用于环氧粘合剂的封闭的聚氨酯增韧剂的制作方法
本发明涉及环氧封闭的聚氨酯增韧剂。这样的增韧剂可用于环氧粘合剂配制品,如可用于汽车工业的单组分结构的粘合剂。
背景技术:
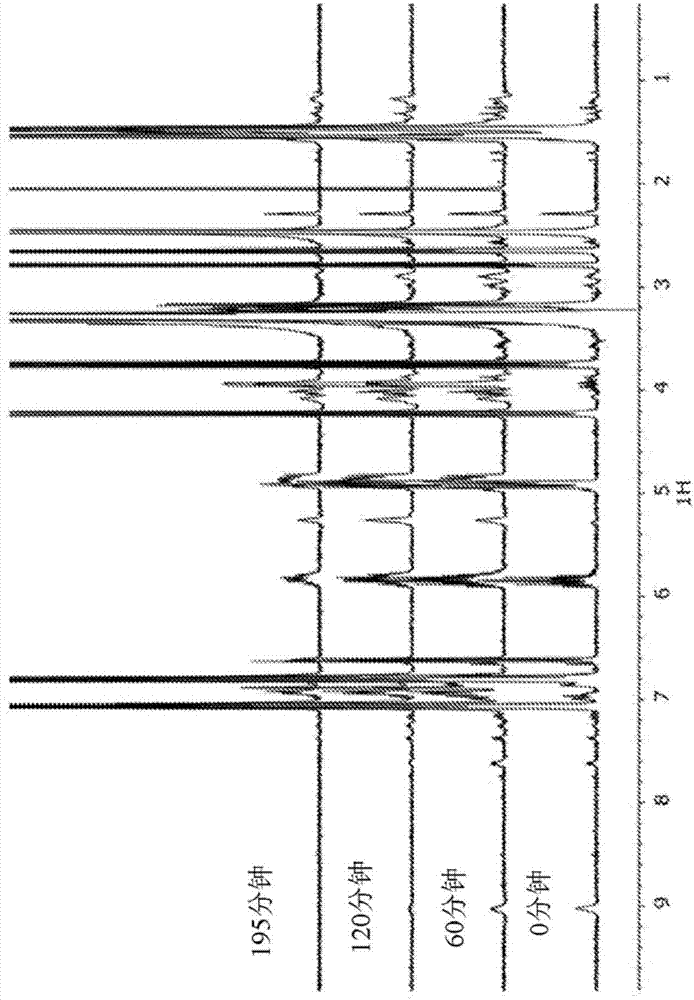
:如美国专利5,278,257和7,557,168中所述的多酚封闭的聚氨酯增韧剂用作非常有效的增韧聚合物,以主要对单组分结构的环氧粘合剂进行增韧。这样的粘合剂可用于若干应用中,如在汽车车身修理店中。这些增韧剂提供高的动态冲击剥离强度值,并提供良好的粘合本体(bulk)性能(例如,弹性模量或断裂伸长率)。通常,将双酚a或改性物如邻,邻'-二烯丙基双酚a用于封闭(或封端)异氰酸酯封端的预聚物。过量使用双酚以保证异氰酸酯官能团完全转化为氨基甲酸酯基团。残留的未反应的双酚含量通常为最终聚合物(增韧剂组合物)的约3%至10%。该过量的芳香族羟基(例如,双酚a)以及增韧聚合物的残留oh官能团限制了最终粘合剂配制品的储存期稳定性。储存期稳定性通常是通过测试作为时间和储存温度的函数的粘度进行的。其它封端基团,如单-酚或仲胺(例如,如美国专利8,404,787和7,625,977中所描述的)由于封闭剂的单官能度,提供了良好的储存期稳定性;与双酚封闭化合物的双官能度相比。单官能封闭基团提供较差(较低)的机械粘合性质,如搭接剪切强度或弹性模量,因为在解封之后,封闭剂的单官能团用作环氧树脂聚合的链终止剂。如本领域普通技术人员所理解的,这些异氰酸酯封闭的增韧剂在粘合剂配制品中很好地工作,因为当粘合剂固化时,封闭基团在热的作用下解封,并且结合到环氧基体中,以在增韧剂相与环氧相之间提供更好的界面。然而,由于其至少双官能度或多官能度,双酚或多酚封闭剂起扩链剂或交联剂的作用。使用双酚封闭的或多酚封闭的增韧剂聚合物的经固化的粘合剂性能表现出较高的弹性模量,结果是较高的机械性能如较高的搭接剪切强度值。在ep1498441a1和ep1741734a1中描述了增韧聚合物,其优选地将双官能酚如双酚m结合到聚合物链中,并通过特定的羟基-缩水甘油基化合物封闭异氰酸酯预聚物。ep1741734a1描述了固体环氧树脂在粘合剂配制品中用于改善动态冲击强度性能的另外用途,其是判断接头的碰撞性能的测试方法。这样的配制品显示出相当高的粘合剂粘度。ep1498441a1描述了用于环氧粘合剂配制品的增韧聚合物,其在聚合物链中使用双酚化合物,并通过使用特定的单羟基-缩水甘油组分来封闭异氰酸酯官能团。ep1900774a1描述了环氧-pu树脂(氨基甲酸酯改性环氧树脂)与己内酰胺封闭的pu聚合物组合用于基于环氧的粘合剂配制品中的用途。异氰酸酯预聚物与环氧树脂直接反应。仍然需要用于环氧粘合剂的增韧剂,所述粘合剂具有良好的储存期稳定性、良好的机械粘合性或两者。仍然需要具有良好的储存期稳定性、良好机械性能或两者的增韧的环氧粘合剂。技术实现要素:已经发现,增韧剂提供了这些和其它益处,所述增韧剂是双酚封闭的pu增韧剂与二缩水甘油醚-双酚产品如液体dgeba的反应产物。本发明的组合物包括增韧剂,所述增韧剂包含通过多酚封闭剂偶联的环氧封端的聚氨酯。本发明提供了异氰酸酯封端的预聚物与具有双官能芳香族部分的封端化合物的第一反应产物,其中所述第一反应产物用所述封端化合物进行封端;与二缩水甘油基双酚环氧树脂的反应产物;其中所述反应产物适于用作环氧粘合剂组合物中的增韧剂。本发明还提供了异氰酸酯封端的预聚物;多酚或二羟基官能苯;与二缩水甘油基双酚环氧树脂的反应产物;其中所述反应产物适于用作环氧粘合剂组合物中的增韧剂。本发明还提供了适于用作环氧粘合剂组合物中的增韧剂的组合物,所述组合物包含通过多酚或其它芳香族二羟基化合物偶联的环氧封端的聚氨酯。附图说明图1示出了在催化剂猝灭之前和之后的eew的演变。图2是一组示出酚-oh官能团的消耗的1hnmr谱。具体实施方式已经发现,双酚封闭的pu增韧剂与这样的dgeba环氧树脂反应封闭了pu聚合物的残留-oh官能团并封闭了聚合物中的残余(过量)双酚。不受理论限制,认为双酚起到异氰酸酯的封闭剂以及聚氨酯与环氧之间的偶联剂的作用。进一步认为这些作用中的一个或两者有助于提高粘合剂储存稳定性和/或提高经固化的粘合剂的机械性能。与包含双酚封闭的增韧剂的粘合剂配制品相比,包含这样的环氧封端的增韧剂的粘合剂配制品提供显著更好的储存期稳定性。增韧剂与环氧基体的反应性是机械性能所必需的。异氰酸酯聚氨酯预聚物通过双酚封端单元与环氧官能团连接。如本领域所知,用于环氧粘合剂的增韧剂(通常包含弹性聚氨酯或聚脲)含有刚性组分和柔性组分(硬链段和软链段)。这些组合物的增韧能力通常归因于软链段。1.本发明的增韧剂本发明涉及与二缩水甘油醚双酚环氧树脂反应的封端的增韧组合物。通常,增韧剂,优选地pu预聚物,优选地用两个(例如,双官能)芳香族-oh基团,更优选地双酚封端单元进行末端封端。使末端封端的预聚物与环氧树脂反应,从而获得本发明的增韧剂。这些反应可以顺序地或同时地进行。无论反应是顺序的还是同时的,所得产物称可以称为环氧树脂与本身为预聚物与封端基团的反应产物的组合物的反应产物。