聚酰亚胺薄膜、有机电致发光元件、透明导电性层叠体、触控面板、太阳能电池及显示设备的制作方法
本发明涉及聚酰亚胺薄膜、有机电致发光元件、透明导电性层叠体、触控面板、太阳能电池及显示设备。
背景技术:
近年来,在使用了有机电致发光元件的显示器或液晶显示器等的显示器机器领域等中,作为其基板等中利用的材料,要求出现一种如玻璃般透光性高且具有充分高的耐热性,并且轻且柔软的材料。而且,作为这样的玻璃替代用途等中所用的材料,具有高耐热性并且轻且柔软的由聚酰亚胺构成的薄膜正在受到关注。
作为这样的聚酰亚胺,例如已知有芳香族聚酰亚胺(例如,dupont公司制造的商品名“kapton”)。然而,这样的芳香族聚酰亚胺虽然是具有充分的柔软性和高耐热性的聚酰亚胺,但其呈现褐色,无法用于需要透光性的玻璃替代用途或光学用途等。
因此,近年来,为了用于玻璃替代用途等,正在进行具有充分的透光性的脂环式聚酰亚胺的开发,例如,在国际公开第2011/099518号(专利文献1)中公开有一种具有特定通式所记载的重复单元的聚酰亚胺。而且,该专利文献1所记载的聚酰亚胺具有充分的透光性和高耐热性。但是,上述专利文献1中,对于聚酰亚胺的拉伸强度或断裂伸长率等机械强度(韧性)方面,没有特别的记载。
现有技术文献
专利文献
专利文献1:国际公开第2011/099518号
技术实现要素:
发明所要解决的技术问题
本发明是鉴于上述现有技术所具有的技术问题而研发的,其目的在于提供一种能够以更高的水平且平衡性良好地具有拉伸强度及伸长特性,并且使以拉伸强度及断裂伸长率为基准的韧性更高的聚酰亚胺薄膜、以及使用其的有机电致发光元件。此外,本发明的目的在于提供一种使用了所述聚酰亚胺薄膜的透明导电性层叠体、以及使用了该透明导电性层叠体的触控面板、太阳能电池及显示设备。
解决技术问题的手段
本发明者们为了达成上述目的,经过重复深入研究,结果发现通过将聚酰亚胺薄膜制成由相对于全部重复单元含有30摩尔%以上的下述通式(1)所表示的重复单元的聚酰亚胺构成的薄膜,可以以更高的水平且平衡性良好地具有薄膜的拉伸强度及伸长特性(显示至断裂为止的充分的伸长的特性),并且使以拉伸强度及断裂伸长率为基准的韧性更高,至此完成了本发明。
即,本发明的聚酰亚胺薄膜由相对于全部重复单元含有30摩尔%以上的下述通式(1)所表示的重复单元的聚酰亚胺构成,其拉伸强度为125mpa以上,并且断裂伸长率为15%以上。
[式(1)中,r1、r2、r3各自独立地表示选自氢原子、碳原子数为1~10的烷基及氟原子中的1种,r10表示选自下述通式(101)~(102)所表示的基团中的1种,n表示0~12的整数。]
上述本发明的聚酰亚胺薄膜中,所述聚酰亚胺优选是相对于全部重复单元含有40摩尔%以上的所述通式(1)所表示的重复单元的聚酰亚胺。
本发明的有机电致发光元件具备上述本发明的聚酰亚胺薄膜。
另外,本发明的透明导电性层叠体具备上述本发明的聚酰亚胺薄膜和层叠于该聚酰亚胺薄膜上的由导电性材料构成的薄膜。
另外,本发明的触控面板、太阳能电池、显示设备分别具备上述本发明的透明导电性层叠体。
发明的效果
根据本发明,可以提供一种能够以更高的水平且平衡性良好地具有拉伸强度及伸长特性,并且使以拉伸强度及断裂伸长率为基准的韧性更高的聚酰亚胺薄膜、以及使用其的有机电致发光元件。另外,根据本发明,可以提一种使用了所述聚酰亚胺薄膜的透明导电性层叠体、以及使用了该透明导电性层叠体的触控面板、太阳能电池及显示设备。
附图说明
[图1]是表示本发明的有机电致发光元件的优选实施方式的概略纵向截面图。
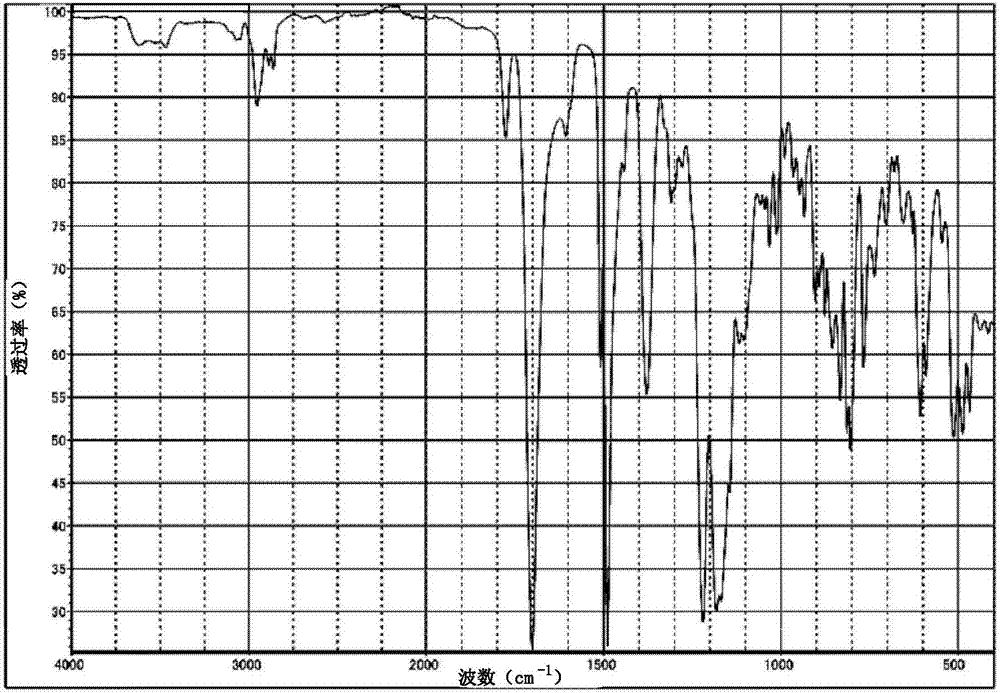
[图2]是表示实施例1中得到的聚酰亚胺薄膜的红外线吸收光谱(ir光谱)的图表。
具体实施方式
以下,就其优选的实施方式来详细地说明本发明。
[聚酰亚胺薄膜]
本发明的聚酰亚胺薄膜由相对于全部重复单元含有30摩尔%以上的下述通式(1)所表示的重复单元的聚酰亚胺构成,其拉伸强度为125mpa以上,并且断裂伸长率为15%以上。
[式(1)中,r1、r2、r3各自独立地表示选自氢原子、碳原子数为1~10的烷基及氟原子中的1种,r10表示选自下述通式(101)~(102)所表示的基团中的1种,n表示0~12的整数。]
能够选择为该通式(1)中的r1、r2、r3的烷基为碳原子数1~10的烷基。该碳原子数超过10时,玻璃化转变温度降低,从而无法达成充分高的耐热性。另外,作为能够选择为这样的r1、r2、r3的烷基的碳原子数,从精制变得更容易的观点出发,优选为1~6,进一步优选为1~5,更优选为1~4,特别优选为1~3。另外,能够选择为这样的r1、r2、r3的烷基可以为直链状,也可以为支链状。另外,作为这样的烷基,从容易精制的观点出发,更优选为甲基、乙基。
作为所述通式(1)中的r1、r2、r3,从在制造聚酰亚胺时,可以得到更高的耐热性的观点出发,更优选各自独立地为氢原子或碳原子数为1~10的烷基,其中,从原料取得容易或精制更容易等观点出发,更优选各自独立地为氢原子、甲基、乙基、正丙基或异丙基,特别优选为氢原子或甲基。另外,从精制的容易度等的观点出发,该式中的多个r1、r2、r3特别优选相同。
另外,能够选择为所述通式(1)中的r10的基团为上述通式(101)~(102)所表示的基团。作为这样的r10,从拉伸强度、断裂伸长率、耐热性的观点出发,优选为上述通式(101)所表示的基团,从拉伸强度、断裂伸长率、厚度方向延迟(retardation)(rth)的观点出发,优选为上述通式(102)所表示的基团。另外,在本发明中,作为所述通式(1)所表示的重复单元,可以含有r10的种类不同的2种以上的重复单元。
另外,所述通式(1)中的n表示0~12的整数。该n的值超过所述上限时,精制变得困难。另外,该通式(1)中的n的数值范围的上限值,从更容易精制的观点出发,进一步优选为5,特别优选为3。另外,该通式(1)中的n的数值范围的下限值,从形成通式(1)所表示的重复单元时所使用的原料化合物的安定性的观点,即从更容易制造聚酰亚胺的观点出发,进一步优选为1,特别优选为2。这样,作为通式(1)中的n,特别优选为2~3的整数。
另外,在本发明中,所述聚酰亚胺为相对于全部重复单元含有30摩尔%以上的上述通式(1)所表示的重复单元的聚酰亚胺。通过将该通式(1)所表示的重复单元的含量(含有多种的通式(1)所表示的重复单元的情况(例如,含有2种以上的选择为r1、r2、r3、r10等的基团的种类不同的式(1)所表示的重复单元的情况等)下,为这些的总量)相对于全部重复单元设定为30摩尔%以上,可以使得到的聚酰亚胺具有平衡性更良好的充分高的拉伸强度及充分高的断裂伸长率的值,由此,可以得到具有更高的韧性(机械强度)的聚酰亚胺薄膜(高韧性聚酰亚胺薄膜)。作为该通式(1)所表示的重复单元的含量(通式(1)所表示的重复单元的总量),从使拉伸强度为更高的值并且使断裂伸长率更高的观点,或从耐热性、透明性、厚度方向延迟(rth)的观点出发,相对于聚酰亚胺中的全部重复单元,进一步优选为40摩尔%以上,更优选为50摩尔%以上,特别优选为70摩尔%以上,特别优选为90摩尔%以上。
这样,在本发明中,所述聚酰亚胺只要相对于全部重复单元含有30摩尔%以上的上述通式(1)所表示的重复单元即可,也可以含有其他的重复单元。作为这样的其他的重复单元,没有特别的限制,可以适当地利用能够构成聚酰亚胺的公知的重复单元。另外,作为可以与该通式(1)所表示的重复单元一起利用的其他的重复单元,从耐热性、透明性、厚度方向延迟(rth)的观点出发,特别优选使用下述通式(2)所表示的重复单元。另外,该通式(2)中的r1、r2、r3及n与上述通式(1)中的r1、r2、r3及n相同含义(其优选的情况也分别与上述通式(1)中的r1、r2、r3及n相同。)。
[式(2)中,r1、r2、r3各自独立地表示选自氢原子、碳原子数为1~10的烷基及氟原子中的1种,ra表示碳原子数为6~40的芳基(但是,不包括上述通式(101)~(102)所表示的基团。),n表示0~12的整数。]
该通式(2)中的ra为上述通式(101)~(102)所表示的基团以外的碳原子数为6~40的芳基(另外,ra为上述通式(101)~(102)所表示的基团时,与上述通式(1)所表示的重复单元相同,因此,从式(2)中的ra中排除上述通式(101)~(102)所表示的基团。)。
能够选择为该通式(2)中的ra的芳基的碳原子数进一步优选为6~40(更优选为6~30、更加优选为12~20)。该碳原子数超过所述上限时,会有耐热性及拉伸强度降低的倾向。
另外,作为所述通式(2)中的ra,从耐热性、拉伸强度的观点出发,优选为下述通式(201)~(204)所表示的基团中的至少1种。
[式(203)中,rb表示选自氢原子、氟原子、甲基、乙基及三氟甲基中的1种,式(204)中,q表示选自式:-o-、-s-、-co-、-conh-、-coo-、-c6h4-、-c10h6-、-nhco-c6h4-conh-、-conh-c6h4-nhco-、-oco-c6h4-coo-、-coo-c6h4-oco-、-o-c10h6-o-、-oco-c10h6-coo-、-coo-c10h6-oco-、-conh-c10h6-nhco-、-nhco-c10h6-conh-、-so2-、-c(cf3)2-、-c(ch3)2-、-ch2-、-o-c6h4-c(ch3)2-c6h4-o-、-o-c6h4-so2-c6h4-o-及-c(ch3)2-c6h4-c(ch3)2-所表示的基团中的1种。]
