利文献1~专利文献3中公开了,在不与上层所形成的光致抗蚀剂膜混合,使用ArF准分子激光进行曝光的情况下,可以获得所期望的光学参数(k值、n值)并且可以获得所期望的干蚀刻速度的抗蚀剂下层膜(防反射膜)。另一方面,作为进一步的微细加工技术的、采用EUV(远紫外线的简称,波长13.5nm)曝光的光刻,虽然没有从基板的反射,但伴随图案微细化,抗蚀剂图案侧壁的粗糙成为问题,因此关于用于形成矩形性高的抗蚀剂图案形状的抗蚀剂下层膜进行了大量的研究。作为形成EUV、X射线、电子束等高能量射线曝光用抗蚀剂下层膜的材料,公开了释气发生减少了的形成抗蚀剂下层膜的组合物(专利文献4)。现有技术文献专利文献专利文献1:国际公开第2005/098542号小册子专利文献2:国际公开第2009/096340号小册子专利文献3:国际公开第2009/104685号小册子专利文献4:国际公开第2010/061774号小册子
技术实现要素:
发明所要解决的课题作为抗蚀剂下层膜所要求的特性,可举出例如,不发生与上层所形成的抗蚀剂膜的混合(在抗蚀剂溶剂中不溶),具有比抗蚀剂膜大的干蚀刻速度。在伴随EUV曝光的光刻的情况下,所形成的图案线宽成为32nm以下,EUV曝光用的抗蚀剂下层膜与以往相比膜厚形成得薄而使用。在形成这样的薄膜时,由于基板表面、所使用的聚合物等的影响,针孔、凝集等易于发生,形成没有缺陷的均匀的膜是困难的。本发明通过解决上述问题,目的在于可以获得可以形成所期望的抗蚀剂图案的用于形成抗蚀剂下层膜的组合物。用于解决课题的手段本发明的第1方式涉及一种形成光刻用抗蚀剂下层膜的组合物,其包含:在聚合物链的末端具有下述式(1)所示结构的聚合物、交联剂、用于促进交联反应的化合物以及有机溶剂。(式中,R1、R2以及R3各自独立地表示氢原子、碳原子数1~13的直链状或支链状的烃基、或羟基,上述R1、R2以及R3中的至少一个为上述烃基,m以及n各自独立地表示0或1,在n表示1时上述聚合物的主链与亚甲基结合,在n表示0时上述聚合物的主链与由-O-表示的基团结合。)上述式(1)中,在m表示0时,不存在羰基,由-O-表示的基团即醚键直接与苯环结合。本发明的第2方式涉及一种抗蚀剂图案的形成方法,其包括下述工序:将本发明的形成光刻用抗蚀剂下层膜的组合物涂布在半导体基板上并进行烘烤,从而形成厚度1nm~20nm的抗蚀剂下层膜的工序;在上述抗蚀剂下层膜上形成抗蚀剂膜的工序;通过选自KrF准分子激光、ArF准分子激光、远紫外线(EUV)以及电子束中的放射线对被上述抗蚀剂下层膜和上述抗蚀剂膜被覆了的半导体基板进行曝光的工序;以及在曝光后通过碱性显影液进行显影的工序。发明的效果本发明的形成光刻用抗蚀剂下层膜的组合物的特征在于,该形成抗蚀剂下层膜的组合物所包含的聚合物的末端被上述式(1)所示结构带帽(capping),是含有这样的聚合物、交联剂、用于促进交联反应的化合物以及有机溶剂的组合物。通过成为这样的构成,则涂布性能提高,可以形成没有缺陷的均匀的抗蚀剂下层膜。附图说明图1为使用由实施例1调制的组合物而形成的抗蚀剂下层膜的FE-SEM像。图2为使用由实施例2调制的组合物而形成的抗蚀剂下层膜的FE-SEM像。图3为使用由实施例3调制的组合物而形成的抗蚀剂下层膜的FE-SEM像。图4为使用由实施例4调制的组合物而形成的抗蚀剂下层膜的FE-SEM像。