本发明的增韧剂也可以称为异氰酸酯封端的预聚物;多酚或二羟基官能苯;与二缩水甘油基双酚环氧树脂的反应产物。当使用顺序方法制备本发明的增韧剂时,可以使用任何酚封端的增韧组合物(即,具有残留的未反应的酚羟基的封端基团),优选为双酚封端的聚氨酯单-预聚物或共-预聚物。合适的封端的增韧组合物的实例在美国专利5,278,257和7,557,168中公开,其公开内容通过引用以其整体并入本文。美国专利5,278,257公开了增韧组合物,包含式i的酚封端的聚氨酯、聚脲或聚脲-氨基甲酸酯:其中m为1或2,n为2至6,r1为在移除末端异氰酸酯、氨基或羟基之后,弹性预聚物的n价基,其可溶于或可分散于环氧树脂中,x和y彼此独立地为-o-或-nr3-,这些基团中的至少一个必须是-nr3-,r2是在分别移除酚羟基或氨基或氨基和酚羟基两者之后的多酚或氨基酚的m+1价基,r3是氢、c1–c6烷基或苯酚。美国专利5,278,257的增韧剂组分是经选择的聚氨酯或经选择的衍生自特定预聚物的聚脲。术语“弹性预聚物基r1”在本说明书的范围内应理解为意指,预聚物的用n-异氰酸酯、n-氨基或n-羟基封端的基,其在将这些基团封端之后,产生式i的酚封端的聚氨酯、聚脲或聚脲-氨基甲酸酯(组分(b)),其与二烯组分a)和环氧树脂c)组合,在固化后产生弹性体相或弹性体相的混合物。这些可以是组分a)、b)和c)的均相或非均相组合。通常,弹性体相的特征在于玻璃化转变温度低于0℃。术语“可溶于或可分散于环氧树脂中的预聚物”在本说明书的范围内应理解为意指,预聚物的由n-异氰酸酯、n-氨基或n-羟基封端的基,其在将这些基团封端之后,产生式i的酚封端的聚氨酯、聚脲或聚脲-氨基甲酸酯,其在没有进一步的辅助(例如,乳化剂)的情况下可溶于或可分散于环氧树脂或环氧树脂与二烯共聚物的组合;因此,在此过程中,形成均相,或至少不进行组分中的一个或组分混合物的宏观相分离。如美国专利5,278,257中所述,式i的酚封端的聚氨酯、聚脲或聚脲-氨基甲酸酯优选为不溶于水的酚封端的聚氨酯、聚脲或聚脲-氨基甲酸酯。这在本说明书的范围内应理解为意指酚封端的聚氨酯、聚脲或聚脲-氨基甲酸酯,其溶于水的程度小于5重量%,优选地小于0.5重量%,并且当储存于水中时,其仅吸收少量的水,优选地小于5重量%,特别地小于0.5重量%,或在其过程中,仅表现出轻微的膨胀。在美国专利5,278,257中,其中r1所基于的预聚物的分子量(数均)通常为150至10,000,优选地1,800至3,000。这些预聚物的平均官能度为至少2,优选为2至3,特别地优选为2至2.5。美国专利7,557,168描述了增韧剂组分,包含一种或多种异氰酸酯封端的预聚物与一种或多种封端剂的反应产物,其中用于制备预聚物的异氰酸酯具有脂族和/或环脂族基团。优选地,预聚物具有分子量以便产生低粘度粘合剂组合物。优选地,预聚物的粘度为约20pas.或更大,更优选为约100pas.或更大。优选地,预聚物的粘度为约1000pas.或更小,更优选为约800pas.或更小。为了获得所期望的增韧剂的粘度,异氰酸酯预聚物中的支链数量和最终反应产物的交联密度必须保持为低的。预聚物的支链数量直接与用于制备异氰酸酯封端的预聚物的原材料的官能度有关。官能度是指反应物中的反应性基团的数量。优选地,预聚物中的支链数量为约6或更少,更优选为约4或更少。优选地,支链数量为约1或更多,更优选为约2或更多。交联密度是聚合物链之间的附接物数量。在较高的交联密度下,反应产物的粘度较高。交联密度受预聚物的官能度和工艺条件的影响。如果制备增韧剂的反应温度保持相对较低,则可使交联最小化。优选地,交联密度为约2或更小,更优选为约1或更小。优选地,预聚物的分子量为约8,000(mw)或更高,更优选为约15,000(mw)或更高。优选地,预聚物的分子量为约40,000(mw)或更低,更优选约30,000(mw)或更低。本文所用的分子量是根据gpc分析测定的重均分子量。与预聚物反应的封端剂的量应该足以对基本上所有的末端异氰酸酯基团进行封端。用封端剂封端异氰酸酯基团意指封端剂与异氰酸酯反应以将封端剂置于聚合物的端部上。基本上所有的意指在预聚物中留有少量的游离异氰酸酯基团。少量意指存在不以任何显著的方式影响组合物的性质的所提及的特征或成分的量。优选地,封端剂当量与异氰酸酯预聚物当量的比值为约1∶1或更大,更优选为约1.5∶1或更大。优选地,封端剂与预聚物的异氰酸酯的当量比为约2.5∶1或更小,更优选为约2∶1或更小。优选地,美国专利7,557,168的反应产物对应于式ii或式iii中之一:其中:在每种情况下,r1独立地为c2-20m-价烷基部分;在每种情况下,r2独立地为聚醚链;在每种情况下,r3独立地为亚烷基、环亚烷基或混合的亚烷基和环亚烷基部分,任选地含有一个或多个氧或硫原子;r4是直接键或亚烷基、羰基、氧、羧基或氨基部分;在每种情况下,r5独立地为烷基、烯基、烷氧基、芳氧基或芳氧基部分,前提条件是,如果p=1,则q=0;x是o或–nr6,前提条件是,x是o,其中p是1;并且在至少一种情况下,其中p是0,x是o;在每种情况下,r6独立地为氢或烷基;在每种情况下,m独立地为约1至约6的数;在每种情况下,n独立地为1或更大;在每种情况下,如果p是0,则o独立地为0或1,并且如果p是1,则为0;在每种情况下,p独立地为0或1;并且在每种情况下,q独立地为0至1的数;美国专利7,557,168的异氰酸酯封端的预聚物对应于式iv和式v中之一:并且封端化合物对应于式vi:其中r1、r2、r3、r4、r5、m、n、o、p和q如上所定义的。