另外,作为该通式(203)中的rb,从耐热性的观点出发,更优选为氢原子、氟原子、甲基或乙基,特别优选为氢原子。另外,作为上述通式(204)中的q,从耐热性、拉伸强度的观点出发,进一步优选为式:-conh-、-coo-、-c6h4-所表示的基团,更优选为-conh-、-c6h4-所表示的基团,特别优选为-conh-所表示的基团。
另外,作为能够选择为该ra的通式(201)~(204)所表示的基团,从耐热性、拉伸强度的观点出发,进一步优选为通式(203)或(204)所表示的基团,更优选为通式(204)所表示的基团。
另外,该通式(2)所表示的重复单元中,从使聚酰亚胺的拉伸强度更高的观点出发,更优选为ra为上述通式(204)所表示的基团且上述通式(204)中的q为-conh-、-c6h4-所表示的基团的重复单元,特别优选为ra为上述通式(204)所表示的基团且上述通式(204)中的q为-conh-所表示的基团的重复单元。
另外,在所述聚酰亚胺含有通式(2)所表示的重复单元的情况下,所述通式(2)所表示的重复单元的含量相对于全部重复单元,优选为10~70摩尔%,进一步优选为20~60摩尔%。该通式(2)所表示的重复单元的含量小于所述下限时,会有通过含有通式(2)所表示的重复单元而得到的效果(特别是使拉伸强度更高的效果等)不充分的倾向。另一方面,所述通式(2)所表示的重复单元的含量超过所述上限时,会有断裂伸长率的值降低、机械强度降低的倾向。
另外,本发明的聚酰亚胺薄膜,其拉伸强度需要为125mpa以上。该拉伸强度小于所述下限时,无法得到具有更高韧性的薄膜。另外,从相同的观点出发,该聚酰亚胺薄膜的拉伸强度进一步优选为130mpa以上,更优选为135mpa以上。另外,作为该聚酰亚胺薄膜的拉伸强度的上限值,没有特别的限制,但优选为1000mpa以下。该拉伸强度为超过所述上限值的值时,会有难以加工的倾向。
另外,本发明的聚酰亚胺薄膜,其断裂伸长率需要为15%以上。该断裂伸长率小于所述下限时,无法得到具有更高韧性的薄膜。另外,从相同的观点出发,该聚酰亚胺薄膜的断裂伸长率进一步优选为20%以上,更优选为25%以上。另外,作为该聚酰亚胺薄膜的断裂伸长率的上限值,没有特别的限制,但优选为300%以下。该断裂伸长率为超过所述上限值的值时,会有难以加工的倾向。
另外,关于本发明的聚酰亚胺薄膜,其拉伸强度及断裂伸长率的值可以采用依以下方法所求得的值。进行该测定时,首先,将dumbbellco.,ltd.制造的商品名“superdumbbellcutter(型号:sdmk-1000-d、按照jisk7139(2009年发行)的a22标准)”安装于sd型杠杆式样品裁剪机(dumbbellco.,ltd.制造的裁剪机(型号sdl-200)),裁剪聚酰亚胺薄膜(厚度:设定为13μm),调制测定样品。另外,对于由此制得的测定样品,除厚度为13μm以外,基本上为符合jisk7139(2009年发行)所记载的a22类型(缩尺试验片)的标准的哑铃形状的测定样品(试验片),对于其大小,全长:75mm、标记(tab)部间距离:57mm、平行部的长度:30mm、肩部的半径:≧30mm、端部的宽度:10mm、中央的平行部的宽度:5mm、厚度:13μm,在使用时,夹具间的宽度:57mm、夹持部分的宽度:10mm(与端部的总宽度为相同的宽度)。接着,使用tensilon型万能试验机(例如,a&dcompany,limited制造的型号“uct-10t”),将所述测定样品以夹具间的宽度为57mm、夹持部分的宽度为10mm(试验片的端部的总宽)的方式配置之后,以载荷满量程:0.05kn、试验速度:300mm/分钟的条件,进行对所述测定样品拉伸的拉伸试验,求得拉伸强度(断裂时的应力[单位:mpa])及断裂伸长率的值(单位:%)(该试验为依照jisk7162(1994年发行)的试验)。另外,对于断裂伸长率的值(%),将拉伸试验开始前的样品的标记部间距离(=夹具间的宽度:57mm)设定为l0、将拉伸试验中发生断裂为止的样品的标记部间距离(断裂时的夹具间的宽度:57mm+α)设定为l时,可以依下述式计算而求得。
[断裂伸长率(%)]={(l-l0)/l0}×100
另外,这样的聚酰亚胺优选酰亚胺化率为90%以上,进一步优选为95%以上,特别优选为96~100%。该酰亚胺化率小于所述下限时,会有耐热性降低、发现有着色、或加热时薄膜产生孔洞或膨胀的倾向。该酰亚胺化率可以通过以下方式算出。即,使测定对象的聚酰亚胺溶解于氘代氯仿等的氘代溶剂(优选为氘代氯仿)中,进行1h-nmr测定,根据1h-nmr的图表求得10ppm附近(10ppm±1ppm)的n-h的h与12ppm附近(12ppm±1ppm)的cooh的h的积分值,由此可以算出。该情况下,积分比(酰亚胺化率)可以采用通过如下方法算出的值,首先,对于原料化合物的酸二酐及二胺,制备使之溶解于对这些可溶的氘代溶剂(dmso-d6等)中而得到的样品,并分别测定这些的1h-nmr谱,在这些的1h-nmr的图表中,求得酸二酐的h的位置(化学位移)和积分值及二胺中的h的位置(化学位移)和积分值,使用该酸二酐的h的位置和积分值及二胺的h的位置和积分值为基准,对所述测定对象的聚酰亚胺的1h-nmr的图表中的10ppm附近的n-h的h和12ppm附近的cooh的h的积分值进行相对比较。另外,测定该酰亚胺化率时,测定1h-nmr谱的聚酰亚胺的量设定为相对于氘代溶剂(优选为氘代氯仿)为0.01~5.0质量%的量,原料化合物的酸二酐的量及二胺的量以相对于对这些可溶的氘代溶剂(dmso-d6等)分别成为0.01~5.0质量%的方式利用。另外,测定该酰亚胺化率时,聚酰亚胺的量、原料化合物的酸二酐的量及二胺的量(上述浓度)设定为相同的浓度进行测定。另外,所述1h-nmr测定中,作为测定装置使用nmr测定机(varian公司制造、商品名:unityinova-600)。
进一步,作为这样的聚酰亚胺,优选5%重量减少温度(td5%)为400℃以上的聚酰亚胺、更优选为450~550℃。这样的5%重量减少温度小于所述下限时,有难以达成充分的耐热性的倾向;另一方面,如果超过所述上限,则有难以制造具有这样的特性的聚酰亚胺的倾向。此外,这样的5%重量减少温度能够通过在氮气气氛下,一边流通氮气、一边将扫描温度设定为30℃~550℃,以升温速度:10℃/min.的条件进行加热,测定所使用的样品的重量减少5%的温度,由此来求得。另外,在这样的测定中,作为测定装置,例如,能够利用热重分析装置(siinanotechnologyinc.制造的“tg/dta220”)。
另外,作为这样的聚酰亚胺,优选玻璃化转变温度(tg)为250℃以上、更优选为300~500℃。该玻璃化转变温度(tg)小于所述下限时,有难以达成充分的耐热性的倾向;另一方面,如果超过上述上限,则有难以制造具有这样的特性的聚酰亚胺的倾向。此外,该玻璃化转变温度(tg)能够使用热机械分析装置(rigaku制造的商品名“tma8311”)作为测定装置,利用与软化温度测定相同的方法同时进行测定。此外,该玻璃化转变温度测定时,优选以升温速度:5℃/分钟的条件,在氮气氛下,通过扫描从30℃到550℃的范围来进行测定。
另外,作为这样的聚酰亚胺,软化温度优选为250~550℃、进一步优选为350~550℃、更加优选为360~510℃。该软化温度小于所述下限时,耐热性降低,例如,在作为太阳能电池、液晶显示设备、有机el显示设备的透明电极用的基板使用聚酰亚胺薄膜的情况下,在该产品的制造过程中的加热工序中,有难以充分抑制所述薄膜(基板)的品质的劣化(破裂的发生等)的倾向;另一方面,如果超过所述上限,则在制造聚酰亚胺时,无法与聚酰胺酸的热闭环缩合反应同时进行充分的固相聚合反应,反而在形成薄膜时有成为脆的膜的倾向。
此外,这样的聚酰亚胺的软化温度能够如下来进行测定。即,作为测定样品,准备长5mm、宽5mm、厚0.013mm(13μm)的大小的由聚酰亚胺构成的薄膜,作为测定装置使用热机械分析装置(rigaku制造的商品名“tma8311”),在氮气氛下,采用升温速度5℃/分钟的条件,在30℃~550℃的温度范围的条件下,用500mn的压力向薄膜中扎入透明石英制针(前端的直径:0.5mm),由此,能够与玻璃化转变温度(tg)同时进行测定(能够通过所谓的针入(penetration)法来进行测定)。另外,在进行该测定时,按照jisk7196(1991年)所记载的方法,基于测定数据来计算软化温度。
另外,作为形成这样的薄膜的聚酰亚胺,热分解温度(td)优选为450℃以上、进一步优选为480~600℃。该热分解温度(td)小于所述下限时,有难以达成充分的耐热性的倾向;另一方面,如果超过所述上限,则会有难以制造具有这样的特性的聚酰亚胺的倾向。另外,该热分解温度(td)可以通过使用tg/dta220热重分析装置(siinanotechnologyinc.制造),在氮气氛下,以升温速度10℃/分钟的条件测定成为热分解前后的分解曲线上画出的切线的交点的温度来求得。
另外,作为这样的聚酰亚胺的数均分子量(mn),以聚苯乙烯换算优选为1000~1000000,进一步优选为10000~500000。该数均分子量小于所述下限时,有不仅难以达成充分的耐热性,还在制造时无法充分地从聚合容器中析出,难以高效地制得聚酰亚胺的倾向;另一方面,如果超过所述上限,则粘性增大,使之溶解需要长时间或需要大量的溶剂,因此,有加工变得困难的倾向。
