制备用于包装应用的开环聚环烯烃的方法与流程
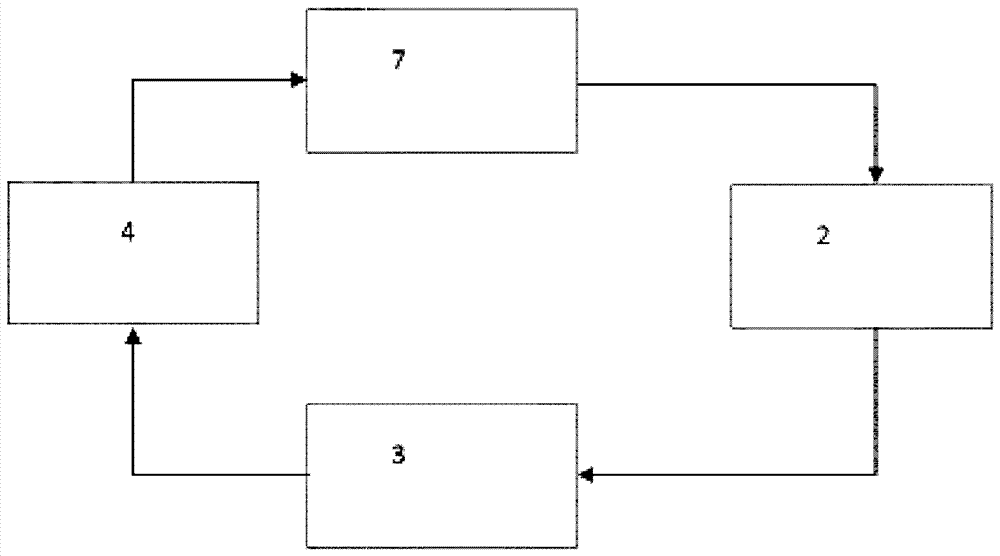
本发明涉及生产含有开环聚环烯烃的组合物的方法。本发明进一步涉及含有开环聚环烯烃的组合物在包装材料、特别是食品用包装材料的领域中的用途。为了增加包装食品的寿命,可以采用主动氧气阻隔的原理。这意味着,除了常规的被动阻隔层(例如尼龙-6、聚对苯二甲酸乙二醇酯或乙烯-乙烯醇共聚物)之外,在包装中还使用通过化学反应(氧化)结合氧气的另外的“主动”组分。这可能首先涉及包装内存在的氧气(气调包装(map)中的残余氧气),其次涉及随着时间的推移穿过被动阻隔扩散到包装中的氧气。该“主动”组分可以存在于包装的不同区域中;例如,它可以是多层包装系统的单独层的一部分,或者可以被直接引入到上文提及的被动阻隔层中。与另外的“主动”组分的化学反应减少了氧气与例如包装食品的成分(脂肪、维生素等)或好氧细菌的任何化学反应以及真菌生长,从而使得食品的品质保存更长时间。这进而又产生了一些益处,因为在销售之前或消耗之前较少的食品被损坏,因此资源在各个方面得以保留。此外,只需要将较低水平的防腐剂添加至食品中,如果有此需要的话。典型地,主动组分含有易氧化的有机化合物,以及另外的其它成分,例如金属盐作为催化剂或光引发剂。为此目的提出的可氧化的化合物是例如开环聚环辛烯;参见例如ep2017308a1、wo9407944a1、wo9407379a1和wo9806779a1。开环聚环辛烯的制备从文献中已知(参见例如us2013/172635),并且其遵循所谓的易位聚合的原理。还已知开环聚环辛烯,与其它的易位聚合物一样,起始于单体,含有一定比例的低分子量环状化合物(低聚物)(参见a.dräxler的“handbookofelastomers”第二版,697-722,2001)。这些分子是相对可移动的,直至特定的分子量,即转化成气相并由于它们的气味活性而导致产生包装材料的不利气味。而且,由于它们的极性导致它们是脂溶性的,因此可以想到它们将进入包装材料中。由于这些性质,通过易位制备的聚合物在包装应用中具有有限的可能用途,这意味着重要的应用领域甚至仍保持紧闭,特别是为了使用开环聚环辛烯作为含有“主动氧气阻隔”的包装的成分的目的。在文献中已经描述了用丙酮或异丙醇从开环聚环辛烯中萃取低分子量环状化合物;参见a.dräxler的“handbookofelastomers”,第2版,697-722,2001。ep2017308a1也描述了用各种溶剂进行相应的萃取。环烯烃的开环易位聚合(romp)本身是已知的(olefinmetathesisandmetathesispolymerization,k.j.irvin,j.c.mol,academicpress1997;handbookofmetathesis,第1-3卷,r.h.grubbs,wiley-vch2003)。该反应通过多种过渡金属或其化合物来催化,通常伴随着助催化剂的使用,该助催化剂与过渡金属或加入的过渡金属化合物一起在反应中形成催化活性的过渡金属物类。合适的助催化剂特别是铝有机物(organyls)和锡有机物。其它催化剂基于限定的过渡金属配合物。最为人所知的化合物包括基于钌的配合物(weskamp,t.kohl,f.j.herrmann,w.a.j.organomet.chem.1999,582,362-365;weskamp,t.kohl,f.j.hieringer,w.,gleich,d.hermann,w.a.angew.chem.int.ed.1999,38,2416-2419;nguyen,s.t.,johnson,l.w.,grubbs,r.h.,ziller,j.w.,j.am.chem.soc.1992,114,3974-3975;bielawski,c.w.,grubbs,r.h.,angew.chem.int.ed.2000,39,2903-2906)。