在美国专利7,557,168的反应产物中,r4优选为直接键或亚烷基、氧、羰基、羰基氧基或氨基部分。更优选地,r4是直接键或c1-3直链或支链亚烷基部分。优选地,在每种情况下,r5独立地为烷基、烯基、烷氧基或芳氧基部分,前提条件是如果p=1,那么q=0。更优选地,r5是c1-20烷基、c1-20烯基、c1-20烷氧基或c6-20芳氧基部分。更优选地,r5是c3-15烷基或c2-15烯基部分。优选地,o是0。更优选地,在每种情况下,n独立地为约1至约3;可以使用任何含有两个或更多个酚羟基的合适化合物,包括在美国专利5,278,257和7,557,168中公开的那些,来对上述增韧组合物进行封端。优选的封端化合物包含恰好两个酚羟基。一些合适的封端化合物包括间苯二酚、儿茶酚、氢醌、联苯-4,4-二醇、双酚a、双酚b、双酚c、双酚e、双酚ap(1,1-双(4-羟基苯基)-1-苯基乙烷)、双酚f、双酚k、双酚m、四甲基双酚和邻,邻'-二烯丙基双酚a(odba)等。odba是优选的。间苯二酚意指间苯二酚及其衍生物,如经取代的间苯二酚。a.r.l.dohme,间苯二酚的酰基和烷基衍生物的制备(thepreparationoftheacylandalkylderivativesofresorcinol),jacs,1926,48(6),第1688至1693页(其全部内容通过引用并入)。如本文所使用的,术语“双官能芳香族部分”旨在包括所有经取代的和经二羟基取代的双官能芳香族化合物及其衍生物,优选为经二羟基取代的及其衍生物。可以在本发明中使用能够与封端的增韧剂反应的任何环氧树脂。低分子量和/或液体环氧树脂是优选的。如果环氧树脂的分子量太高,则这会导致处理问题,并且随着反应的进行,粘度过度增加。优选的环氧树脂包括液体环氧树脂,包括下文第2部分中更详细地描述的那些。用于封闭增韧剂的一些优选的环氧树脂包括d.e.r.330、d.e.r.331和d.e.r.383。使用具有反应性芳香族羟基的基团封端的增韧剂与环氧树脂反应以提供本发明的化合物。可以使用,并且可以由本领域普通技术人员使用本公开作为动机和/或指导来设计用于进行反应的任何方法。可以使用催化剂来促进封端的pu预聚物与环氧树脂之间的反应。一些优选的催化剂包括乙基三苯基乙酸磷(etpac)、四丁基溴化铵(tbab)和三苯基膦(tpp)。为了在反应进行时限制环氧当量(eew)的增加,可以在反应期间添加催化剂猝灭剂,如甲基甲苯-4-磺酸酯(mpts)。当使用时,例如,在预定时间(例如,6小时的反应时间)之后,在达到一定的目标eew(例如,300g/当量、350g/当量、400g/当量或500g/当量),或者当满足一些其它标准(例如,颜色变化)时,可以添加淬灭剂。本发明的增韧剂可以具有适于用作增韧剂的任何分子量,如本领域普通技术人员所确定的。如果分子量太高,则增韧剂可能变得太粘或凝固,这会损害其有效性和易用性。通常对分子量没有优选的下限,但是增韧剂应当具有足够高的分子量以具有足够的软链段来用作增韧剂。例如,如果双酚封端的pu增韧剂具有足够的分子量来用作增韧剂,则其应当具有足够的分子量以适用于本发明。通常,较高分子量赋予经固化的粘合剂更好的机械性能。优选的质量加权分子量(mw)为或高于5,000da、6,000da、10,000da、14,000da或17,000da。虽然没有特别的上限,但mw通常将小于或等于30,000da、25,000da或20,000da。来自实施例的mw也是优选的。优选的数量加权分子量(mn)为或高于3,000da、4,000da或6,000da。虽然没有特别的上限,但mn通常将小于或等于15,000da或10,000da。来自实施例的mw和mn也是优选的。由成对的这些值的mw或mn形成的范围也是优选的。根据本发明的本发明的粘合剂和方法包括根据本发明的一种或多种本发明的增韧剂。本发明的粘合剂组合物和方法可包含任何量的本发明的增韧剂。优选地,本发明的粘合剂组合物包含,按环氧粘合剂组合物的重量计,大于或约20重量%,更优选地大于或约25重量%,或30重量%的本发明的增韧剂。优选地,本发明的粘合剂组合物包含,按环氧粘合剂组合物的重量计,小于或约60重量%,更优选地小于或约50重量%或45重量%的本发明增韧剂。其它优选的量在实例中示出。还优选的是,由成对的这些值(例如,20重量%至60重量%和30重量%至40重量%的(粘合剂bh))形成的范围。应当理解,本发明的增韧剂组合物可以包含与环氧反应的经转化的封闭的pu,并且通常还将包括未反应的环氧树脂。本发明的增韧剂的上述百分比包括未反应的环氧树脂。