另外,作为形成这样的薄膜的聚酰亚胺的重均分子量(mw),以聚苯乙烯换算优选为1000~5000000。另外,作为该重均分子量(mw)的数值范围的下限值,进一步优选为5000,更加优选为10000,特别优选为20000。另外,作为重均分子量(mw)的数值范围的上限值,进一步优选为5000000,更加优选为500000,特别优选为100000。该重均分子量小于所述下限时,有不仅难以达成充分的耐热性,还在制造时无法充分地从聚合容器中析出,难以高效地制得聚酰亚胺的倾向;另一方面,如果超过所述上限,则粘性增大,使之溶解需要长时间或需要大量的溶剂,因此,有加工变得困难的倾向。
进一步,作为这样的聚酰亚胺的分子量分布(mw/mn)优选为1.1~5.0,进一步优选为1.5~3.0。该分子量分布小于所述下限时,有难以制造的倾向;另一方面,如果超过所述上限,则有难以得到均匀的薄膜的倾向。此外,这样的聚酰亚胺的分子量(mw或mn)、分子量的分布(mw/mn)能够通过将作为测定装置使用凝胶渗透色谱(gpc)测定装置(脱气机:jasco公司制造的dg-2080-54,送液泵:jasco公司制造的pu-2080,接口:jasco公司制造的lc-netll/adc,柱:shodex公司制造的gpc柱kf-806m(×2根),柱温箱:jasco公司制造的860-co,ri检测器:jasco公司制造的ri-2031,柱温度:40℃,氯仿溶剂(流速1ml/min.))测得的数据用聚苯乙烯换算而求得。
另外,这样的聚酰亚胺其线膨胀系数优选为0~100ppm/k,进一步优选为10~70ppm/k。该线膨胀系数超过所述上限时,在与线膨胀系数的范围为5~20ppm/k的金属或无机物组合复合化的情况下,会有在热履历中容易产生剥离的倾向。另外,所述线膨胀系数小于所述下限时,会有溶解性降低或薄膜特性降低的倾向。
作为该聚酰亚胺的线膨胀系数的测定方法,在形成了长:76mm、宽52mm、厚度13μm的大小的聚酰亚胺薄膜之后,将该薄膜真空干燥(120℃下1小时),在氮气氛下以200℃进行1小时热处理,将得到的干燥薄膜用作测定用样品,作为测定装置使用热机械分析装置(rigaku制造的商品名“tma8310”),在氮气氛下,使用拉伸模式(49mn)、升温速度5℃/分钟的条件,测定50℃~200℃下的所述样品的纵向的长度的变化,求得50℃~200℃的温度范围内每1℃的长度变化的平均值,采用由此得到的值。
进一步,这样的薄膜中的聚酰亚胺优选可以使之溶解于低沸点的铸造溶剂。只要是由这样的聚酰亚胺构成的薄膜,都可以更容易地调制。另外,作为这里所说的铸造溶剂,从溶解性、挥发性、蒸腾性、去除性、成膜性、生产性、工业取得性、可回收性、现有设备的有无、价格的观点出发,优选为沸点为200℃以下(进一步优选为20~150℃、更优选为30~120℃、特别优选为40~100℃、最优选为60℃~100℃)的溶剂。另外,作为这样的沸点为200℃以下的溶剂,进一步优选为沸点为200℃以下的卤素类溶剂,更加优选为二氯甲烷、三氯甲烷(氯仿)、四氯化碳、二氯乙烷、三氯乙烯、四氯乙烯、四氯乙烷、氯苯、邻二氯苯,特别优选为二氯甲烷、三氯甲烷(氯仿)。另外,这样的铸造溶剂可以单独使用1种,或者也可以将2种以上组合使用。
另外,作为这样的聚酰亚胺薄膜,优选透明性充分高,进一步优选总透光率为80%以上(更优选为85%以上,特别优选为87%以上)。另外,作为这样的聚酰亚胺薄膜,从优选透明性更高的观点出发,更优选雾度(浊度)为5以下(更加优选为4以下,特别优选为3以下)。另外,作为这样的聚酰亚胺薄膜,从优选透明性更高的观点出发,更优选黄度(yi)为10以下(更加优选为8以下,特别优选为6以下)。这样的总透光率、雾度(浊度)及黄度(yi)可以通过适当选择聚酰亚胺的种类等而容易地达成。另外,作为这样的总透光率、雾度(浊度)及黄度(yi),能够采用如下测得的值:作为测定用的样品,形成长:76mm、宽52mm、厚度13μm的大小的聚酰亚胺薄膜,作为测定装置,使用日本电色工业株式会社制造的商品名“雾度计ndh-5000”来进行测定。
这样的聚酰亚胺薄膜的形态只要是薄膜状即可,没有特别的限制,可以适当地设计为各种形状(圆盘状、圆筒状(将薄膜加工成筒状)等)。
进一步,本发明的聚酰亚胺薄膜的厚度没有特别的限制,优选为1~500μm,进一步优选为10~200μm。该厚度小于所述下限时,有强度降低,难以进行处理的倾向;另一方面,如果超过所述上限,则有产生需要进行多次涂布的情况或产生加工复杂化的情况的倾向。
另外,本发明的聚酰亚胺薄膜在用于显示机器时,从可以得到抑制对比度的降低及改善视角的效果出发,波长590nm下所测定的厚度方向的延迟(rth)换算为厚度10μm时,优选为-1000~1000nm(进一步优选为-500~500nm、更加优选为-250~250nm)的薄膜。另外,这样的本发明的聚酰亚胺薄膜的“厚度方向的延迟(rth)”可以通过使用axometrics公司制造的商品名“axoscan”作为测定装置,将以后述方式测定的聚酰亚胺薄膜的折射率(589nm)的值输入所述测定装置之后,在温度:25℃、湿度:40%的条件下,使用波长590nm的光,测定聚酰亚胺薄膜的厚度方向的延迟,基于所求得的厚度方向的延迟的测定值(通过测定装置的自动测定(自动计算)得到的测定值),换算为薄膜的每10μm厚度的延迟值而求得。另外,测定样品的聚酰亚胺薄膜的尺寸,只要比测定器平台的测光部(直径:约1cm)大即可,因此,没有特别的限制,但优选设为长:76mm、宽52mm、厚度13μm的大小。
另外,测定厚度方向的延迟(rth)中使用的“所述聚酰亚胺薄膜的折射率(589nm)”的值可以通过在形成由与形成成为延迟的测定对象的薄膜的聚酰亚胺相同种类的聚酰亚胺构成的未拉伸薄膜之后,将该未拉伸的薄膜用作测定样品(另外,作为测定对象的薄膜为未拉伸的薄膜时,可以将该薄膜直接用作测定样品。),作为测定装置使用折射率测定装置(atago株式会社制造的商品名“nar-1tsolid”),使用589nm的光源,在23℃的温度条件下测定测定样品的面内方向(与厚度方向垂直的方向)相对于589nm的光的折射率而求得。另外,由于测定样品未拉伸,因此,对于薄膜的面内方向的折射率,在面内的任意方向中皆为固定的值,通过测定该折射率,可以测定该聚酰亚胺的固有的折射率(另外,由于测定样品并未拉伸,因此,在将面内的延迟轴方向的折射率设为nx,将与延迟轴方向垂直的面内方向的折射率设为ny时,成为nx=ny)。这样,利用未拉伸的薄膜测定聚酰亚胺的固有的折射率(589nm),将所得到的测定值用于上述厚度方向的延迟(rth)的测定。在此,测定样品的聚酰亚胺薄膜的尺寸只要为可以用于所述折射率测定装置的大小即可,没有特别的限制,可以为1cm见方(长宽1cm)且厚度为13μm的大小。
这样的本发明的聚酰亚胺薄膜由于平衡性更良好地具有充分高的拉伸强度和充分高的断裂伸长率的值(以这些为基准,具有更高的韧性),并且机械强度更高,因此,在例如挠性配线基板用薄膜、耐热绝缘胶带、漆包线漆、半导体的保护涂敷剂、液晶取向膜、有机el用透明导电性薄膜、显示器的基板材料(tft基板、透明电极基板(例如,有机el用透明电极薄膜等)等显示器用基板)、有机el照明用薄膜、挠性基板薄膜、挠性有机el用基板薄膜、挠性透明导电性薄膜、有机薄膜型太阳能电池用透明导电性薄膜、染料敏化型太阳能电池用透明导电性薄膜、挠性气体阻隔膜、触控面板用的基板材料(触控面板用薄膜等)、挠性显示器用前膜、挠性显示器用背膜等中特别有用。另外,本发明的聚酰亚胺薄膜,如上所述,具有更高的韧性、更高的机械强度,因此,在用于如上所述的用途(其中,显示器的基板材料(tft基板、透明电极基板等的显示器用基板)、或触控面板用的基板材料(触控面板用薄膜等)等的用途)时,由来于其机械强度,也可以充分地改善最终产品(例如有机el元件等)的产率。
用于制造这样的聚酰亚胺薄膜的方法没有特别的限制,为了制造由含有上述重复单元的聚酰亚胺构成的薄膜,可以采用:使用下述通式(3)所表示的四羧酸二酐与含有下述通式(301)~(302)所表示的二胺化合物中的至少1种的芳香族二胺,以使在所得到的聚酰亚胺中上述通式(1)所表示的重复单元的含量相对于全部重复单元为30摩尔%以上,并适当地采用公知的方法(例如,国际公开第2011/099518号、国际公开第2014/034760号所记载的聚酰亚胺的制造方法)使之反应来制造聚酰亚胺薄膜的方法。
[式(3)中,r1、r2、r3、n与所述通式(1)中的r1、r2、r3、n相同意义。]
另外,作为用于制造这样的聚酰亚胺薄膜的方法,例如,可以适当地利用:在聚合溶剂的存在下,使上述通式(3)所表示的四羧酸二酐与含有所述通式(301)~(302)所表示的二胺化合物中的至少1种的芳香族二胺(根据目标的聚酰亚胺的设计,也可以仅含有所述通式(301)~(302)所表示的二胺化合物中的至少1种,或者也可以将所述通式(301)~(302)所表示的二胺化合物中的至少1种与其他的二胺化合物混合使用)进行反应,形成相对于全部重复单元含有30摩尔%以上(进一步优选为40摩尔%以上,更加优选为50摩尔%以上,特别优选为70摩尔%以上,最优选为90摩尔%以上)的下述通式(4)所表示的重复单元的聚酰胺酸之后,将含有该聚酰胺酸的聚酰胺酸的溶液涂布于基材(例如玻璃基材等)的表面上,接着,将该聚酰胺酸酰亚胺化,形成层叠于所述基材上的由相对于全部重复单元含有30摩尔%以上(进一步优选为40摩尔%以上,更加优选为50摩尔%以上,特别优选为70摩尔%以上,最优选为90摩尔%以上)的所述通式(1)所表示的重复单元的聚酰亚胺构成的薄膜的方法(以下,根据情况简称为“方法(a)”)。