然而,这里的缺点是它们的成本高,特别是难以将其从反应产物中分离。钌的残留物导致产品的颜色通常是不可接受的。在这些情况下,聚合物必须通过复杂的方法(例如再沉淀)来纯化,这对于经济型制备而言是一个障碍。所得的聚合物的性质可以通过参数(诸如温度、单体浓度、催化剂浓度和反应时间)来调节。分子量可以通过加入链转移剂来控制,其任务是终止增长的链。由于该过程是统计过程,分子量相当近似地与链转移剂的浓度呈反比关系。由于二次易位(链转移或“反咬”)而导致的分子量分布变宽在这里未被考虑。因此,可以通过加入链转移剂来影响分子量,而不是分子量分布的宽度。之后在该反应中,存在二次易位,其中被添加到增长链的活性末端上的不是另外的单体分子,而是现有的聚合物链。结果是链转移,这导致多分散性(表示为(mw/mn)-1或mw/mn)的增加。随着反应推进的进一步观察结果是顺式/反式比朝有利于反式构型而移动。这个效应同样可以归因于二次易位。为了在聚合物中构建特定的性质,由此需要的是对各种各样的不同工艺参数的精确控制。通过romp聚合环烯烃构成了制备开环聚环烯烃的重要方法。其中一个实例是环辛烯的聚合,得到开环聚环辛烯(例如来自evonikindustries,de的vestenamer®)。通常,开环聚环烯烃以固体形式使用;但是对于某些应用而言,需要使聚合物在室温下处于液态。开环聚环烯烃的一个重要应用是用于包装中,例如用于包装膜中,以改善膜的阻隔性质,尤其是在氧气方面,还在于其它物质方面,例如co2或水。更具体地说,通过由开环聚环烯烃化学结合氧气(主动阻隔效果)改善了阻隔性质。在这种情况下,通常将加速开环聚环烯烃与氧气反应的过渡金属化合物加入到开环聚环烯烃中(ep2017308a1)。环烯烃的聚合在所获得的产物混合物中留下单体和单体的低聚物。研究已表明,这些化合物尤其具有增强的气味活性。若干位作者报导了,气味活性与除其它性质以外的摩尔质量相关。根据来源的不同,这类有气味的物质(气味活性有机化合物,ovoc)具有不大于350g/mol或小于300g/mol的摩尔质量,以便具有充分的挥发性并且可以作为气味来感知(m.schlegelmilch,geruchsmanagement:methodenzurbewertungandverminderungvongeruchsemissionen[odourmanagement:methodsofassessingandreducingodouremissions],来自hamburg-harburguniversityoftechnology的hamburgerberichte32,abfallaktuellpublishers2009,isbn978-3-9810064-9-0;m.schön,r.hübner,geruch-messung和beseitigung[odour–measurementandelimination],vogelpublisherswürzburg,1996年第1版,isbn3-8023-1561-8;umweltbundesamt,innenraumlufthygiene-kommissiondesumweltbundesamtes,leitfadenfürdieinnenraumhygieneinschulgebäuden[germanenvironmentagency,indoorairhygienecommissionofthegermanenvironmentagency,guidelinesforindoorairhygieneinschoolbuildings],第47页,2008;g.scharfenberger,papier+kunststoff-verarbeiter10,1990)。