2.环氧树脂可用于根据本发明的粘合剂组合物的环氧树脂包括多种可固化的环氧化合物及其组合。可用的环氧树脂包括液体、固体及其混合物。通常,环氧化合物是环氧树脂,其也称为聚环氧化物。可用于本文的聚环氧化物可以是单体的(例如,双酚a的二缩水甘油醚、双酚f的二缩水甘油醚、四溴双酚a的二缩水甘油醚、基于酚醛清漆的环氧树脂和三官能环氧树脂)、较高分子量的树脂(例如,与双酚a一起的双酚a的二缩水甘油醚)或将不饱和单环氧化物(例如,丙烯酸缩水甘油酯、甲基丙烯酸缩水甘油酯、烯丙基缩水甘油醚等)聚合为均聚物或共聚物。最期望的是,环氧化合物每分子平均含有至少一个侧基或末端l,2-环氧基团(即,邻位环氧基团)。可用于本发明的固体环氧树脂优选地可以包括或优选地基于主要的双酚a。然而,为了实现本发明的粘度分布,所使用的双酚a的量应当保持在低于粘合剂组合物的0.5重量%。一些优选的环氧树脂包括,例如,d.e.r.330、d.e.r.331和d.e.r.671,所有的均可从陶氏化学公司(dowchemicalcompany)购得。一种可优选的环氧树脂具有通式:其中n通常在0至约25的范围内。碱性液体树脂,例如d.e.r.331的环氧当量在约180g/mol至195g/mol的范围内。环氧树脂的组合可用于调节环氧粘合剂的性质。在本发明的组合物和方法中,环氧粘合剂可以包含任何量的环氧树脂。优选地,液体环氧树脂和/或固体环氧树脂占环氧粘合剂的大于或约20重量%,更优选地大于或约25重量%、30重量%或35重量%。优选地,液体环氧树脂和/或固体环氧树脂占环氧粘合剂的小于或约65重量%,更优选地小于或约55重量%或45重量%。其它优选的量在实例中示出。还优选的是,由成对的这些值(例如,25重量%至35重量%,25重量%至65重量%,30重量%至38重量%(粘合剂aa))形成的范围。当使用液体环氧树脂与固体环氧树脂的组合时,可以使用任何比例,并且可以由本领域普通技术人员确定。为了获得合适的粘度,通常优选的是,液体环氧树脂与固体环氧树脂的重量比大于50:50。本发明的环氧粘合剂组合物优选地包含比值为或大于55:45、65:35或70:30的液体环氧树脂与固体环氧树脂。本发明的环氧粘合剂组合物优选地包含比值为或小于100:0、99:1、90:10或85:10的液体环氧树脂与固体环氧树脂。其它优选的比值在实例中示出。还优选的是,由成对的这些值(例如,50:50至100:0、65:35至82:18(粘合剂au))形成的范围。3.硬化剂硬化剂,优选地适于1k粘合剂组合物,优选地包括潜在硬化剂。可以使用在环境条件下(“环境条件”意指,例如,通常的室温和正常照明条件)不会导致硬化的任何潜在硬化剂。通过施加热使环氧粘合剂可固化的潜在硬化剂是优选的。一些优选的硬化剂包括双氰胺、咪唑、胺、酰胺、多元酚和聚酐。双氰胺(也称为dicy,双氰胺和1-氰基胍或2-氰基胍)是优选的。dicy(cas461-58-5)具有经验式c2n4h4,分子量84以及结构式:根据本发明的任何特定组合物可以适当使用任何量的硬化剂。硬化剂的量优选为环氧粘合剂的至少1重量%,更优选为至少2重量%,更优选为至少3重量%。环氧硬化剂的量优选地高达环氧粘合剂的约6重量%,更优选地高达约6重量%、5重量%或4重量%。其它优选的量在实例中示出。还优选的是,由成对的这些值(例如,1重量%至3重量%或3重量%至6重量%)形成的范围。4.固化促进剂一种或多种固化促进剂(催化剂)可以任选地用于,例如,改变条件,在所述条件下使潜在催化剂变成催化活性的。当使用时,优选的固化促进剂可以包括胺,如氨基酚、脲和咪唑。更优选的促进剂包括胺,如氨基酚。如ep-a-0197892中所述的,优选的促进剂包括整合到聚(对乙烯基苯酚)基体中的2,4,6-三(二甲基氨基甲基)苯酚。如在wo2012000171中所述的,更优选的促进剂包括整合到多酚树脂基体中的2,4,6-三(二甲基氨基甲基)苯酚。其它促进剂可以包括整合到聚(对-乙烯基苯酚)基体,或如美国专利第4,659,779号(及其同族成员美国专利第4,713,432号和第4,734,332号;以及ep-a-0197892)中所述的rezicure基体中的2,4,6-三(二甲基氨基甲基)苯酚。其它优选的固化促进剂包括脲如omicureu52和omicure405(emeraldperformance)。当使用时,固化促进剂可以以适当地调节潜在催化剂的活化条件的任何量存在。固化促进剂可以完全省略,或者以大于或约为环氧粘合剂的0.1重量%、0.2重量%、0.3重量%或0.4重量%的量存在。优选地,固化促进剂可以以小于或约为环氧粘合剂的4重量%、3重量%、2重量%或1重量%的量存在。其它优选的量在实例中示出。还优选的是,由成对的这些值(例如,0重量%至3重量%,0.1重量%至3重量%或1重量%至3重量%)形成的范围。5.橡胶组分包括液体橡胶或核-壳橡胶的橡胶组分可以任选地用于本发明中。一些优选的液体橡胶和核-壳橡胶组合物公开于美国专利第7,642,316号和第7,625,977号中,这两个专利以其整体并入本文。