[式(4)中,r1、r2、r3、n与所述通式(1)中的r1、r2、r3、n相同意义(其优选的情况也与所述通式(1)中的r1、r2、r3、n相同。),r10与所述通式(1)中的r10相同意义(其优选的情况也与所述通式(1)中的r10相同。)。]
关于该通式(3)所表示的四羧酸二酐,式(3)中的r1、r2、r3各自独立地为选自氢原子、碳原子数为1~10的烷基及氟原子中的1种,n为0~12的整数。该通式(3)中的r1、r2、r3、n与上述通式(1)中的r1、r2、r3、n相同,其优选的情况也与上述通式(1)中的r1、r2、r3、n的优选的情况相同。作为用于制造该通式(3)所表示的四羧酸二酐的方法,没有特别的限制,可以适当采用公知的方法,例如,也可以采用国际公开第2011/099517号所记载的方法或国际公开第2011/099518号所记载的方法等。
另外,作为这样的通式(3)所表示的四羧酸二酐,从调整薄膜特性、热物性、机械物性、光学特性、电特性的观点出发,优选含有下述通式(5)所表示的化合物(a)及下述通式(6)所表示的化合物(b)中的至少1种,并且所述化合物(a)及(b)的总量为90摩尔%以上。
[式(5)中,r1、r2、r3、n与所述通式(3)中的r1、r2、r3、n相同意义。]
[式(6)中,r1、r2、r3、n与所述通式(3)中的r1、r2、r3、n相同意义。]
该通式(5)所表示的化合物(a)是2个降冰片烷基(norbornanegroup)反式构型,并且环烷酮的羰基相对于该2个降冰片烷基分别成为内位(endo)的立体构型的上述通式(3)所表示的四羧酸二酐的异构体。另外,该通式(6)所表示的化合物(b)是2个降冰片烷基顺式构型,并且环烷酮的羰基相对于该2个降冰片烷基分别成为内位的立体构型的上述通式(3)所表示的四羧酸二酐的异构体。另外,以上述比例含有这样的异构体的四羧酸二酐的制造方法也没有特别的限制,可以适当采用公知的方法,例如,也可以适当采用国际公开第2014/034760号所记载的方法等。
另外,作为与通式(3)所表示的四羧酸二酐进行反应的芳香族二胺,需要以在所制得的聚酰亚胺中相对于全部重复单元含有30摩尔%以上(进一步优选为40摩尔%以上,更加优选为50摩尔%以上,特别优选为70摩尔%以上,最优选为90摩尔%以上)的所述通式(1)所表示的重复单元的方式,以适当的比例利用通式(301)所表示的二胺化合物(1,4-双(4-氨基苯氧基)苯)及通式(302)所表示的二胺化合物(4,4’-双(4-氨基苯氧基)联苯)中的至少1种。从这样的观点出发,通式(301)~(302)所表示的二胺化合物中的至少1种的含量优选为30摩尔%以上(进一步优选为40摩尔%以上,更加优选为50摩尔%以上,特别优选为70摩尔%以上,最优选为90摩尔%以上)。另外,作为这样的通式(301)~(302)所表示的二胺化合物,也可以分别使用市售品。
进一步,在用于制造这样的聚酰亚胺薄膜的方法中,在作为其他重复单元含有所述通式(2)所表示的重复单元时,作为与通式(3)所表示的四羧酸二酐进行反应的芳香族二胺,优选使用将上述通式(301)~(302)所表示的二胺化合物中的至少1种与其他的二胺化合物混合而成的混合物。
作为这样的其他的二胺化合物,没有特别的限制,可以适当地利用制造聚酰亚胺时所使用的公知的芳香族二胺化合物。这样的其他的二胺化合物中,从耐热性、透明性的观点出发,更优选为式:h2n-ra-nh2[式中,ra表示碳原子数为6~40的芳基(但是,不包括上述通式(101)~(102)所表示的基团。)。]所表示的二胺化合物。另外,这样的其他的二胺化合物的式中的ra与上述通式(2)中的ra相同意义(其优选的情况也相同)。另外,作为这样的其他的二胺化合物,从耐热性、透明性的观点出发,更加优选为4,4’-二氨基苯甲酰苯胺、4,4”-二氨基对三联苯、4-氨基苯甲酸4-氨基苯基酯,特别优选为4,4’-二氨基苯甲酰苯胺。这些其他的二胺化合物可以单独使用1种或将2种以上组合使用。另外,作为这样的其他的芳香族二胺,也可以适当地使用市售品。
另外,作为所述方法(a)中使用的所述聚合溶剂,优选能够溶解上述通式(3)所表示的四羧酸二酐和所述芳香族二胺(含有所述通式(301)~(302)所表示的二胺化合物中的至少1种的芳香族二胺(也可以是所述通式(301)~(302)所表示的二胺化合物中的至少1种与其他的二胺化合物的混合物))两者的有机溶剂。
作为这样的有机溶剂,例如,可以列举n-甲基-2-吡咯烷酮、n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、二甲基亚砜、γ-丁内酯、碳酸丙烯酯、四甲基脲、1,3-二甲基-2-咪唑啉酮、六甲基磷酰三胺、吡啶等的非质子类极性溶剂;间甲酚、二甲酚、苯酚、卤代苯酚等的酚类溶剂;四氢呋喃、二恶烷、溶纤剂、乙二醇二甲醚、二甘醇二甲醚(diglyme)等的醚类溶剂;苯、甲苯、二甲苯等的芳香族类溶剂;环戊酮或环己酮等的酮类溶剂;乙腈、苄腈等的腈类溶剂等。这样的聚合溶剂(有机溶剂)可以单独使用1种或将2种以上混合使用。
另外,在所述方法(a)中,对于上述通式(3)所表示的四羧酸二酐与所述芳香族二胺的使用比例,相对于所述芳香族二胺所具有的氨基1当量,优选将上述通式(3)所表示的四羧酸二酐的酸酐基设定为0.2~2当量,进一步优选为0.8~1.2当量。该使用比例小于所述下限时,聚合反应无法有效率地进行,会有无法得到高分子量的聚酰胺酸的倾向;另一方面,如果超过所述上限,则与上述同样地有无法得到高分子量的聚酰胺酸的倾向。
进一步,在所述方法(a)中,作为所述聚合溶剂(有机溶剂)的使用量,优选以使上述通式(3)所表示的四羧酸二酐与所述芳香族二胺的总量相对于反应溶液的总量成为0.1~50质量%(进一步优选为10~30质量%)的量。该有机溶剂的使用量小于所述下限时,会有无法有效率地制得聚酰胺酸的倾向;另一方面,如果超过所述上限,则有由于高粘度化而难以搅拌的倾向。
另外,在所述方法(a)中,作为使上述通式(3)所表示的四羧酸二酐与所述芳香族二胺进行反应的方法,没有特别的限制,可以适当地采用能够进行四羧酸二酐与所述芳香族二胺的反应的公知的方法,例如,也可以采用:在大气压的条件下,在氮、氦、氩等的惰性气氛下,使所述芳香族二胺溶解于溶剂中之后,添加上述通式(3)所表示的四羧酸二酐,其后,使之反应10~48小时的方法。另外,在进行该反应时,优选将温度条件设定为-20~100℃左右。该反应时间或反应温度小于所述下限时,会有难以充分地进行反应的倾向;另一方面,如果超过所述上限,则使聚合物劣化的物质(氧等)的混入几率提高,有分子量降低的倾向。
另外,在所述方法(a)中,关于作为中间体形成的所述聚酰胺酸,所述通式(4)中的r1、r2、r3、n与所述通式(1)中的r1、r2、r3、n相同意义,其优选的情况也与所述通式(1)中的r1、r2、r3、n相同意义。另外,所述通式(4)中的r10与所述通式(1)中的r10相同意义,其优选的情况也相同。这样,所述通式(4)中的r10为选自上述通式(101)~(102)所表示的基团中的1种基团。
进一步,在所述方法(a)中,作为形成为中间体的所述聚酰胺酸,其特性粘度[η]优选为0.05~3.0dl/g,进一步优选为0.1~2.0dl/g。如果该特性粘度[η]小于0.05dl/g,则使用其制造薄膜状的聚酰亚胺时,有所得到的薄膜变脆的倾向;另一方面,如果超过3.0dl/g,则粘度过高加工性降低,例如在制造薄膜时,难以制得均匀的薄膜。
另外,该聚酰胺酸的特性粘度[η]可以以如下的方式进行测定。即,首先使用n,n-二甲基乙酰胺作为溶剂,将所述聚酰胺酸溶解于该n,n-二甲基乙酰胺中使浓度为0.5g/dl,得到测定样品(溶液)。接着,使用所述测定样品,在30℃的温度条件下使用运动粘度计测定所述测定样品的粘度,将求得的值作为特性粘度[η]使用。另外,作为该运动粘度计,使用离合公司制造的自动粘度测定装置(商品名“vmc-252”)。
另外,作为所述方法(a)中使用的基材,没有特别的限制,可以根据作为目标的由聚酰亚胺构成的薄膜的形状等,适当使用能够用于薄膜的形成的由公知的材料构成的基材(例如,玻璃板或金属板)。
进一步,在所述方法(a)中,作为在所述基材上涂布所述聚酰胺酸溶液的方法,没有特别的限定,例如,可以适当采用旋涂法、喷涂法、浸涂法、滴加法、凹版印刷法、丝网印刷法、凸版印刷法、模具涂布法、淋幕式涂布法、喷墨法等公知的方法。
所述方法(a)中,将所述聚酰胺酸酰亚胺化的方法也没有特别的限制,只要是能够使聚酰胺酸酰亚胺化的方法即可,没有特别的限制,可以适当采用公知的方法(国际公开第2011/099518号、国际公开第2014/034760号所记载的酰亚胺化的方法等)。作为使该聚酰胺酸酰亚胺化的方法,例如,优选采用将含有上述通式(4)所表示的重复单元的聚酰胺酸在60~400℃(进一步优选为60~370℃、更加优选为150~360℃)的温度条件下施以加热处理由此进行酰亚胺化的方法,或使用所谓的“酰亚胺化剂”进行酰亚胺化的方法。