超临界气体用于固体萃取的用途长久以来都是已知的(munshi等人在currentscience97(1),2009,63-72中;thepharmainnovationjournal2014,3(5)19-24)。还已经描述了从聚合物中萃取低分子量成分(us4306058;journalofchromatographya,855(1999)715-721;journalofappliedpolymerscience,第101卷,4487-4492(2006);bartle等人在anal.chem.1991,63,2371-2377中)。这里也可以使用助溶剂。由于与使用有机溶剂相关的已知缺点以及该气体是易燃的或只能以相当大的难度从最终产物中再次分离的事实,二氧化碳是现有技术中优选的萃取剂。然而,已经发现,使用超临界二氧化碳的常规萃取对所萃取的产物的材料一致性具有不利影响,因为所萃取的产物在用标准工艺参数萃取的过程中部分烧结或至少部分压缩。这具有渗透性降低的效应,其可能导致边缘流和通道的形成。这造成萃取性能降低。此外,萃取材料在萃取后可能不再是原来的粉末或颗粒形式。因此所解决的问题是提供生产含有开环聚环烯烃的组合物的方法,该方法产生具有降低的气味活性的产物。与现有技术的方法相比,获得具有合适的降低的单体和低聚物含量的聚合物。通过温和的co2萃取除去单体和低聚物,但不引起开环聚环烯烃的烧结或压缩。开环聚环烯烃类化合物具有至少相等的主动阻隔效果(例如在氧气的化学结合方面具有相等的效果)。这将确保在食品部门中的使用。通过分两个阶段进行co2萃取来实现该目的。因此,通过生产含有开环聚环烯烃的组合物的方法来实现该目的,所述方法包括以下步骤:a)通过开环易位聚合转化至少一种环烯烃以获得含有开环聚环烯烃的产物混合物,以及b)通过用co2萃取(co2萃取)对所述产物混合物进行后处理以除去环烯烃的单体和低聚物,以便获得含有开环聚环烯烃的组合物,其中所述萃取包括至少两个阶段:b1)用液态co2萃取,然后b2)用超临界co2萃取。以这种方式,通过co2作为萃取剂从产物混合物中萃取单体和低聚物。根据本发明的两阶段方法防止了萃取材料的烧结或压缩。调整萃取条件,从而使得阶段b1中不存在超临界co2。因此,可以避免烧结和压缩。该萃取因而采用液态二氧化碳开始。为此目的,液态二氧化碳在其可以在超临界条件下工作之前首先必须至少一次穿过萃取装置。之后,调整条件,从而使超临界co2得以存在。本领域技术人员通过简单的初步实验根据低聚物分布确定阶段b2中用于除去单体和低聚物的最佳温度和压力条件。优选地,首先引入co2,在亚临界条件(压力低于73.8巴或温度低于31℃)下转化为液态物质(阶段b1),例如通过热交换器、泵或压缩机。液态co2优选具有大于0℃至99℃的温度和10巴至1000巴的压力(这里压力和温度是相互匹配的,从而使得co2为液体形式)。因此,可以调整阶段b1的条件,从而使得温度或压力已经高于临界点。但是,优选至少使温度保持低于31℃的临界温度。在这方面,阶段b1中的压力或温度设定如下:i)压力和温度低于co2的临界值,或ii)压力高于co2的临界值,温度低于co2的临界值,或iii)压力低于co2的临界值,温度高于co2的临界值。这里优选选择变型ii或iii,因为在步骤b2中已经为萃取构建了一个参数(压力或温度),并且不需要再调整。变型ii是特别优选的,因为它在装置方面不那么复杂,并且提高温度但不增加压力以构建临界条件所耗费的时间较短。在压力增加的情况下,还会产生压缩热;必须另外移除该热量。随后在超临界条件下引入超临界气体,例如通过热交换器、泵或压缩机(阶段b2)。为此目的,co2优选具有31℃至99℃的温度和74巴至1000巴的压力。优选40℃至90℃的温度和100巴至500巴的压力。特别优选40℃至70℃的温度和200巴至500巴的压力;甚至更优选40℃至50℃的温度和250巴至500巴的压力。压力越高,萃取结果通常应该越好。