图5为使用由比较例1调制的组合物而形成的抗蚀剂下层膜的FE-SEM像。图6为使用由比较例2调制的组合物而形成的抗蚀剂下层膜的FE-SEM像。具体实施方式[聚合物]本发明的形成光刻用抗蚀剂下层膜的组合物中所包含的聚合物在聚合物链的末端具有上述式(1)所示结构。该式(1)中,R1、R2以及R3中的至少1个为碳原子数1~13的直链状或支链状的烃基。作为该烃基,从提高聚合物的溶解性和组合物的涂布性方面出发,优选为烷基,例如,叔丁基、异丙基、甲基,其中特别优选为叔丁基和异丙基。而且,上述聚合物具有例如下述式(2)和式(3)所示的结构单元。(式中,Q1和Q2各自独立地表示具有碳原子数1~13的直链状或支链状的烃基的二价有机基团、具有脂环式烃基的二价有机基团、具有芳香环或包含1~3个氮原子的杂环的二价有机基团,上述烃基、上述脂环式烃基、上述芳香环和上述杂环可以具有至少1个取代基。)上述式(2)所示的结构单元由例如下述式(2’)表示。(式中,Q3表示碳原子数1~13的直链状或支链状的烃基、脂环式烃基或芳香环,上述烃基、上述脂环式烃基和上述芳香环可以具有至少1个取代基,2个v各自独立地表示0或1。)上述烃基、上述脂环式烃基和上述芳香环可以具有至少1个甲基、羟基或氟基作为取代基。此外,上述式(3)所示的结构单元由例如下述式(3’)表示。(式中,Q4表示碳原子数1~13的直链状或支链状的烃基、脂环式烃基或芳香环,上述烃基、上述脂环式烃基和上述芳香环可以具有至少1个取代基,上述烃基在主链中可以具有1个或2个硫原子,也可以具有双键,2个w各自独立地表示0或1。)作为上述直链状或支链状的烃基的取代基,可举出例如,羟基、氟基。作为上述脂环式烃基、上述芳香环和上述杂环的取代基,可举出例如,甲基、乙基、叔丁基、烯丙基、羟基、氟基。作为该脂环式烃基,可举出例如,环亚丁基、环亚戊基、环亚己基。作为上述芳香环,可举出例如,苯环、萘环、蒽环。作为上述杂环,可举出例如,三嗪三酮环、嘧啶三酮环、咪唑烷二酮环、咪唑烷酮环、吡啶酮环。作为形成上述式(1)所示结构的单体,可举出例如,下述式(1-1)~式(1-9)所示的化合物。作为形成上述式(2)所示的结构单元的单体,可举出例如,下述式(2-1)~式(2-16)所示的具有2个环氧基的化合物。作为形成上述式(3)所示的结构单元的单体,可举出例如,下述式(3-1)~式(3-10)所示的化合物。本发明的形成光刻用抗蚀剂下层膜的组合物中所包含的聚合物例如由下述式(4)表示。(式中,R1、R2以及R3与上述式(1)含义相同,2个m和2个n各自独立地表示0或1,X表示具有上述式(2)和式(3)所示的结构单元的聚合物链。)上述式(4)表示通过上述式(1)所示结构,上述聚合物链的末端被带帽。关于形成为了获得上述式(4)所示的聚合物而必要的上述式(1)所示结构的单体,如果将形成式(2)和式(3)所示的结构单元的单体的总计设为100质量%,则为例如1质量%~30质量%(单体的加入比换算),优选为2质量%~20质量%。本发明的形成光刻用抗蚀剂下层膜的组合物中包含的聚合物可以为无规共聚物、嵌段共聚物、交替共聚物、接枝共聚物中的任一种。作为聚合物的聚合方法,能够为溶液聚合、悬浮聚合、乳液聚合、本体聚合等各种方法,可以使用适宜聚合催化剂等。