另外,该方法(a)中,也可以采用:在将所述聚酰胺酸酰亚胺化之前,不分离含有上述通式(4)所表示的重复单元的聚酰胺酸,而在聚合溶剂(有机溶剂)中使上述通式(3)所表示的四羧酸二酐类与所述芳香族二胺反应,直接使用所得到的反应液(包含含有上述通式(4)所表示的重复单元的聚酰胺酸的反应液),将所述反应液涂布在基材上之后,施以干燥处理去除溶剂,其后,施以所述加热处理由此进行酰亚胺化的方法。作为该干燥处理的方法中的温度条件,优选为0~180℃,进一步优选为60~150℃。另外,也可以从所述反应液中将含有上述通式(4)所表示的重复单元的聚酰胺酸分离,该情况下,作为聚酰胺酸的分离方法,没有特别的限制,可以适当使用能够将聚酰胺酸分离的公知的方法,例如,也可以采用作为再沉淀物进行分离的方法等。
经由该方法(a),可以以层叠于基材上的状态得到本发明的聚酰亚胺薄膜。另外,将由此得到的聚酰亚胺薄膜由基材剥离并回收时,该剥离方法没有特别的限制,可以适当采用公知的方法,例如,也可以采用通过将基材上层叠有聚酰亚胺薄膜的层叠体浸渍于高温的水(例如80℃以上的水)中,从基材上将聚酰亚胺薄膜剥离的方法等。
[有机电致发光元件]
本发明的有机电致发光元件具备上述本发明的聚酰亚胺薄膜。
作为这样的有机电致发光元件,例如,除了具备上述本发明的聚酰亚胺薄膜以外,其他的结构没有特别的限制,可以适当地利用公知的结构。另外,作为这样的有机电致发光元件,没有特别的限制,例如,从提高生产时的产率的观点出发,优选具备上述本发明的聚酰亚胺薄膜作为透明电极层叠用的基板。
以下,一边参照附图,一边对能够适用作这样的本发明的有机电致发光元件(有机el元件)的有机el元件的一个实施方式进行简单地说明。另外,在以下的说明及附图中,对相同或相当的要素赋予相同的符号,省略重复的说明。
图1为本发明的有机电致发光元件(有机el元件)的优选的一个实施方式的概略纵向截面图。图1所示的实施方式的有机el元件1具备聚酰亚胺薄膜11、气体阻隔层12、透明电极层13、有机层14和金属电极层15。
该有机el元件中的聚酰亚胺薄膜11由上述本发明的聚酰亚胺薄膜构成。在本实施方式中,该聚酰亚胺薄膜11作为有机el元件的基板(透明电极层叠用的基板)使用。
另外,气体阻隔层12使防止气体(包含水蒸气)的透过性能更高,是适合用于抑制气体向元件内部穿透的层。作为该气体阻隔层12,没有特别的限制,例如,可以适当利用由sin、sio2、sic、sioxny、tio2、al2o3等的无机物构成的层、超薄板玻璃等。对于该气体阻隔层12,也可以在聚酰亚胺薄膜11上适当配置(形成)并层叠公知的具有气体阻隔性的层。
另外,气体阻隔层12的厚度没有特别的限制,优选为0.01~5000μm的范围,进一步优选为0.1~100μm的范围。该厚度小于所述下限时,有无法得到充分的气体阻隔性的倾向;另一方面,超过所述上限时,有重厚化,挠性或柔软性等特长消失的倾向。
透明电极层13为用作有机el元件的透明电极的层。作为该透明电极层13的材料,只要是能够利用于有机el元件的透明电极的即可,没有特别的限制,例如,可以使用氧化铟、氧化锌、氧化锡、及这些的复合体即铟·锡·氧化物(ito)、金、铂、银、铜。这些中,从兼有透明性与导电性的观点出发,优选为ito。
另外,透明电极层13的厚度优选为20~500nm的范围。厚度小于所述下限时,有导电性变得不充分的倾向;另一方面,超过所述上限时,有透明性不充分,未使发光的el光充分地射向外部的倾向。
另外,也可以在气体阻隔层12与透明电极层13之间形成所谓的薄膜晶体管(tft)层。通过这样设置tft层,也可以形成具有连接于tft的透明电极的装置(tft元件)。该tft层的材料(氧化物半导体、非晶硅、多晶硅、有机晶体管等)或结构没有特别的限制,可以基于公知的tft的结构作适当设计。另外,在聚酰亚胺薄膜11与气体阻隔层12的层叠体上设置tft层时,也可以将这些的层叠体用作所谓的tft基板。另外,作为这样的tft层的制造方法,没有特别的限制,可以适当采用公知的方法,例如,也可以采用低温多晶硅法、高温多晶硅法、非晶硅法、氧化物半导体法等制造方法。
有机层14只要是可以用于形成有机el元件的即可,其结构没有特别的限制,可以适当利用公知的有机el元件的有机层中能够使用的物质。另外,该有机层14的结构也没有特别的限制,可以采用公知的结构,例如,也可以将由空穴传输层、发光层及电子传输层构成的层叠体作为有机层。
作为该空穴传输层的材料,可以适当地使用能够形成空穴传输层的公知的材料,例如,可以使用萘二胺(α-npd)、三苯胺、三苯基二胺衍生物(tpd)、联苯胺、吡唑啉、苯乙烯胺、腙、三苯基甲烷、咔唑等的衍生物等。
另外,发光层为由电极层等注入的电子和空穴再结合并发光的层,作为该发光层的材料没有特别的限制,可以适当地使用能够形成有机el元件的发光层的公知的材料,例如,可以适当地使用在4,4’-n,n’-二咔唑-联苯(cbp)中掺杂有三苯基吡啶铱(iii)配合物(ir(ppy)3)而成的材料,或由8-羟基喹啉铝(alq3、绿色、低分子)、双(8-羟基)喹哪啶铝苯氧化物(bis-(8-hydroxy)quinaldinealuminumphenoxide)(alq’2oph、蓝色、低分子)、5,10,15,20-四苯基-21h,23h-卟吩(5,10,15,20-tetraphenyl-21h,23h-porphine)(tpp、红色、低分子)、聚(9,9-二辛基芴-2,7-二基)(poly(9,9-dioctylfluorene-2,7-diyl))(pfo、蓝色、高分子)、聚[2-甲氧基-5-(2’-乙基己氧基)-1,4-(1-氰基亚乙烯基)亚苯基](poly[2-methoxy-5-(2’-ethylhexyloxy)-1,4-(1-cyanovinylene)phenylene])(meh-cn-ppv、红色、高分子)、蒽等的荧光性的有机固体构成的材料等通过施加电压而发光的公知的材料。
进一步,作为电子传输层的材料没有特别的限制,可以适当地使用能够形成电子传输层的公知的材料,例如,可以使用铝喹啉醇配合物(alq)、邻二氮杂菲衍生物、恶二唑衍生物、三唑衍生物、苯基喹喔啉(phenylquinoxaline)衍生物、噻咯(silole)衍生物等。
另外,有机层14为由空穴传输层、发光层及电子传输层构成的层叠体的情况下,空穴传输层、发光层及电子传输层的各层的厚度没有特别的限制,分别优选为1~50nm的范围(空穴传输层)、5~200nm的范围(发光层)及5~200nm的范围(电子传输层)。另外,作为有机层14的整体的厚度,优选为20~600nm的范围。
金属电极层15为由金属构成的电极。作为该金属电极的材料,可以使用功函数较小的物质,没有特别的限定,例如可以列举铝、mgag、mgin、alli。另外,金属电极层15的厚度优选为50~500nm的范围。厚度小于所述下限时,会有导电性降低的倾向;另一方面,超过所述上限时,会有容易剥离或产生裂缝的倾向。
另外,这样的有机el元件的制造方法没有特别的限制,例如,可以采用:通过在准备上述本发明的聚酰亚胺薄膜之后,在该聚酰亚胺薄膜的表面上依次层叠所述气体阻隔层、所述透明电极、所述有机层及所述金属电极来进行制造的方法。
作为在该聚酰亚胺薄膜11的表面上层叠气体阻隔层12的方法,没有特别的限制,可以适当使用蒸镀法、溅射法等公知的方法,其中,从制成致密的膜的观点出发,优选采用溅射法。另外,作为在气体阻隔层12的表面上层叠透明电极层13的方法,可以适当使用蒸镀法、溅射法等公知的方法,其中,从制成致密的膜的观点出发,优选采用溅射法。
另外,在透明电极层13的表面上层叠有机层14的方法也没有特别的限制,例如,如上所述,在将有机层制成由空穴传输层、发光层及电子传输层构成的层叠体时,只要将这些层依次层叠于透明电极层13上即可。另外,作为层叠该有机层14中的各层的方法,没有特别的限制,可以适当地利用公知的方法,例如,可以采用蒸镀法、溅射法、涂布法等。这些方法中,从充分防止有机层的分解、劣化及变性的观点出发,优选采用蒸镀法。
进一步,作为在有机层14上层叠金属电极层15的方法,没有特别的限制,可以适当地利用公知的方法,例如,可以采用蒸镀法、溅射法等。这些方法中,从充分防止先形成的有机层14分解、劣化及变性的观点出发,优选采用蒸镀法。
另外,通过这样制造有机el元件,可以形成将所述聚酰亚胺薄膜11用作支持所谓的元件部的基板的有机el元件,因此,可以来源于其机械强度而实现产率的提高,并且可以使挠性充分高。
以上,说明了本发明的有机el元件的优选的一个实施方式,本发明的有机el元件不限定于上述实施方式。例如,在上述实施方式中,有机层14由空穴传输层、发光层及电子传输层的层叠体构成,但对有机层的实施方式没有特别的限制,可以适当采用公知的有机层的结构,例如,可以制成由空穴注入层与发光层的层叠体构成的有机层;由发光层与电子注入层的层叠体构成的有机层;由空穴注入层、发光层与电子注入层的层叠体构成的有机层;或由缓冲层、空穴传输层与电子传输层的层叠体构成的有机层等。另外,该有机层的其他实施方式中的各层的材料没有特别的限制,可以适当使用公知的材料。例如,作为电子注入层的材料,可以使用苝衍生物等;作为空穴注入层的材料,可以使用三苯胺衍生物等;作为阳极缓冲层的材料,可以使用铜酞菁、pedot等。另外,即使是未在上述实施方式中配置的层,只要是能够用于有机el元件的层,也可以适当地配置,例如,从容易对有机层14进行电荷注入或空穴注入的观点出发,也可以在透明电极层13上或有机层14上设置由氟化锂(lif)、li2o3等的金属氟化物、ca、ba、cs等活性高的碱土金属、有机绝缘材料等构成的层。