压力可以通过所用的萃取装置来限制。将超临界二氧化碳通入萃取容器中,其中存在含有开环聚环烯烃的产物混合物。优选地,使超临界二氧化碳连续地通过萃取材料。在根据本发明的方法中,相对质量通量(在下文中称为s/f=溶剂/进料比)可以为10kg直至500kg的co2/kg的含有开环聚环烯烃的产物混合物。理想地,s/f值为25kg至250kg的co2/kg的含有开环聚环烯烃的产物混合物。在低于10kg的co2/kg的含有开环聚环烯烃的产物混合物的s/f值下,没有观察到单体和低聚物的显著耗尽。适合地,在根据步骤b2)的萃取之后,从萃取材料(单体和低聚物)中分离co2萃取剂。这可以通过本领域技术人员熟知的工艺来完成(工艺i;参见例如ep0154258a2)。例如,这可以通过分离器来实施,由此使得co2萃取物经受使萃取物所包含的co2转化成气态并且使包含所萃取的单体和低聚物的相以液态存在的温度-压力条件。压力和/或温度条件的变化随后也会导致气体的溶液性质的变化,并且随后在分离容器中使之前溶解的物质例如从气体中分离出来。然后气态co2可以被转化回到液态,转移到储存容器中,然后以超临界状态再循环到萃取回路中(例如通过泵)。co2可以通过可以是气态、液态或超临界形式的吸附剂来纯化。合适的吸附剂例如选自活性炭、氧化铝、二氧化硅或混合物,所述混合物包括例如硅铝酸盐,如沸石。优选地,负载萃取物的co2被减压至低于临界压力(73.8巴)的压力。这使气体冷却,然后其呈湿蒸气的形式。形成富含萃取物的液化气相和基本上不含萃取物的气相,该比率取决于起始压力或起始温度。为了分离,使液态二氧化碳蒸发,优选连续地使液态二氧化碳蒸发,然后以等压方式使其达到分离温度。为此目的,优选的温度在相应的主要分离压力下比沸点高至少1k-50k。特别优选比沸点高5k-40k的温度,非常特别优选比沸点高10-20k的温度。在分离之后,可以在压力相关的浓度温度下使再生的气体液化并且将其送回至工艺中。或者,根据本发明的方法以及因此还有所述分离可以在临界co2条件下进行,其中吸附剂吸收或结合所萃取的材料(单体和低聚物)(工艺ii)。这里优选使压力相对于步骤b2保持,并降低温度(仍然是临界温度)或保持温度(等压条件),由此保持温度是优选的。合适的吸附剂例如选自活性炭、氧化铝、二氧化硅或混合物,所述混合物包括例如硅铝酸盐,如沸石。活性炭是优选的吸附剂。这里可以在各种情况下采用所选择的吸附材料影响含有开环聚环烯烃的产物混合物,例如通过均质或异质混合,通过将吸附材料分层引入含有开环聚环烯烃的产物混合物中,或通过吸附材料的下游连接。当将吸附剂分层引入到床中时或者当在待萃取的床的下游连接吸附剂时是优选的。已经发现,当所述分离通过工艺ii进行时,尤其是在等压条件下进行时,是特别有利的。特别是在相对高的s/f值(>50)下,工艺ii明显优于工艺i(通过分离器萃取)。由于显著较低的能量消耗和较低的资金成本所产生的优势显然超过了吸附材料的附加成本。优选将工艺ii的s/f值设定为50-400kg的co2/kg的含有开环聚环烯烃的产物混合物。更优选设定为50-200kg的co2/kg的含有开环聚环烯烃的产物混合物。图1示出了其中进行工艺i的设备的示意性构造。co2保留在储存容器(1)中。下游设置的是高压泵或压缩机(2)。高压釜(萃取容器)(4)的上游是热交换器(3)。在高压釜的下游,将co2引导至分离容器(5)中,其中收集单体和低聚物z。使从分离容器(5)中移除的气体在冷凝器(6)中液化并且将其送回至储存容器(1)。图2示出了用于工艺ii的设备构造。与工艺i的装置不同,分离容器被压力容器(7)所替代,co2被引导至所述压力容器(7)中;该装置含有吸附剂。随后经由泵(2)和热交换器(3)将气体传导至高压釜(4)。图2中未示出储存容器(1);它在回路的外部以确保等压操作模式。可以连续地实施所述方法。