作为聚合方法的一例,可以在有机溶剂中,在形成上述式(2)所示的结构单元的单体和形成式(3)所示的结构单元的单体中,添加形成上述式(1)所示结构的单体和聚合催化剂进行加热聚合,从而合成。这里所使用的有机溶剂可以从后述的作为本发明的形成光刻用抗蚀剂下层膜的组合物所包含的有机溶剂而优选的例示中适宜选择。作为该聚合催化剂,可举出例如,苄基三乙基氯化铵、乙基三苯基溴化,可以加热至例如50℃~160℃,优选为70℃~130℃来进行聚合。作为反应时间,为例如1小时~50小时,优选为2小时~12小时。上述聚合物的重均分子量为例如1000~100000,优选为1000~10000。如果该重均分子量的值过高,则本发明的形成光刻用抗蚀剂下层膜的组合物的涂布性恶化。如果将本发明的形成光刻用抗蚀剂下层膜的组合物设为100质量%,则该组合物中所包含的上述聚合物为例如0.01质量%~3质量%,优选为0.1质量%~2质量%。另外,重均分子量为通过凝胶渗透色谱(GPC),使用聚苯乙烯作为标准试样而得的值。[交联剂]本发明的形成光刻用抗蚀剂下层膜的组合物还包含交联剂。作为该交联剂,没有特别限制,优选使用具有至少二个交联形成取代基(例如,羟甲基、甲氧基甲基、丁氧基甲基)的含氮化合物。作为上述交联剂,可举出例如,六甲氧基甲基三聚氰胺、四甲氧基甲基苯胍胺、1,3,4,6-四(甲氧基甲基)甘脲、1,3,4,6-四(丁氧基甲基)甘脲、1,3,4,6-四(羟基甲基)甘脲、1,3-双(羟基甲基)脲、1,1,3,3-四(丁氧基甲基)脲、1,1,3,3-四(甲氧基甲基)脲。关于本发明的形成光刻用抗蚀剂下层膜的组合物中所包含的上述交联剂,如果将该组合物中的聚合物设为100质量%,则为例如1质量%~100质量%,优选为10质量%~50质量%。这些交联剂有时由于自缩合而发生交联反应,但可以与上述聚合物,特别是作为与交联剂反应而形成交联的结构单元的式(2)和式(3)所示的结构单元中的交联官能团(羟基)发生交联反应。[用于促进交联反应的化合物]为了促进交联反应,本发明的形成光刻用抗蚀剂下层膜的组合物还包含用于促进交联反应的化合物。作为这样的化合物,可以使用例如,对甲苯磺酸、三氟甲磺酸、吡啶-对甲苯磺酸盐、水杨酸、樟脑磺酸、5-磺基水杨酸、4-氯苯磺酸、4-羟基苯磺酸、苯二磺酸、1-萘磺酸、柠檬酸、苯甲酸、羟基苯甲酸等磺酸化合物和羧酸化合物。这些用于促进交联反应的化合物可以仅使用一种,此外,也可以二种以上组合使用。关于本发明的形成光刻用抗蚀剂下层膜的组合物中所包含的上述用于促进交联反应的化合物,如果将该组合物中的聚合物设为100质量%,则为例如0.1质量%~25质量%,优选为1质量%~10质量%。[有机溶剂]本发明的形成光刻用抗蚀剂下层膜的组合物还包含有机溶剂。作为在本发明中所使用的有机溶剂,只要可以溶解上述聚合物,就没有特别限制,可以使用例如,乙二醇单甲基醚、乙二醇单乙基醚、甲基溶纤剂乙酸酯、乙基溶纤剂乙酸酯、二甘醇单甲基醚、二甘醇单乙基醚、丙二醇、丙二醇单甲基醚、丙二醇单丙基醚、1-乙氧基-2-丙醇、丙二醇单甲基醚乙酸酯、丙二醇丙基醚乙酸酯、1-甲氧基-2-丁醇、2-甲氧基-1-丁醇、3-甲氧基-3-甲基丁醇、3-甲氧基-1-丁醇、甲苯、二甲苯、甲基乙基酮、环戊酮、环己酮、γ-丁内酯、N-甲基-2-吡咯烷酮、2-羟基异丁酸甲酯、2-羟基丙酸乙酯、2-羟基-2-甲基丙酸乙酯、乙氧基乙酸乙酯、羟基乙酸乙酯、2-羟基-3-甲基丁酸甲酯、3-甲氧基丙酸甲酯、3-甲氧基丙酸乙酯、3-乙氧基丙酸乙酯、3-乙氧基丙酸甲酯、丙酮酸甲酯、丙酮酸乙酯、乙酸乙酯、乙酸丁酯、乳酸乙酯、乳酸丁酯。