[透明导电性层叠体]
本发明的透明导电性层叠体具备上述本发明的聚酰亚胺薄膜、和层叠于该聚酰亚胺薄膜上的由导电性材料构成的薄膜。
这样,本发明的透明导电性层叠体为在上述本发明的聚酰亚胺薄膜上层叠由所述导电性材料构成的薄膜而成的。在该透明导电性层叠体中,从透明的观点出发,优选总透光率为78%以上(进一步优选为80%以上,更加优选为82%以上)。该总透光率可以通过适当地选择上述本发明的聚酰亚胺薄膜的种类或所述导电性材料的种类等而容易地达成。另外,作为该总透光率,可以采用使用日本电色工业株式会社制造的商品名“浊度仪ndh-5000”作为测定装置所测定的值。
另外,作为所述导电性材料,只要是具有导电性的材料即可,没有特别的限制,可以适当利用能够用于太阳能电池或有机el元件、液晶显示设备的透明电极等的公知的导电性材料,例如,可以列举金、银、铬、铜、钨等的金属;在锡、铟、锌、镉、钛等金属氧化物中掺杂有其他的元素(例如,锡、碲、镉、钼、钨、氟、锌、锗、铝等)而得到的复合体(例如,氧化铟锡(ito(in2o3:sn))、氟掺杂氧化锡(fto(sno2:f))、铝掺杂氧化锌(azo(zno:al))、铟掺杂氧化锌(izo(zno:i))、锗掺杂氧化锌(gzo(zno:ge))等);等。另外,这些导电性材料中,从可以以更高的水平且平衡性良好地发挥透明性和导电性的观点出发,优选使用ito(特别优选为含有锡3~15质量%的ito)。
作为由该导电性材料构成的薄膜(导电性薄膜)的膜厚,可以根据用途等适当地变更设计,没有特别的限制,优选为1~2000nm,进一步优选为10nm~1000nm,更优选为20~500nm,特别优选为20~200nm。该导电性薄膜的厚度小于所述下限时,表面电阻值没有充分地变低,在用于太阳能电池的情况等时,会有光电变换效率降低的倾向;另一方面,超过所述上限时,有透过率降低或成膜时间变长,造成生产效率降低的倾向。
作为将由该导电性材料构成的薄膜层叠于上述本发明的聚酰亚胺薄膜上的方法,没有特别的限制,可以适当地利用公知的方法,例如,可以采用:在所述聚酰亚胺薄膜上通过溅射法、真空蒸镀法、离子镀法、等离子体cvd法等气相堆积法形成所述导电性材料的薄膜,由此将所述薄膜层叠于所述聚酰亚胺薄膜上的方法。另外,这样,在所述聚酰亚胺薄膜上层叠所述薄膜时,也可以预先在所述聚酰亚胺薄膜上形成气体阻隔膜,经由该气体阻隔膜在所述聚酰亚胺薄膜上层叠所述薄膜。另外,作为该气体阻隔膜,没有特别的限制,可以适当地使用能够用于太阳能电池或有机el元件、液晶显示设备的透明电极等的公知的膜,其形成方法也可以适当地利用公知的方法。
这样的本发明的透明导电性层叠体由于所述聚酰亚胺薄膜具有更高的韧性,因此,对于例如太阳能电池的透明电极、显示设备(有机el显示设备、液晶显示设备等)的透明电极等特别有用,可以更充分地改善这些的最终产品的产率。
[触控面板、太阳能电池、显示设备]
本发明的触控面板、太阳能电池、显示设备分别具备上述本发明的透明导电性层叠体。
此处所说的“显示设备”只要是能够利用透明导电性层叠体的即可,没有特别的限制,可以列举液晶显示设备、有机el显示设备。另外,作为这样的触控面板、太阳能电池、显示设备,除了分别具备上述本发明的透明导电性层叠体以外,其他的结构没有特别的限制,可以根据作为目标的设计,适当地采用公知的结构。作为这样的结构,例如,作为触控面板,可以列举如包含夹持透明电极和空隙地配置的其他的透明电极的结构,作为太阳能电池,可以列举如包含透明电极、半导体层及对电极用导电层的结构,作为有机el显示设备,可以列举如包含透明电极、有机层及对电极用导电层的结构,作为液晶显示设备,可以列举如包含透明电极、液晶层及对电极用导电层的结构。另外,作为这样的有机层、液晶层或半导体层等各层的材料,没有特别的限制,可以适当地使用公知的材料。另外,本发明的触控面板、太阳能电池、显示设备中,优选分别将上述本发明的透明导电性层叠体作为所述透明电极利用。这样,通过使用上述本发明的透明导电性层叠体作为所述透明电极,即使暴露在触控面板、太阳能电池、显示设备(液晶显示设备、有机el显示设备)的制造过程中通常使用的高温条件下,也可以充分抑制透明电极层(由导电性材料构成的薄膜)产生裂痕等,因此,可以以良好的产率制造品质充分高的触控面板、太阳能电池、显示设备。
实施例
以下,基于实施例及比较例对本发明进行更具体地说明,但本发明不限定于以下的实施例。
首先,将各实施例、各比较例中所使用的芳香族二胺的化学式和该化合物的简称示于以下。
接着,说明各实施例、各比较例中所得到的聚酰亚胺薄膜等的特性的评价方法。
<分子结构的鉴定>
各实施例和各比较例中所得到的化合物的分子结构的鉴定通过利用ir测定机(日本分光株式会社制造、商品名:ft/ir-4100)进行ir测定来进行。
<特性粘度[η]的测定>
在各实施例和各比较例中作为中间体而得到的聚酰胺酸的特性粘度[η]的值(单位:dl/g),如上所述,利用离合公司制造的自动粘度测定装置(商品名“vmc-252”),利用以n,n-二甲基乙酰胺为溶剂的浓度为0.5g/dl的测定样品,在30℃的温度条件下进行测定。
<拉伸强度及断裂伸长率的测定>
各实施例及各比较例中,聚酰亚胺薄膜(厚度:13μm)的拉伸强度(单位:mpa)及断裂伸长率(单位:%)以如下方式测定。即,首先,在sd型杠杆式样品裁剪机(dumbbellco.,ltd.制造的裁剪机(型号sdl-200))上安装dumbbellco.,ltd.制造的商品名“superdumbbellcutter(型号:sdmk-1000-d、按照jisk7139(2009年发行)的a22标准)”,以所述聚酰亚胺薄膜的大小成为全长:75mm、标记部间距离:57mm、平行部的长度:30mm、肩部的半径:30mm、端部的宽度:10mm、中央的平行部的宽度:5mm、厚度:13μm的方式进行剪裁,制得哑铃形状的试验片(除了将厚度设为13μm以外,其他按照jisk7139类型a22(缩尺试验片)的标准)作为测定样品。接着,使用tensilon型万能试验机(例如,a&dcompany,limited制造的型号“uct-10t”),将所述测定样品配置为夹具间的宽度57mm、夹持部分的宽度10mm(端部的总宽)之后,以载荷满量程:0.05kn、验速度:300mm/分钟的条件进行对所述测定样品拉伸的拉伸试验,求得拉伸强度及断裂伸长率的值。另外,这样的试验为按照jisk7162(1994年发行)进行的试验。另外,对于断裂伸长率的值(%),将拉伸试验开始前的样品的标记部间距离(=夹具间的宽度:57mm)设定为l0、将拉伸试验中发生断裂为止的样品的标记部间距离(断裂时的夹具间的宽度:57mm+α)设定为l时,以下述式计算而求得。
[断裂伸长率(%)]={(l-l0)/l0}×100
<玻璃化转变温度(tg)的测定>
各实施例及各比较例中所得到的化合物(形成薄膜的化合物)的玻璃化转变温度(tg)的值(单位:℃)通过使用各实施例及各比较例中制得的聚酰亚胺薄膜,使用热机械分析装置(rigaku制造的商品名“tma8311”)作为测定装置,并采用与下述软化温度的测定方法相同的方法(相同条件),与测定软化温度同时进行测定。
<软化温度的测定>
各实施例及各比较例中所得到的化合物(形成薄膜的化合物)的软化温度(软化点)的值(单位:℃)通过使用各实施例和各比较例中制得的聚酰亚胺薄膜,作为测定装置利用热机械分析装置(rigaku制造的商品名“tma8311”),在氮气氛下,以升温速度5℃/分钟、30℃~550℃的温度范围(扫描温度)的条件以500mn压对薄膜扎入透明石英制针(前端的直径:0.5mm),由此来测定(通过所谓的针入法来测定)。在该测定时,除了利用上述测定样品以外,依据jisk7196(1991年)中所记载的方法,根据测定数据来计算软化温度。
<5%重量减少温度(td5%)的测定>
各实施例等中所得到的化合物的5%重量减少温度(td5%)的值(单位:℃)通过利用在各实施例和各比较例中制造得到的聚酰亚胺薄膜,使用热重分析装置(siinanotechnologyinc.制造的“tg/dta220”),将扫描温度设定为30℃~550℃,在氮气氛下、一边流通氮气一边以10℃/min.的条件进行加热,测定所使用的样品的重量减少5%的温度,由此来求得。
<总透光率、雾度(浊度)和黄度(yi)的测定>
总透光率的值(单位:%)、雾度(浊度:haze)和黄度(yi)通过使用各实施例和各比较例中制造得到的聚酰亚胺薄膜,作为测定装置使用日本电色工业株式会社制造的商品名“雾度计ndh-5000”,进行按照jisk7361-1(1997年发行)的测定,由此来求得。
<折射率的测定>
各实施例及各比较例中制得的聚酰亚胺薄膜的折射率(对589nm的光的折射率)通过从用与各实施例及各比较例中采用的方法相同的方法制得的聚酰亚胺薄膜(未拉伸的薄膜)切取1cm见方(长宽1cm)且厚度13μm的薄膜,将其用作测定样品,作为测定装置使用折射率测定装置(atago株式会社制造的商品名“nar-1tsolid”),使用589nm的光源,在23℃的温度条件下,测定相对于589nm的光的面内方向(与厚度方向垂直的方向)的折射率(聚酰亚胺的固有的折射率),由此来求得。