当使用液体橡胶时,优选的类型是基于dgeba的丁腈橡胶改性的环氧树脂。一些优选的橡胶改性的环氧树脂可以以商品名例如,3604或3614从schill&seilacher商购获得。当使用核壳橡胶时,优选的类型是在u.s.2007/0027233(ep1632533a1)中所述的那种,所述专利通过引用以其整体并入本文。如在文件中所述的核-壳橡胶颗粒包括交联的橡胶核,所述核在大多数情况下为丁二烯的交联共聚物,以及壳,所述壳优选为苯乙烯、甲基丙烯酸甲酯、甲基丙烯酸缩水甘油酯和任选地丙烯腈的共聚物。核-壳橡胶优选地分散在聚合物或环氧树脂中,也如在文件中所述的。优选的核-壳橡胶(csr)包括由陶氏化学公司以名称fortegra出售的那些,优选地包括300系列,如fortegra301。其它优选的核壳橡胶包括以名称kanekakaneace从钟渊化学工业株式会社(kanekacorporation)获得的产品,包括kanekakaneace15和120系列的产品,包括kanekakaneacemx153、kanekakaneacemx156和kanekakaneacemx120核-壳橡胶分散体及其混合物。所述产品包含以约为33%或25%的浓度预分散在环氧树脂中的核-壳橡胶颗粒。6.其它组分可以将其它组分任选地用于根据本发明的粘合剂中,如填充剂、间隔物、促粘剂、颜料、触变剂、润湿剂、反应性稀释剂、抗氧化剂等。可以将产品如玻璃珠用于填充粘合剂和/或作为间隔件,例如,以帮助控制施加到表面的粘合剂的层厚度。这样的产品的类型和尺寸可由本领域普通技术人员针对预期的应用来确定。一些优选的产品包括spheriglass(波特工业(potterindustries))。任选的填充剂包括矿物填充剂,如空心玻璃球、碳酸钙、氧化钙和滑石。填充剂确保良好的失效模式行为、增加的耐湿性、改进的抗腐蚀性、增加的模量和/或优越的可加工性。碳酸钙(例如,以商品名出售的)可用于减少收缩和增加抗腐蚀性。氧化钙(例如,以商品名chauxvive出售的)是湿度清除剂,其可有助于在最终固化之前保护部分固化的环氧粘合剂。滑石可以例如以商品名或获得,硅酸铝镁(硅灰石)可以例如以商品名200获得。还可以使用二氧化硅,优选为疏水性烟雾硅胶,如aerosilr202或aerosilr805。一些优选的空心玻璃球包括玻璃泡(glassbubbles)(3m)。当使用时,填充剂和间隔物(例如,玻璃球)可以以任何有用的量存在,并且可以由本领域普通技术人员使用该文件作为指导来确定。通常,填充剂可以以大于或约为环氧粘合剂的5重量%、10重量%或15重量%的量存在。填充剂可以以小于或约为环氧粘合剂的30重量%、25重量%或20重量%的量存在。其它优选的量在实例中示出。由成对的这些值形成的范围也是优选的。也可以任选使用地反应性稀释剂和非反应性稀释剂。一些优选的反应性稀释剂包括新癸酸的单缩水甘油酯,其也可用作降粘剂。其可以,例如,以商品名erisysgs-110(emerald)和cardurae10(momentive)商购获得。还可以任选使用地触变剂和其它粘度调节剂。一个这样的优选实例包括烟雾硅胶(例如,以商品名出售的)。还改进耐洗涤性的优选的触变剂是聚酯和液体环氧树脂(ler)的混合物,如dynacol(25%的聚酯7330和75%的ler330)。还可以使用具有聚酰胺的蓖麻油蜡,并且可以以商品名rheotix,例如,rheotix240从rockwood商购获得。当使用时,可以包括任何合适的胶凝剂。优选的胶凝剂应该包含能够与环氧树脂反应的官能团。这些包括热塑性化合物如聚酯二醇、聚酰胺或聚乙烯醇缩丁醛。合适的胶凝剂的实例包括聚酯二醇,例如,可从evonik获得的7330。也可以使用具有聚酰胺的蓖麻油蜡,并且可以以商品名rheotix,例如,rheotix240从rockwood商购获得。其它合适的胶凝剂包括由lehmann提供的luvotix等级(如luvotixhp)和由kusumotochemicalsltd.提供的voss,其是不含蜡或disparlon等级的聚酰胺。合适的聚乙烯醇缩丁醛包括来自kuraray的mowitalb60h和mowitalb60hh。可以在粘合剂组合物中单独或者彼此组合地使用这些胶凝剂。当使用时,触变剂和/或胶凝剂可各自以大于或约为环氧粘合剂的0.5重量%或1重量%的量,和/或小于或约为环氧粘合剂的5重量%或3重量%的量存在。其它优选的量在实例中示出。由成对的这些值形成的范围也是优选的。本发明的增韧剂可用于1-组分(1k)或2-组分(2k)环氧粘合剂组合物,优选为1k组合物。本发明包括包含本发明的增韧剂的粘合剂、使用增韧剂和包含其的产品(例如,粘合剂组合物)的方法以及经固化的本发明粘合剂和包含其的产品。实例表1中列出了以下实例中使用的一些材料以及一些已知的供应商或制造建议。表1实例1(环氧封端的增韧剂)顺序路径增韧剂odba-封端的pu使用以下程序用环氧官能团(der338)进行封端。