这些有机溶剂可以单独使用,或2种以上组合使用。上述有机溶剂中,优选为丙二醇单甲基醚、丙二醇单甲基醚乙酸酯、1-乙氧基-2-丙醇、乳酸乙酯、乳酸丁酯和环己酮。关于本发明的形成光刻用抗蚀剂下层膜的组合物中所包含的上述有机溶剂,如果将该组合物设为100质量%,则为例如90质量%~99.99质量%,优选为98质量%~99.9质量%。[产酸剂]本发明的形成光刻用抗蚀剂下层膜的组合物还可以包含产酸剂。作为这样的产酸剂,可举出例如,双(4-羟基苯基)砜。在本发明的形成光刻用抗蚀剂下层膜的组合物包含上述产酸剂的情况下,如果将该组合物中的聚合物设为100质量%,则包含例如0.1质量%~5质量%,优选为0.2质量%~3质量%。[其它添加剂]本发明的形成光刻用抗蚀剂下层膜的组合物中,只要不损害本发明的效果,可以根据需要进一步包含表面活性剂等各种添加剂。表面活性剂为用于提高该组合物对基板的涂布性的添加物。可以使用非离子系表面活性剂、氟系表面活性剂那样的公知的表面活性剂。作为上述表面活性剂的具体例,可举出例如,聚氧乙烯月桂基醚、聚氧乙烯硬脂基醚、聚氧乙烯鲸蜡基醚、聚氧乙烯油基醚等聚氧乙烯烷基醚类、聚氧乙烯辛基苯酚醚、聚氧乙烯壬基苯酚醚等聚氧乙烯烷基芳基醚类、聚氧乙烯-聚氧丙烯嵌段共聚物类、失水山梨糖醇单月桂酸酯、失水山梨糖醇单棕榈酸酯、失水山梨糖醇单硬脂酸酯、失水山梨糖醇单油酸酯、失水山梨糖醇三油酸酯、失水山梨糖醇三硬脂酸酯等失水山梨糖醇脂肪酸酯类、聚氧乙烯失水山梨糖醇单月桂酸酯、聚氧乙烯失水山梨糖醇单棕榈酸酯、聚氧乙烯失水山梨糖醇单硬脂酸酯、聚氧乙烯失水山梨糖醇三油酸酯、聚氧乙烯失水山梨糖醇三硬脂酸酯等聚氧乙烯失水山梨糖醇脂肪酸酯类等非离子系表面活性剂、エフトップ〔注册商标〕EF301、エフトップEF303、エフトップEF352〔三菱マテリアル电子化成(株)(旧(株)ジェムコ)制〕、メガファック〔注册商标〕F171、メガファックF173、メガファックR30(DIC(株)制)、フロラードFC430、フロラードFC431(住友スリーエム(株)制)、アサヒガード〔注册商标〕AG710、サーフロン〔注册商标〕S-382、サーフロンSC101、サーフロンSC102、サーフロンSC103、サーフロンSC104、サーフロンSC105、サーフロンSC106(旭硝子(株)制)等氟系表面活性剂、有机硅氧烷聚合物KP341(信越化学工业(株)制)。这些表面活性剂可以单独添加,此外也可以2种以上组合添加。在本发明的形成光刻用抗蚀剂下层膜的组合物包含上述表面活性剂的情况下,如果将该组合物中的聚合物设为100质量%,则包含例如0.1质量%~5质量%,优选为0.2质量%~3质量%。接下来对本发明的抗蚀剂图案形成方法进行说明。首先,在精密集成电路元件的制造中所使用的基板〔例如,被氧化硅膜、氮化硅膜或氧化氮化硅膜被覆了的硅晶片等半导体基板、氮化硅基板、石英基板、玻璃基板(包含无碱玻璃、低碱玻璃、结晶玻璃)、形成有ITO膜的玻璃基板〕上通过旋涂器、涂布机等适当的涂布方法来涂布本发明的形成光刻用抗蚀剂下层膜的组合物,然后,使用电热板等加热手段进行烘烤,进行固化来制作抗蚀剂下层膜。