<厚度方向的延迟(rth)>
厚度方向的延迟(rth)的值(单位:nm)通过直接使用各实施例及各比较例中制得的聚酰亚胺薄膜(长:76mm、宽:52mm、厚度:13μm)作为测定样品,作为测定装置使用axometrics公司制造的商品名“axoscan”,输入各个聚酰亚胺薄膜的折射率(通过上述折射率测定所求得的薄膜对589nm的光的折射率)的值之后,在温度:25℃、湿度:40%的条件下,使用波长590nm的光,测定厚度方向的延迟之后,使用所求得的厚度方向的延迟的测定值(通过测定装置的自动测定所得到的测定值),换算为薄膜的厚度每10μm的延迟值,由此而求得。
(实施例1)
<四羧酸二酐的准备工序>
按照国际公开第2011/099518号的合成例1、实施例1及实施例2所记载的方法,准备下述通式(7)所表示的四羧酸二酐(降冰片烷-2-螺-α-环戊酮-α’-螺-2”-降冰片烷-5,5”,6,6”-四羧酸二酐)。
<聚酰胺酸的调制工序>
首先,将30ml的三口烧瓶使用加热枪进行加热,使其充分干燥。接着,将充分干燥后的所述三口烧瓶内的气氛以氮气置换,使所述三口烧瓶内为氮气氛。接着,在所述三口烧瓶内添加作为芳香族二胺的1,4-双(4-氨基苯氧基)苯0.2631g(0.90mmol:和歌山精化工业株式会社制造:4,4-bab)之后,再添加n,n-二甲基乙酰胺2.7g,进行搅拌,由此使芳香族二胺(4,4-bab)溶解于所述n,n-二甲基乙酰胺中,得到溶解液。
接着,在含有所述溶解液的三口烧瓶内,在氮气氛下,添加上述通式(7)所表示的化合物0.3459g(0.90mmol)之后,在氮气氛下、在室温(25℃)下搅拌12小时而制得反应液。由此在反应液中形成聚酰胺酸。
另外,利用该反应液(聚酰胺酸的n,n-二甲基乙酰胺溶液:聚酰胺酸溶液)的一部分,调制聚酰胺酸的浓度为0.5g/dl的n,n-二甲基乙酰胺溶液,如上所述测定作为反应中间体的聚酰胺酸的特性粘度[η],结果聚酰胺酸的特性粘度[η]为0.98dl/g。
<由聚酰亚胺构成的薄膜的调制工序>
作为玻璃基板准备大型载玻片(松浪硝子工业株式会社制造的商品名“s9213”、长:76mm、宽52mm、厚度1.3mm),将如上所述得到的反应液(聚酰胺酸溶液)旋涂于所述玻璃基板的表面上以使加热固化后的涂膜的厚度为13μm,在所述玻璃基板上形成涂膜。其后,将形成有所述涂膜的玻璃基板放置在60℃的加热板上,静置2小时,使溶剂从所述涂膜中蒸发并除去(溶剂去除处理)。
在实施这样的溶剂去除处理之后,将形成有所述涂膜的玻璃基板投入以3l/分钟的流量流通氮气的惰性气氛烘箱(inertoven)内,在惰性气氛烘箱内,在氮气氛下在25℃的温度条件下静置0.5小时之后,在135℃的温度条件下加热0.5小时,进一步在340℃的温度条件(最终加热温度)下加热1小时,使所述涂膜固化,得到在所述玻璃基板上涂布有由聚酰亚胺构成的薄膜(聚酰亚胺薄膜)的聚酰亚胺涂覆玻璃。
接着,将由此制得的聚酰亚胺涂覆玻璃浸渍于90℃的热水中,将聚酰亚胺薄膜从所述玻璃基板上剥离,由此得到聚酰亚胺薄膜(长76mm、宽52mm、厚度13μm的大小的薄膜)。
另外,为了鉴定由此得到的形成聚酰亚胺薄膜的化合物的分子结构,使用ir测定机(日本分光株式会社制造、商品名:ft/ir-4100),测定ir光谱。将作为这样的测定结果得到的ir光谱示于图2中。由图2所示的结果也可以明确,对于构成实施例1中所形成的薄膜的化合物,在1701cm-1处观察到酰亚胺羰基的c=o伸缩振动。由基于这样的结果等所鉴定的分子结构,确认所得到的薄膜由聚酰亚胺构成。
另外,根据所使用的单体的种类,所制得的聚酰亚胺为上述通式(1)所表示的重复单元(式中的r1、r2、r3都为氢原子,n为2,r10为上述通式(101)所表示的基团的重复单元)的含有率相对于全部重复单元为100摩尔%的聚酰亚胺。另外,有关所得到的聚酰亚胺薄膜,将特性的评价结果(由上述特性的评价方法所求得的tg和软化温度等)示于表1中。
(实施例2)
除了在聚酰胺酸的调制工序中,作为芳香族二胺,替代1,4-双(4-氨基苯氧基)苯而使用4,4’-双(4-氨基苯氧基)联苯0.3316g(0.90mmol:日本纯良药品株式会社制造:apbp),在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为320℃以外,采用与实施例1相同的方法制得聚酰亚胺薄膜。另外,鉴定形成与实施例1同样地得到的薄膜的化合物的分子结构,结果确认其为聚酰亚胺。另外,根据所使用的单体的种类,所制得的聚酰亚胺为上述通式(1)所表示的重复单元(式中的r1、r2、r3都为氢原子,n为2,r10为上述通式(102)所表示的基团的重复单元)的含有率相对于全部重复单元为100摩尔%的聚酰亚胺。另外,将聚酰胺酸的特性粘度[η]、聚酰亚胺薄膜的特性的评价结果(由上述特性的评价方法所求得的tg和软化温度等)示于表1中。
(比较例1)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用4,4’-二氨基苯甲酰苯胺0.2045g(0.90mmol:日本纯良药品株式会社制造:daban)以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例2)
除了在聚酰胺酸的调制工序中,作为芳香族二胺,替代1,4-双(4-氨基苯氧基)苯而使用4,4’-二氨基-2,2’-双(三氟甲基)联苯0.2882g(0.90mmol:东京化成株式会社制造:6fda),另外,在聚酰胺酸的调制工序中,制得反应液时,替代在氮气氛下在室温(25℃)下进行12小时的搅拌,变更温度条件而在氮气氛下在60℃下进行12小时搅拌,进一步,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为360℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例3)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用1,3-双(4-氨基苯氧基)苯0.2631g(0.90mmol:和歌山精化工业株式会社制造:tpe-r),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为300℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例4)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用2,2-双[4-(4-氨基苯氧基)苯基]丙烷0.3695g(0.90mmol:和歌山精化工业株式会社制造:bapp),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为300℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例5)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用双[4-(4-氨基苯氧基)苯基]砜0.3892g(0.90mmol:和歌山精化工业株式会社制造:baps),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为300℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例6)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用4,4’-二氨基二苯基醚0.1802g(0.90mmol:东京化成株式会社制造:4,4’-dde),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为300℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例7)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用双[4-(3-氨基苯氧基)苯基]砜0.3892g(0.90mmol:和歌山精化工业株式会社制造:baps-m),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为300℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例8)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用2,2-双[4-(4-氨基苯氧基)苯基]六氟丙烷0.4666g(0.90mmol:东京化成株式会社制造:bis-af)以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例9)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用1,3-双(3-氨基苯氧基)苯0.2631g(0.90mmol:mitsuifinechemicals,inc.制造:3,3-bab),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为300℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(比较例10)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用间甲苯胺0.1911g(0.90mmol:和歌山精化工业株式会社制造:m-tol),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为350℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,将聚酰胺酸及聚酰亚胺薄膜的特性的评价结果示于表1中。