当含有开环聚环烯烃的产物混合物已经在第一高压釜中萃取时,第二高压釜可以配备有其它产物混合物。在处理第一高压釜之后,将超临界co2导入第二高压釜。第一高压釜中的压力得以释放。该方法具有使用超临界co2或压缩co2而无需任何额外的能量输入的优点。co2例如以径向或轴向方式流过含有开环聚环烯烃的产物混合物。在径向流入的情况下,已经发现当流动方向从外部向内引导时是有利的。这产生了返混(backup),结果是co2更均匀地分布在床中。超临界co2的量是不受限制的。超临界co2的总重量基于含有开环聚环烯烃的产物混合物的总重量的比率优选为1:1至500:1,优选为10:1至200:1,更优选为20:1至50:1。co2可以含有助溶剂。合适的助溶剂选自芳族烃如甲苯、烷烃、氯代烷烃、醇、链烷羧酸酯、脂族酮、脂族醚、水及其混合物。优选的助溶剂是己烷。基于co2和助溶剂的质量,优选co2含有小于10重量%、更优选0.5-7.5重量%、最优选1-6重量%的助溶剂。本发明的上下文中的低聚物尤其包括所用环烯烃的二聚体、三聚体和四聚体。本发明的上下文中的开环聚环烯烃是包含至少五个环烯烃单体单元的环烯烃的聚合物。优选地,基于所述组合物的总重量,含有开环聚环烯烃的组合物中的单体、二聚体、三聚体和四聚体(杂质)的总和小于20000ppm。更优选存在小于10000ppm、甚至更优选小于3500ppm、尤其是小于1000ppm的杂质。按如下定量地测定二聚体、三聚体和四聚体:样品制备:在每种情况下将400mg的样品精确地称取到10ml标准烧瓶中并加入约8ml的二氯甲烷。借助于搅拌器,使样品溶解(理想地过夜);随后,用二氯甲烷将标准烧瓶定容至刻度并再次混合。用微升注射器将50μl的由此获得的样品溶液注入到tds管内的一块硅烷化玻璃棉中。将该管放置在通风橱中约30分钟,从而使溶剂可以蒸发。外部标准溶液:将50mg的十六烷精确地称取到100ml标准烧瓶中,用甲醇定容至刻度并通过振摇均化。将5μl的该溶液(相当于约2.5μg)施加至tenax管。该外部标准物在测定次序(sequence)开始时分析一次并在测定次序结束时分析一次。通过配有chemstation软件的agilent6890气相色谱仪实施该测定;参数:rtx-5分离柱;长度:60m;内径:250μm;膜厚度:0.25μm;载气:氦气;柱供应压力:203.1kpa;柱温箱温度:50℃-10℃/分钟-320℃(33分钟);分流比:50:1;检测器温度:280℃(thermalaux)。热解吸单元按如下设置:gersteltdsa;tds炉(初始温度:20℃;平衡时间:1分钟;初始时间:0.5分钟;加热速率:60℃/分钟;最终温度:280℃;保持时间:10分钟);冷应用系统(初始温度:-150℃(采用液态n2冷却);平衡时间:1分钟;初始时间:0.5分钟;加热速率:10℃/秒;最终温度:300℃;保持时间:5分钟)。另外,使用以下设置:转移温度:300℃;解吸模式:溶剂排气-干吹扫;排气时间:0.5分钟;进样模式:移取管(removetube)。针对外部标准物十六烷实施半定量评估。响应因子假定为1。仅积分对应于低聚物的峰组。二聚体在约20分钟时洗脱,三聚体在约28分钟时洗脱,四聚体在约37分钟时洗脱。使用质谱确定峰是否属于积分区域,低聚物可容易地通过离子质量来表征(例如,m/z=220、m/z=330和m/z=440:分别为开环聚环辛烯的二聚体、三聚体和四聚体)。按如下测定单体:样品制备:将300mg的样品精确地称取到6个顶空瓶的每一个中,加入5ml的十二烷并通过搅拌使混合物均化。将两个混合物作为样品分析。向另外两个混合物的每一个中加入5μl的加标溶液。向其余两个混合物的每一个中加入10μl的加标溶液。加标溶液:将300mg的环辛烷和40mg的环辛烯精确地称取到25ml标准烧瓶中,用十二烷定容至刻度并通过振摇均化。将5ml的该溶液用移液管移入25ml标准烧瓶中,用十二烷定容至刻度并通过振摇均化。