作为在涂布后进行烘烤的条件,从例如烘烤温度80℃~250℃、烘烤时间0.3分钟~60分钟的范围适宜选择,优选为150℃~250℃、0.5分钟~5分钟。通过在这样的条件下进行烘烤,则聚合物的结构单元中的羟基等交联部位与交联剂反应而形成交联结构。特别是通过使本发明的形成光刻用抗蚀剂下层膜的组合物中所包含的聚合物进行交联,从而可以提高交联聚合物的交联密度。此外,作为抗蚀剂下层膜的膜厚,为例如0.001μm(1nm)~0.1μm,优选为0.001μm~0.02μm,进一步优选为0.003μm~0.01μm。接下来,在所制作的抗蚀剂下层膜上形成抗蚀剂膜。抗蚀剂膜的形成可以通过一般的方法来进行,即,通过将抗蚀剂溶液在抗蚀剂下层膜上涂布和烘烤来进行。作为被涂布的抗蚀剂溶液,只要对例如KrF准分子激光、ArF准分子激光、EUV、电子束感光,就没有特别限定,负型、正型都可以使用。作为能够使用的抗蚀剂溶液,可举出例如,住友化学(株)制;商品名PAR710,同PAR855,JSR(株)社制;商品名AR2772JN,信越化学工业(株)制;商品名SEPR430,ダウケミカル社(旧ローム·アンド·ハース·エレクトロニック·マテリアルズ社)制;商品名APEX-X。接着,对于抗蚀剂下层膜的上层所形成的抗蚀剂膜,通过规定的掩模(中间掩模)进行曝光。曝光时,可以使用例如,KrF准分子激光、ArF准分子激光、EUV。但是,在电子束曝光的情况下,掩模(中间掩模)不是必要的。此外曝光后,还可以根据需要进行曝光后加热(PEB:PostExposureBake)。作为曝光后加热的条件,从加热温度80℃~150℃、加热时间0.3分钟~60分钟的范围适宜选择。通过在曝光后进行显影、冲洗和干燥,来获得良好的抗蚀剂图案。作为抗蚀剂膜的显影液,可以使用碱类,例如,氢氧化钠、氢氧化钾、碳酸钠、硅酸钠、偏硅酸钠、氨水等无机碱类、乙基胺、正丙基胺等伯胺类、二乙基胺、二-正丁基胺等仲胺类、三乙胺、甲基二乙基胺等叔胺类、二甲基乙醇胺、三乙醇胺等醇胺类、氢氧化四甲铵、氢氧化四乙铵、胆碱等季铵盐、吡咯、哌啶等环状胺类、的水溶液。此外,还可以在上述碱类的水溶液中添加适当量的异丙醇等醇类、非离子系等表面活性剂来使用。其中优选的显影液为季铵盐的水溶液,进一步优选为氢氧化四甲铵的水溶液。作为显影的条件,从显影温度5℃~50℃、显影时间10秒~300秒的范围适宜选择。而且,可以将通过利用上述工序显影除去抗蚀剂膜而露出的部分的抗蚀剂下层膜,利用干蚀刻来除去,在基板上形成所期望的图案。实施例以下,举出合成例和实施例对本发明进行详述,但本发明不受下述记载任何限定。本说明书的下述合成例1~合成例6中所示的重均分子量为通过凝胶渗透色谱(以下,在本说明书中简称为GPC。)得到的测定结果。测定时使用东ソー株式会社制GPC装置,测定条件如下所述。此外,本说明书的下述合成例中所示的分散度由测定的重均分子量、和数均分子量算出。GPC柱:Shodex〔注册商标〕·Asahipak〔注册商标〕(昭和电工(株))柱温度:40℃溶剂:N,N-二甲基甲酰胺(DMF)流量:0.6ml/分钟标准试样:聚苯乙烯(东ソー株式会社制)检测器:RI检测器(东ソー株式会社制,RI-8020)<合成例1>将对苯二甲酸二缩水甘油基酯(ナガセケムテックス株式会社制,商品名:デナコール〔注册商标〕EX711)5g、5-羟基间苯二甲酸3.146g、3,5-二-叔丁基水杨酸水合物0.562g和苄基三乙基氯化铵0.199g添加至丙二醇单甲基醚35.6g中并溶解。