(实施例3)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用1,4-双(4-氨基苯氧基)苯0.2105g(0.72mol:4,4-bab)与4,4’-二氨基苯甲酰苯胺0.0409g(0.18mol:daban)的混合物(4,4-bab与daban的摩尔比([4,4-bab]:[daban])为80:20),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为350℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,鉴定形成用与实施例1相同的方法得到的薄膜的化合物的分子结构,结果确认其为聚酰亚胺。另外,根据所使用的单体的种类,由此制得的聚酰亚胺为上述通式(1)所表示的重复单元(式中的r1、r2、r3都为氢原子,n为2,r10为上述通式(101)所表示的基团的重复单元)的含有率相对于全部重复单元为80摩尔%的聚酰亚胺。另外,将聚酰胺酸的特性粘度[η]、聚酰亚胺薄膜的特性的评价结果(由上述特性的评价方法所求得的tg和软化温度等)示于表2中。
(实施例4)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用4,4’-双(4-氨基苯氧基)联苯0.2653g(0.72mol:apbp)与4,4’-二氨基苯甲酰苯胺0.0409g(0.18mol:daban)的混合物(apbp与daban的摩尔比([apbp]:[daban])为80:20),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为350℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,鉴定形成用与实施例1相同的方法得到的薄膜的化合物的分子结构,结果确认其为聚酰亚胺。另外,根据所使用的单体的种类,由此制得的聚酰亚胺为上述通式(1)所表示的重复单元(式中的r1、r2、r3都为氢原子,n为2,r10为上述通式(102)所表示的基团的重复单元)的含有率相对于全部重复单元为80摩尔%的聚酰亚胺。另外,将聚酰胺酸的特性粘度[η]、聚酰亚胺薄膜的特性的评价结果(由上述特性的评价方法所求得的tg和软化温度等)示于表2中。
(实施例5)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用4,4’-双(4-氨基苯氧基)联苯0.1990g(0.54mol:apbp)与4,4’-二氨基苯甲酰苯胺0.0818g(0.36mol:daban)的混合物(apbp与daban的摩尔比([apbp]:[daban])为60:40),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为350℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,鉴定形成用与实施例1相同的方法得到的薄膜的化合物的分子结构,结果确认其为聚酰亚胺。另外,根据所使用的单体的种类,由此制得的聚酰亚胺为上述通式(1)所表示的重复单元(式中的r1、r2、r3都为氢原子,n为2,r10为上述通式(102)所表示的基团的重复单元)的含有率相对于全部重复单元为60摩尔%的聚酰亚胺。另外,将聚酰胺酸的特性粘度[η]、聚酰亚胺薄膜的特性的评价结果(由上述特性的评价方法所求得的tg和软化温度等)示于表2中。
(实施例6)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用4,4’-双(4-氨基苯氧基)联苯0.1326g(0.36mol:apbp)与4,4’-二氨基苯甲酰苯胺0.1227g(0.54mol:daban)的混合物(apbp与daban的摩尔比([apbp]:[daban])为40:60),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为350℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,鉴定形成用与实施例1相同的方法得到的薄膜的化合物的分子结构,结果确认其为聚酰亚胺。另外,根据所使用的单体的种类,由此制得的聚酰亚胺为上述通式(1)所表示的重复单元(式中的r1、r2、r3都为氢原子,n为2,r10为上述通式(102)所表示的基团的重复单元)的含有率相对于全部重复单元为40摩尔%的聚酰亚胺。另外,将聚酰胺酸的特性粘度[η]、聚酰亚胺薄膜的特性的评价结果(由上述特性的评价方法所求得的tg和软化温度等)示于表2中。
(比较例11)
除了在聚酰胺酸的调制工序中,作为芳香族二胺替代1,4-双(4-氨基苯氧基)苯而使用4,4’-双(4-氨基苯氧基)联苯0.0663g(0.18mol:apbp)与4,4’-二氨基苯甲酰苯胺0.1636g(0.72mol:daban)的混合物(apbp与daban的摩尔比([apbp]:[daban])为20:80),另外,在由聚酰亚胺构成的薄膜的调制工序中,将惰性气氛烘箱内的所述最终加热温度由340℃变更为350℃以外,采用与实施例1相同的方法,制得聚酰亚胺薄膜。另外,根据所使用的单体的种类,由此制得的聚酰亚胺为上述通式(1)所表示的重复单元(式中的r1、r2、r3都为氢原子,n为2,r10为上述通式(102)所表示的基团的重复单元)的含有率相对于全部重复单元为20摩尔%的聚酰亚胺。另外,将聚酰胺酸的特性粘度[η]、聚酰亚胺薄膜的特性的评价结果(由上述特性的评价方法所求得的tg和软化温度等)示于表2中(另外,表2中,为了参照,也一并示出实施例1及2的结果。)。
[表2]
由表1及表2所示的结果也可以明显确认,本发明的聚酰亚胺薄膜(实施例1~6)都拉伸强度为125mpa以上,并且断裂伸长率为15%以上,以更高的水平且平衡地具有拉伸强度及伸长特性(至断裂为止的伸长特性),具有更高的韧性。另外,本发明的聚酰亚胺薄膜(实施例1~8)都tg为295℃以上,软化温度为470℃以上,5%重量减少温度为494℃以上,因此,确认其具有非常高的耐热性。另外,本发明的聚酰亚胺薄膜(实施例1~8)其总透光率都为87%以上,也确认了其具有充分高的透明性。由这样的结果可知,各实施例中所得到的聚酰亚胺薄膜(本发明的聚酰亚胺薄膜)具有高耐热性和充分的透明性,并且显示更高的韧性(高韧性:更高的机械强度)。另外,由表2所示的结果也明显可知,通过含有40摩尔%以上的上述通式(1)所表示的重复单元,可以以更高的水平且平衡性良好地发挥拉伸强度与伸长特性,并且显示更高的韧性(更高的机械强度)。
另一方面,比较例1~11中制得的聚酰亚胺薄膜,其拉伸强度小于125mpa或断裂伸长率小于15%,未必可以以充分高的水平平衡性良好地发挥拉伸强度与伸长特性。由这样的结果可知,各比较例中所得到的聚酰亚胺薄膜与本发明的聚酰亚胺薄膜(实施例1~6)比较时,其韧性不一定充分,且机械强度也不一定充分。
产业上利用性的可能性
如以上所说明的,根据本发明,可以提供一种能够以更高的水平且平衡性良好地具有拉伸强度及伸长特性,并且使以拉伸强度及断裂伸长率为基准的韧性更高的聚酰亚胺薄膜、以及使用其的有机电致发光元件。另外,根据本发明,可以提一种使用了所述聚酰亚胺薄膜的透明导电性层叠体、以及使用了该透明导电性层叠体的触控面板、太阳能电池及显示设备。
另外,本发明的聚酰亚胺薄膜由于具有更高的韧性,并且其机械强度更优异,因此,在用于例如有机el显示器、液晶显示器或触控面板等的基板材料的情况下,由来于其优异的机械强度,也可以充分地改善最终产品的产率。从这样的观点出发,本发明的聚酰亚胺薄膜作为例如挠性配线基板用薄膜、耐热绝缘胶带、漆包线漆、半导体的保护涂敷剂、液晶取向膜、有机el(有机电致发光)用透明导电性薄膜、有机el照明用薄膜、挠性基板薄膜、挠性有机el用基板薄膜、挠性透明导电性薄膜、有机薄膜型太阳能电池用透明导电性薄膜、染料敏化型太阳能电池用透明导电性薄膜、挠性气体阻隔膜、触控面板用薄膜、挠性显示器用前膜、挠性显示器用背膜等特别有用。
符号的说明:
1…有机el元件、11…聚酰亚胺薄膜、12…气体阻隔层、13…透明电极层、14…有机层、15…金属电极层。
- 还没有人留言评论。精彩留言会获得点赞!