通过配有chemstation软件的agilent7890气相色谱仪实施该测定;参数:分离柱1:熔融二氧化硅cp-sil8cb;长度:50m;内径:530μm;膜厚度:1μm;分离柱2:熔融二氧化硅db-wax;长度:60m;内径:530μm;膜厚度:1μm;载气:氮气;柱供应压力:10.15psi;柱温箱温度:50℃(4分钟)-5℃/分钟-130℃-30℃/分钟-180℃(10分钟);进样口温度:160℃;检测器温度:230℃;检测器h2流量:40ml/分钟;检测器空气流量:400ml/分钟;尾吹流量(n2):25ml/分钟;顶空进样器:turbomatrix40perkinelmer;柱温箱温度:100℃;进样针温度:120℃;转移温度:150℃;顶空压力:130kpa;恒温时间:60分钟;压力积聚时间:0.5分钟;进样时间:0.16分钟;进样针停留时间:0.2分钟;样品瓶排气(vialvent):有。通过标准物加入方法在两个分离柱上并且经两个加标操作用验证过的excel表格进行定量评估。一种或多种环烯烃的转化可以在没有溶剂的情况下进行。或者,该反应可以在至少一种溶剂中进行。合适的溶剂是例如饱和脂族烃,如己烷、庚烷、辛烷、壬烷、癸烷、十二烷、环己烷、环庚烷或环辛烷;芳族烃,如苯、甲苯、二甲苯或均三甲苯;卤代烃,如氯甲烷、二氯甲烷、氯仿或四氯化碳;醚,如乙醚、四氢呋喃或1,4-二噁烷;酮,如丙酮或甲基乙基酮;酯,如乙酸乙酯;和上文提及的溶剂的混合物。更优选地,用于该反应的溶剂选自脂族烃和芳族烃,这里特别优选己烷和甲苯,尤其是己烷。另外优选的选择是四氢呋喃、甲基乙基酮、氯甲烷、二氯甲烷、氯仿或其混合物,非常特别优选己烷或甲苯。基于环烯烃和溶剂的总重量,溶剂的含量可以设定为例如20重量%至60重量%、优选40重量%至60重量%的值。在选择用于开环易位反应的溶剂时,应注意该溶剂不应使催化剂或催化活性物类失活。本领域技术人员可以通过简单的实验或通过研究文献来认识到这一点。在含有铝有机物的催化剂体系的情况下,不含杂原子的芳族烃或脂族烃是尤其合适的。在本发明的另一个实施方案中,溶剂混合物可以含有稳定剂。这可以扩散到开环聚环烯烃中并增加其储存稳定性和/或加工稳定性。合适的稳定剂可以选自位阻酚,例如2,5-二叔丁基氢醌、2,6-二叔丁基对甲酚、4,4'-硫代双(6-叔丁基苯酚)、2,2'-亚甲基双(4-甲基-6-叔丁基苯酚)、3-(3',5'-二叔丁基-4'-羟基苯基)丙酸十八烷基酯、4,4'-硫代双(6-叔丁基苯酚)、2-叔丁基-6-(3-叔丁基-2-羟基-5-甲基苄基)-4-甲基苯基丙烯酸酯、2,6-二(叔丁基)-4-甲基苯酚(bht)、2,2-亚甲基双(6-叔丁基-对甲酚),选自有机亚磷酸酯,例如亚磷酸三苯酯、亚磷酸三(壬基苯基)酯,选自有机硫代化合物,例如硫代二丙酸二月桂酯、季戊四醇四(3-月桂基硫代丙酸酯)和抗坏血酸及其混合物。在每种情况下,基于开环聚环烯烃、优选开环聚环辛烯的重均分子量,稳定剂可以在5至7500ppm、优选25至750ppm的范围内存在。可以根据以下步骤之一来添加稳定剂:可以将稳定剂掺入聚合物的熔体中,例如经由在挤出机中配混。稳定剂可以直接计量加入或经由母料添加。这也可以仅在形成与聚合物的共混物的进一步加工过程中发生和/或在生产成型体(例如膜)的过程中发生。另一种选择是将稳定剂溶解在合适的溶剂中,并将其施加至开环聚环烯烃的颗粒。随后,除去溶剂,例如通过干燥步骤,其中使用升高的温度和/或降低的压力。然后稳定剂保留在颗粒的表面上和/或在干燥过程中被吸收到颗粒中。另一种选择是将稳定剂作为粉末涂料施加至颗粒。也可以制备这样一种混合物,其中以相对高的浓度包含稳定剂的开环聚环烯烃颗粒与不含稳定剂或含有较低浓度的稳定剂的开环聚环烯烃颗粒同时存在。此外,开环聚环烯烃组合物、优选开环聚环辛烯组合物可以含有染料(可溶性着色剂)。