在100℃下,以300g/当量的环氧当量(eew)(11:1摩尔比的der383:odba-封端的pu)为目标运行反应。用etpac(乙基三苯基乙酸磷)进行一次运行,用tpp(三苯基膦)作为催化剂以0.05重量%的负载进行另一次运行。tpp催化剂是控制体系。der330的分子量为360,ram965的分子量为1900,der330和odba-封端的pu两者的官能度为2或更高。当达到目标eew时,用甲基甲苯-4-磺酸酯(mpts)淬灭催化剂。作为测试,继续加热过夜以观察是否发生了环氧树脂的消耗。观察到猝灭催化剂停止了反应的进一步进行,如图1中稳定的eew所示。在来自图1的etpac-催化的材料上收集nmr谱。如图2所示,1hnmr谱可以用来表示在反应期间消耗了酚-oh官能团。随着反应的进行,酚-oh信号(9.0ppm)降低,当达到目标eew时,信号不再显著。从上到下,图2中的线对应于t=0hr、t=1hr、t=2hr和t=3.25hr。实例2(本发明的增韧剂)原位偶合用1,6-六亚甲基二异氰酸酯(hdi)作为异氰酸酯化合物来制备系列a的本发明的增韧剂(增韧剂a至增韧剂e、增韧剂u和增韧剂v)。用异佛尔酮二异氰酸酯(ipdi)代替hdi来制备系列b的本发明的增韧剂(增韧剂f至增韧剂j和增韧剂t)。使用omnisec软件通过具有viscotekdual检测器(viscotek)的gpc分析(dionex)来测量质量加权(mw)分子量和数量加权(mn)分子量以获得绝对分子量。表2:本发明的增韧剂原位偶合:为了制备本发明的增韧剂a至增韧剂j,将指定重量%的聚四氢呋喃二醇(聚thf)添加到实验室反应器中并加热至120℃。将聚四氢呋喃二醇在120℃在真空下混合30min,然后冷却至60℃。一旦温度达到60℃,就加入指定重量%的二异氰酸酯(hdi或idpi)并混合2min。加入指定重量%的dbtl(西格玛奥德里奇(sigmaaldrich)),并使混合物在85℃(浴温)下在氮气下反应45min。加入指定重量%的邻,邻'-二烯丙基双酚a和der331(ler)并搅拌混合物直到材料温度达到80℃为止。根据astmd2572-97测量混合物的样品以进行nco测定。nco应当为0%。如果nco大于0%,则继续反应,直到nco达到0%为止。一旦nco为0%,就加入指定重量%的催化剂tpp并将混合物加热至110℃(材料温度),并在真空下混合直到颜色从橙色变为深红色为止,约30分钟(例如,20分钟至40分钟)。将油浴设定为100℃,并将混合物混合另外30min。通过与增韧剂a至增韧剂j相同的环氧化方法来制备增韧剂t、增韧剂u和增韧剂v,但进行另外的催化剂失活。顺序偶合:通过将指定量的聚四氢呋喃二醇、聚丁二烯二醇和三羟甲基丙烷混合并在真空下在120℃下加热直到均质为止来制备增韧剂t。然后将混合物冷却至60℃。加入二异氰酸酯并混合。加入月桂酸二丁基锡(dbtl)催化剂,并使混合物在氮气下于85℃下反应45min。在混合下加入邻,邻'-二烯丙基双酚a。使混合物在90℃下在氮气下反应90min,然后在真空下混合10min。加入der330和乙基三苯基乙酸磷,并使混合物在100℃(材料温度)下反应150min。然后加入甲基甲苯-4-磺酸酯并混合另外10min。将混合物在真空下混合至少10min以上。顺序偶合:通过将指定量的聚四氢呋喃二醇和三羟甲基丙烷混合并在真空下在120℃下加热直到均质为止来制备增韧剂u。然后将混合物冷却至60℃。加入二异氰酸酯并混合。加入催化剂(dbtl),使混合物在氮气下于85℃下反应45min。在混合下加入邻,邻'-二烯丙基双酚a。使混合物在90℃下在氮气下反应45min,然后在真空下混合10min。加入der330和四丁基溴化铵,并使混合物在90℃(材料温度)下反应360min。然后加入甲基甲苯-4-磺酸酯并混合另外10min。将混合物在真空下混合至少10min以上。顺序偶合:通过将指定量的聚四氢呋喃二醇和三羟甲基丙烷混合并在真空下加热至120℃直到均质为止来制备增韧剂v。然后将混合物冷却至60℃。加入二异氰酸酯并混合。加入催化剂(dbtl),使混合物在氮气下于85℃下反应45min。在混合时加入邻,邻'-二烯丙基双酚a。使混合物在90℃下在氮气下反应45min,然后在真空下混合10min。加入der330和三苯基膦并将混合物加热到110℃(材料温度),将其在真空下混合,直到颜色由橙色变为深红色为止。将油浴设定为100℃并将混合物混合另外30min。然后加入甲基甲苯-4-磺酸酯并混合另外10min。将混合物在真空下混合至少10min以上。实例3(比较增韧剂)比较增韧剂q和比较增韧剂r使用odba封闭,并且不与液体环氧树脂进一步反应。将其稳定性与所制备的粘合剂配制品中的本发明的增韧剂配制品进行比较。比较增韧剂q是直链odba-封端的pu聚合物,比较增韧剂r是支链odba-封端的pu聚合物,并且比较增韧剂s是支链dipa-封端的pu聚合物。