将反应容器氮气置换后,在135℃反应4小时,从而获得聚合物溶液。该聚合物溶液即使冷却至室温也不产生白浊等,在丙二醇单甲基醚中的溶解性良好。进行GPC分析得到的结果是,所得的溶液中的聚合物以标准聚苯乙烯换算重均分子量为6066,分散度为2.64。<合成例2>将对苯二甲酸二缩水甘油基酯(ナガセケムテックス株式会社制,商品名:デナコール〔注册商标〕EX711)5g、5-羟基间苯二甲酸3.146g、3,5-二-叔丁基水杨酸水合物0.864g和苄基三乙基氯化铵0.197g添加至丙二醇单甲基醚36.8g中并溶解。将反应容器氮气置换后,在135℃反应4小时,从而获得聚合物溶液。该聚合物溶液即使冷却至室温也不产生白浊等,在丙二醇单甲基醚中的溶解性良好。进行GPC分析得到的结果是,所得的溶液中的聚合物以标准聚苯乙烯换算重均分子量为4746,分散度为2.68。<合成例3>将对苯二甲酸二缩水甘油基酯(ナガセケムテックス株式会社制,商品名:デナコール〔注册商标〕EX711)5g、5-羟基间苯二甲酸3.146g、3,5-二异丙基水杨酸水合物0.575g和苄基三乙基氯化铵0.197g添加至丙二醇单甲基醚35.7g中并溶解。将反应容器氮气置换后,在135℃反应4小时,从而获得聚合物溶液。该聚合物溶液即使冷却至室温也不产生白浊等,在丙二醇单甲基醚中的溶解性良好。进行GPC分析得到的结果是,所得的溶液中的聚合物以标准聚苯乙烯换算重均分子量为5634,分散度为2.70。<合成例4>将对苯二甲酸二缩水甘油基酯(ナガセケムテックス株式会社制,商品名:デナコール〔注册商标〕EX711)5g、5-羟基间苯二甲酸3.146g、3,5-二异丙基水杨酸水合物0.767g和苄基三乙基氯化铵0.197g添加至丙二醇单甲基醚35.7g中并溶解。将反应容器氮气置换后,在135℃反应4小时,从而获得聚合物溶液。该聚合物溶液即使冷却至室温也不产生白浊等,在丙二醇单甲基醚中的溶解性良好。进行GPC分析得到的结果是,所得的溶液中的聚合物以标准聚苯乙烯换算重均分子量为4669,分散度为2.50。<合成例5>将对苯二甲酸二缩水甘油基酯(ナガセケムテックス株式会社制,商品名:デナコール〔注册商标〕EX711)5g、5-羟基间苯二甲酸3.146g和苄基三乙基氯化铵0.202g添加至丙二醇单甲基醚35.6g中并溶解。将反应容器氮气置换后,在135℃反应4小时,从而获得聚合物溶液。该聚合物溶液即使冷却至室温也不产生白浊等,在丙二醇单甲基醚中的溶解性良好。进行GPC分析得到的结果是,所得的溶液中的聚合物以标准聚苯乙烯换算重均分子量为15673,分散度为3.39。<合成例6>将对苯二甲酸二缩水甘油基酯(ナガセケムテックス株式会社制,商品名:デナコール〔注册商标〕EX711)5g、5-羟基间苯二甲酸3.146g、水杨酸0.357g和苄基三乙基氯化铵0.202g添加至丙二醇单甲基醚37.5g中并溶解。将反应容器氮气置换后,在135℃反应4小时,从而获得聚合物溶液。该聚合物溶液即使冷却至室温也不产生白浊等,在丙二醇单甲基醚中的溶解性良好。