在根据本发明的方法的一个优选的实施方案中,所述环烯烃选自环丁烯、环戊烯、环庚烯、环辛烯、环壬烯、环癸烯、环十二碳烯、环辛-1,5-二烯、1,5-二甲基环辛-1,5-二烯、环癸二烯、降冰片二烯、环十二碳-1,5,9-三烯、三甲基环十二碳-1,5,9-三烯、降冰片烯(双环[2.2.1]庚-2-烯)、5-(3'-环己烯基)-2-降冰片烯、5-乙基-2-降冰片烯、5-乙烯基-2-降冰片烯、5-乙叉基-2-降冰片烯、二环戊二烯及其混合物。特别优选环戊烯、环庚烯、环辛烯和环十二碳烯。环辛烯是非常特别优选的环烯烃,因为它可获得且易于处理。可以使用两种或更多种环烯烃,以形成开环聚环烯烃的共聚物。所述环烯烃可以被烷基、芳基、烷氧基、羰基、烷氧基羰基和/或卤素原子取代。在根据本发明的方法的一个实施方案中,标准溶剂萃取可以在co2萃取之前或在co2萃取之后进行。这可以进一步降低单体和低聚物的比例。所述溶剂萃取可以在20℃直至溶剂混合物的沸程(回流),优选至60℃,更优选30℃至50℃,甚至更优选35℃至45℃的温度范围内进行。然而,在提及的值的范围内的温度受溶剂的沸点和开环聚环烯烃的性质限制。例如,所述温度应不高于半结晶聚合物的熔点或无定形聚合物的玻璃化转变温度,优选比沸程低至少5℃。原则上可以萃取处于熔融状态的开环聚环烯烃。然而,这是不太优选的,因为原始存在的离散颗粒可能形成团块或聚结。这减少了萃取材料的表面积,并导致萃取率下降。结果是,必须在萃取后将所获得的产物转变回到均匀的颗粒物形式,例如通过粒化或研磨。用于所述溶剂萃取的示意性溶剂可选自己烷、庚烷、二戊醚、乙醚、丁酸丁酯、乙基戊基酮、乙酸丁酯、甲基异丁基酮、甲基戊基酮、乙酸戊酯、正丁酸乙酯、四氯化碳、碳酸二乙酯、乙酸丙酯、二乙基酮、二甲醚、甲苯、乙酸乙酯、四氢呋喃、苯、四氯乙烯、氯仿、甲基乙基酮、氯苯、二氯甲烷、氯甲烷、1,1,2,2-四氯乙烷、二氯乙烷、丙酮、1,2-二氯苯、二硫化碳、1,4-二噁烷、甲酚、苯胺、吡啶、n,n-二甲基乙酰胺、环己醇、环己酮、丁醇、2-丁醇、乙腈、二甲基亚砜、n,n-二甲基甲酰胺、糠醇、丙二醇、碳酸-1,2-亚丙酯、乙醇、甲醇、丙醇、异丙醇、丁醇、乙二醇、碳酸亚乙酯、甘油、水或其混合物。本领域技术人员将能够通过简单的初步实验找到合适的溶剂或混合物。所述溶剂萃取可以以各种形式进行;例如,可以采用索氏提取的原理,从而使得待萃取的材料与新鲜的溶剂半连续地接触。所述溶剂萃取也可以以这样的方式进行:例如在搅拌槽中,在特定的时间将溶剂的体积全部或部分替换为新体积的溶剂,在这种情况下,这可以重复若干次。另外,所述溶剂萃取可以以这样的方式进行:将溶剂再循环操作集成,在这种情况下,再循环可能涉及混合物的一种或多种组分。视具体情况而定,随后可能需要将更多的一种或多种组分计量加入回收物中,以重新建立原来的混合比。此外,所述溶剂萃取也可以以这样的方式进行:在溶剂萃取的过程中组分的比率改变,在这种情况下,所述改变可以是恒定的或者是跳跃式地发生。所述溶剂萃取优选在惰性气体下进行。在溶剂萃取的过程中温度和/或压力可以保持恒定。还可以想到的是,在萃取操作的过程中温度或压力是变化的。在所述溶剂萃取之后,可以将含有开环聚环烯烃的组合物与剩余的溶剂分离,例如通过将其滗析或过滤。可替代地或另外,可以进行干燥操作以除去溶剂,例如在降低的压力下和/或在升高的温度下。在a)中获得的含有开环聚环烯烃的产物混合物可以是固体形式或溶解在溶剂中。优选地,除去溶剂。这可以通过加热或降低压力来进行,例如通过真空脱气。在进行步骤b)(co2萃取)之前或在进行任选的溶剂萃取之前,优选将产物混合物造粒成颗粒,例如通过线料造粒(strandpelletization)或水下造粒或粉碎,例如通过喷雾干燥或研磨。在一个优选的实施方案中,a)中获得的含有开环聚环烯烃的产物混合物为固体形式,并在步骤b)之前造粒或粉碎成颗粒。