表3:比较增韧剂qrs重量%[%][%][%]聚thf200063.1164.8979.31hdi10.619.9813.29三羟甲基丙烷0.330.54dbtl0.060.050.06odba93%26.2224.75二异丙基胺6.8总量100.00100.00100mn[da]440062005100mw[da]82001390010500通过混合指定重量%的聚四氢呋喃二醇和三羟甲基丙烷并在真空下加热至120℃直到均质为止来制备比较增韧剂q和比较增韧剂r。将该混合物冷却至60℃。加入指定重量%的二异氰酸酯,同时混合。加入指定重量%的催化剂,并使混合物在85℃下反应25min。然后使混合物冷却至60℃,并使混合物反应另外20min。然后通过与指定重量%的邻,邻-二烯丙基双酚a反应30min来封端/扩链所得预聚物。然后在真空下将所得混合物混合至少10min。通过将指定量的聚四氢呋喃二醇和三羟甲基丙烷混合并在真空下加热至120℃下直到均质为止来制备比较增韧剂s。将该混合物冷却至60℃。加入指定重量%的二异氰酸酯,同时混合。加入指定重量%的催化剂,并使混合物在85℃下反应25min。然后将混合物冷却至60℃,并反应另外20min。然后通过与指定重量%的二异丙基胺反应30min来封端所得预聚物,然后在真空下混合至少10min。实例4(粘合剂配制品)使用本发明的增韧剂a至增韧剂e、增韧剂u和增韧剂v制备在配制品系列a中的称为aa至ae、au和av的七种粘合剂配制品。在表4中总结了配制品的细节和描述。表4:使用增韧剂a至增韧剂e的配制品系列a*这表示有助于增韧性能的增韧剂的柔性部分(软链段)(例如,增韧剂的聚氨酯部分)。例如,包含50重量%增韧剂组分(其中增韧剂组分包含50重量%的软pu部分)的组合物将具有25%的总增韧剂含量。**氧化钙、碳酸钙和滑石的组合。使用本发明的增韧剂f至增韧剂j和增韧剂t制备在配制品系列b中的称为bf至bj和bt的六种粘合剂配制品。使用来自实例2的比较增韧剂q至比较增韧剂s制备称为rq至rs的三种比较粘合剂配制品。在表5中总结了配制品的细节和描述。表5:使用增韧剂f至增韧剂j和增韧剂t,以及比较增韧剂q、比较增韧剂r和比较增韧剂s的配制品系列b将液体环氧树脂d.e.r.tm330、液体环氧树脂/固体环氧树脂共混物、反应性稀释剂、硅烷促粘剂、润湿剂、着色剂和增韧剂在50℃(50rpm)下合并并剧烈混合5分钟,随后在相同温度下在真空下混合20分钟(150rpm)。然后加入烟雾硅胶混合物以及滑石(矿物填充剂的部分),随后在冷却至室温时将组合物混合5分钟(50rpm),接着在真空下混合另外20分钟(150rpm)。然后加入cg1200g、固化促进剂、矿物填充剂的其它部分、任选的玻璃泡和mowital,随后以50rpm的混合速度混合3分钟,然后在降低的大气条件下以150rpm混合15分钟。实例5(粘合剂测试)流变性能测试如下。旋转粘度/屈服应力:bohlincs-50流变仪,c/p20,上/下0.1s/1至20s/1;根据casson模型进行评估。粘度casson:使用casson公式数学计算粘度系数。在45℃下,以10s-1的剪切速率测量实际粘度值。在钢上进行机械测试,例如,hc220b-ze-b(如可从thysssenkrupp获得的)。在180℃下固化30分钟。根据dinen1465测量搭接剪切强度:10mmx25mm的粘合区域,0.2mm粘合剂层厚度,拉伸速率为10mm/min。根据iso11343测量冲击剥离强度:20mmx30mm的粘合区域,0.2mm粘合剂层厚度,以2m/sec。根据dineniso527-1用哑铃状试样测量e-模量测试。表6示出了制备配制品之后不久,未经固化的粘合剂配制品系列a的流变学性质。表6:配制品系列a的流变测试结果aaabacadaeauav粘度,casson,45℃[pas]323327507439430152299在10s-1下的粘度[pas]577575819799754300490屈服应力,casson,45℃[pa]375357371461340245231未经固化的配制品aa、au和av经过1周至4周的时间范围产生的在多个温度下的储存数据,并且提供在表7和表8中。在表9中给出了一些系列a粘合剂的机械强度和粘合剂本体(bulk)数据。表7:配制品aa的储存性质表8:配制品au和配制品av的储存性质表9:配制品系列a(经固化的)的机械性能aaabacadaeauav在rt下的冲击剥离强度[n/mm]32342817303128在-40℃下的冲击剥离强度[n/mm]23249024搭接剪切强度[mpa]1919201917e-模量[mpa]1940178022842717148020921992拉伸强度[mpa]36.738.746.654.9333734伸长率[%]6.57.46.25.811.74.96.1tg,dsc[℃]10810310093112表10示出了制备配制品之后不久,未经固化的粘合剂配制品系列b的流变学性质。表10:配制品系列b的初始流变学性质bfbgbhbibjbt粘度,casson,45℃[pas]221286229529302180在10s-1下的粘度[pas]455580519958573308屈服应力,casson,45℃[pa]358438528505331163未经固化的配制品bf和bt经过1周至4周的时间范围产生在多个温度下的储存数据,并提供在表11和表12中。