进行GPC分析得到的结果是,所得的溶液中的聚合物以标准聚苯乙烯换算重均分子量为5745,分散度为2.67。<实施例1>在由上述合成例1获得的包含聚合物0.07g的溶液0.4g中,混合四甲氧基甲基甘脲(日本サイテックインダストリーズ〔旧三井サイテック〕株式会社制,商品名:POWDERLINK〔注册商标〕1174)0.0184g和5-磺基水杨酸0.0018g,添加丙二醇单甲基醚21.27g和1-乙氧基-2-丙醇9.33g并溶解。然后使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,制成形成光刻用抗蚀剂下层膜的组合物。<实施例2>在由上述合成例2获得的包含聚合物0.07g的溶液0.4g中,混合四甲氧基甲基甘脲(日本サイテックインダストリーズ〔旧三井サイテック〕株式会社制,商品名:POWDERLINK〔注册商标〕1174)0.0184g和5-磺基水杨酸0.0018g,添加丙二醇单甲基醚21.27g和1-乙氧基-2-丙醇9.33g并溶解。然后使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,制成形成光刻用抗蚀剂下层膜的组合物。<实施例3>在由上述合成例3获得的包含聚合物0.07g的溶液0.4g中,混合四甲氧基甲基甘脲(日本サイテックインダストリーズ〔旧三井サイテック〕株式会社制,商品名:POWDERLINK〔注册商标〕1174)0.0184g和5-磺基水杨酸0.0018g,添加丙二醇单甲基醚21.27g和1-乙氧基-2-丙醇9.33g并溶解。然后使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,制成形成光刻用抗蚀剂下层膜的组合物。<实施例4>在由上述合成例4获得的包含聚合物0.07g的溶液0.4g中,混合四甲氧基甲基甘脲(日本サイテックインダストリーズ〔旧三井サイテック〕株式会社制,商品名:POWDERLINK〔注册商标〕1174)0.0184g和5-磺基水杨酸0.0018g,添加丙二醇单甲基醚21.27g和1-乙氧基-2-丙醇9.33g并溶解。然后使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,制成形成光刻用抗蚀剂下层膜的组合物。<比较例1>在由上述合成例5获得的包含聚合物0.07g的溶液0.4g中,混合四甲氧基甲基甘脲(日本サイテックインダストリーズ〔旧三井サイテック〕株式会社制,商品名:POWDERLINK〔注册商标〕1174)0.0184g、5-磺基水杨酸0.0018g,添加丙二醇单甲基醚21.27g和1-乙氧基-2-丙醇9.33g并溶解。然后使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,制成形成光刻用抗蚀剂下层膜的组合物。<比较例2>在由上述合成例6获得的包含聚合物0.07g的溶液0.4g中,混合四甲氧基甲基甘脲(日本サイテックインダストリーズ〔旧三井サイテック〕株式会社制,商品名:POWDERLINK〔注册商标〕1174)0.0184g和5-磺基水杨酸0.0018g,添加丙二醇单甲基醚21.27g和1-乙氧基-2-丙醇9.33g并溶解。