在表13中给出了一些系列b粘合剂的机械强度和粘合剂本体数据。表11:配制品bf的储存性质30℃40℃50℃60℃1周(所计算的粘度casson)247291311360系数增加1.11.31.41.6差值2670901391周(在10s-1下的粘度)465524593638系数增加11.21.31.4差值10691381832周(所计算的粘度casson)257293358592系数增加1.21.31.62.7差值36721373712周(在10s-1下的粘度)4965406511036系数增加1.11.21.42.3差值41851965813周(所计算的粘度casson)273306412999系数增加1.21.41.94.5差值52851917783周(在10s-1下的粘度)5165806931466系数增加1.11.31.53.2差值611251.510114周(所计算的粘度casson)2813174571773系数增加1.31.42.18差值609623615524周(在10s-1下的粘度)4936117852137系数增加1.11.31.74.7差值381563301682表12:配制品bt的储存性质表13:配制品系列b(经固化的)的机械性能bfbgbhbibjbt在rt下的冲击剥离强度[n/mm]292622122627在-40℃下的冲击剥离强度[n/mm]17181120搭接剪切强度[mpa]1919191818e-模量[mpa]229624462670322017732389拉伸强度[mpa]454654593638伸长率[%]6.85.36.64.08.04.6tg,dsc[℃]98909192106可以将本发明配制品aa至ae和配制品bf至bj的储存数据与下表中总结的参考粘合剂配制品相比较。特别地,以类似的方式,在表14至表16中提供了比较粘合剂配制品随着时间推移的初始流变学性质和流变学性质。在表17中提供了经固化的比较配制品的机械性能。表14:比较配制品的初始流变学性质rqrrrs粘度,casson,45℃[pas]7310239在10s-1下的粘度[pas]219247817屈服应力,casson,45℃[pa]367301430表15:配制品rq的储存性质表16:配制品rr的储存性质表17:比较配制品(经固化的)的机械性能rqrrrs在rt下的冲击剥离强度[n/mm]343038在-40℃下的冲击剥离强度[n/mm]182221搭接剪切强度[mpa]192420e-模量[mpa]230521521844拉伸强度[mpa]423733伸长率[%]5.03.84.1tg,dsc[℃]939994机械准静态搭接剪切强度值非常相似,几乎与增韧剂配制品无关(除了使用比较增韧剂r的配制品rr之外)。在23℃测试温度下的冲击剥离强度值似乎取决于增韧剂组合物的分子量。所使用的pthf(ptmeg)的分子量越低,所述值越低,其在-40℃测试温度下甚至更为显著。比较配制品rq示出了稍高的23℃冲击剥离强度值。与比较配制品相比,本发明的配制品的粘合剂粘度较高,这可能是由于本发明的增韧剂的较高分子量。通过简单地改变粘合剂配制品和使用有利于更多液体环氧树脂的更少的液体/固体环氧树脂共混物,可以将粘合剂粘度调节到更低的值。与比较配制品相比,本发明配制品的casson粘度以及在给定剪切速率下的实际粘度随着温度和时间的增加显著地较低。在40℃的测试温度下已经非常显著,在50℃以上的温度下变得非常明显。将ipdi用于增韧剂组合物(b-系列)的本发明的粘合剂配制品的e-模量通常高于将hdi用于增韧剂组合物(a-系列)的粘合剂配制品。将ipdi用于增韧剂组合物(b-系列)的本发明的增韧剂配制品的分子量低于将hdi用于增韧剂组合物(a-系列)的增韧剂配制品的分子量。通过使增韧剂与环氧树脂反应来提高稳定性的概念似乎是独立于增韧剂组合物而有效的。如上所述,可以使用任何合成方法,例如,同时或顺序。本发明包括增韧剂,其可以在结构上视为通过多酚基团或至少二羟基官能苯(两个或更多个羟基)连接到环氧基的异氰酸酯封端的pu预聚物。系列a和系列b的本发明的粘合剂配制品在粘合剂本体稳定性方面显示出比参考配制品显著的改善。机械强度值是相当的并且在高水平上。在实例中,观察到聚thf的分子量对本发明的增韧剂的性能有影响。由于当聚thf的分子量为1400da以下时冲击剥离强度降低,所以对于在23℃下改善的冲击剥离强度值而言,聚thf的分子量高于1400da是优选的。由于当聚thf的分子量为1700da以下时在-40℃测试温度下的冲击剥离强度值较低,所以聚thf的分子量高于1700da是更优选的。对于增韧剂组合物而言,ipdi优于hdi,因为其似乎赋予粘合剂配制品更高的模量。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1