然后使用孔径0.05μm的聚乙烯制微型过滤器进行过滤,制成形成光刻用抗蚀剂下层膜的组合物。<比较例3>准备包含下述式(5)所示的共聚物作为聚合物,此外作为添加物包含下述式(6)所示的交联剂、和吡啶-对甲苯磺酸盐的形成抗蚀剂下层膜的组合物。(在光致抗蚀剂溶剂中的溶出试验)将由实施例1~实施例4、比较例1和比较例2调制的形成抗蚀剂下层膜的组合物分别通过旋涂器涂布在作为半导体基板的硅晶片上。将该硅晶片配置在电热板上,在205℃烘烤1分钟,形成抗蚀剂下层膜(膜厚0.05μm)。将这些抗蚀剂下层膜浸渍在作为光致抗蚀剂所使用的溶剂的乳酸乙酯和丙二醇单甲基醚中,确认了在这些溶剂中不溶。(干蚀刻速度的测定)将由实施例1~实施例4和比较例3调制的形成抗蚀剂下层膜的组合物分别使用旋涂器涂布在硅晶片上。将该硅晶片配置在电热板上,在205℃烘烤1分钟,形成抗蚀剂下层膜(膜厚0.10μm)。然后,使用日本サイエンティフィック社制,RIE系统ES401来测定干蚀刻速度。同样地,将抗蚀剂溶液(住友化学(株)制,PAR855)使用旋涂器涂布在硅晶片上,制成抗蚀剂膜。然后,使用日本サイエンティフィック社制RIE系统ES401来测定干蚀刻速度,与由实施例1~实施例4和比较例3的形成抗蚀剂下层膜的组合物获得的抗蚀剂下层膜的干蚀刻速度进行比较。将结果示于表1中。表1中,本发明的涂布型抗蚀剂下层膜相对于抗蚀剂膜的干蚀刻速度的选择比(抗蚀剂下层膜/抗蚀剂膜)的测定使用CF4气体作为蚀刻气体来进行。由各实施例的形成抗蚀剂下层膜的组合物获得的抗蚀剂下层膜与由比较例3的形成抗蚀剂下层膜的组合物获得的抗蚀剂下层膜相比,干蚀刻速度的选择比成为大的值。[表1](涂布后缺陷数的测定)将由实施例1~实施例4、比较例1和比较例2调制的形成抗蚀剂下层膜的组合物分别使用旋涂器涂布在硅晶片上。将该硅晶片配置在电热板上,在205℃烘烤1分钟,形成抗蚀剂下层膜(膜厚0.005μm)。然后,使用株式会社日立ハイテクノロジーズ制,高分辨率场致发射型扫描电子显微镜(FE-SEM)S-4800,进行作为白色点而观察到的抗蚀剂下层膜上的缺陷数的比较。将结果示于表2中。表2中,抗蚀剂下层膜上的缺陷数通过计数在倍率15000倍时的16.7μm×12.6μm的范围内的缺陷数来测定。图1~图6表示利用上述FE-SEM,从上面观察使用由各实施例和各比较例调制的组合物形成的抗蚀剂下层膜时的SEM像。[表2]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 形成抗蚀剂下层膜的组合物及使用该组合物的抗蚀剂图案的形成方法与流程
- 窗膜用组成物、由其形成的柔性窗膜以及包括其的柔性显示器装置的制造方法
- 铜膜形成用组合物和使用其的铜膜的制造方法与流程
- 生物膜形成抑制剂和/或除去剂、以及生物膜形成抑制方法和/或除去方法与流程
- 抗蚀剂下层膜形成用组合物的制造方法与工艺
- 窗膜用组成物、由其形成的柔性窗膜以及包括其的柔性显示器装置的制造方法
- 用于窗膜的组合物、由此形成的可挠式窗膜以及包括所述可挠式窗膜的可挠式显示装置的制造方法
- 一种生物膜成膜过程在线监控系统及监控方法与流程
- 上层膜形成用组合物以及使用了该上层膜形成用组合物的图案形成方法及电子器件的制造方法与流程
- 固化膜形成用组合物、取向材料和相位差材料的制造方法与工艺