优选地,所述颗粒的平均质量小于100g/1000,更优选小于10g/1000,特别优选小于1g/1000。这包括直到1000g/1000的最大规格的平均质量。所述颗粒优选具有至少0.3mm、更优选至少0.5mm、最优选至少0.8mm的直径。为了测定平均质量,将约2-3g的颗粒施加至干净的底层(例如一张纸)上。随后,将该样品中的所有颗粒计数并转移至培养皿中;长度>1.0cm的尖状物或丸粒>1.0cm的链状物被排除(弃去),在这里不予评估。注意到丸粒的数量;它必须是最少150。随后,将丸粒精确地称取至0.1g,并基于1000个丸粒表示。如果存在少于150个丸粒,则必须采用新的相应较大的颗粒体积作为样品。根据本发明的方法可以连续或间歇地进行。开环聚环烯烃、优选开环聚环辛烯优选具有5000g/mol至500000g/mol、优选10000g/mol至250000g/mol、更优选20000至200000g/mol的重均分子量(mw)。通过凝胶渗透色谱法(gpc)相对于苯乙烯标准物测定分子量。测量基于din55672-1。样品制备:在室温下将样品以5g/l的含量溶解在四氢呋喃中。在注入gpc系统之前将其过滤(0.45μm注射器式过滤器)。测量在室温下实施。柱组合:1×5cm,5μm,100å,(苯乙烯-二乙烯基苯共聚物)1×30cm,5μm,50å,(苯乙烯-二乙烯基苯共聚物)1×30cm,5μm,1000å,(苯乙烯-二乙烯基苯共聚物)1×30cm,5μm,100000å,(苯乙烯-二乙烯基苯共聚物)流动相:超纯四氢呋喃,经稳定化流量:1ml/分钟检测:折射率检测器校准:聚苯乙烯。可以构建所需的摩尔质量,例如,在至少一种链转移剂的存在下,这导致链增长停止。合适的链转移剂是例如具有一个或多个可以在末端或内部位置处的非共轭双键的非环状烯烃,并且其优选不带有任何取代基。这样的化合物是例如戊-1-烯、己-1-烯、庚-1-烯、辛-1-烯或戊-2-烯。另外,可以使用其侧链上具有双键的环状化合物,例如乙烯基环己烯。可以通过本领域技术人员熟知的方法来调节开环聚环烯烃(cycloalkenamers)的顺式/反式比。例如,该比率取决于催化剂、溶剂、搅拌强度或温度或反应时间。优选地,反式含量为至少55%。通过氘代氯仿中的1hnmr测定顺式/反式比。环烯烃的转化可以在至少一种催化剂的存在下实施。合适的催化剂是例如过渡金属卤化物,其与作为助催化剂的有机金属化合物一起形成对聚合有催化活性的物类。在这里,有机金属化合物中的金属与卤化物中的过渡金属不同。或者,可以使用过渡金属-碳烯配合物。可用的过渡金属包括第4族至第8族的金属,例如钼、钨、钒、钛或钌。有机金属化合物中的金属例如是铝、锂、锡、钠、镁或锌。例如,在ep-a-2017308中详细描述了合适的催化剂及其用量。优选使用含有至少一种烷基氯化铝、六氯化钨或其混合物的催化剂体系。合适的烷基氯化铝是乙基二氯化铝(etalcl2)和乙基铝倍半氯化物,其也可以以混合物使用。优选的催化剂体系含有六氯化钨和乙基二氯化铝,或者在一个特别优选的实施方案中由这两种化合物组成。氯化铝对六氯化钨的质量比优选为1-6。特别优选的是2-5的比率。为了活化催化剂,可以使用酸性化合物,例如醇。基于所用的环烯烃,六氯化钨可以在0.1-0.04mol%、更优选0.1-0.01mol%的范围内使用。基于环烯烃,烷基氯化铝优选在0.2-0.08mol%、更优选0.2-0.02mol%的范围内。环烯烃的转化可以等温或绝热地进行。温度优选在-20至120℃的范围内。这具体取决于所用的单体和任何存在的溶剂。特别优选的温度为10-60℃。反应优选在保护气氛中进行。在绝热方法体制的情况下,温度可以经由参数(例如催化剂的量、催化剂添加速率、反应的终止时间等)来确定。在这里,优选的温度范围为20-50℃。在达到所需的反应时间时,可以通过使催化剂体系失活来终止聚合。为此目的,例如可以加入适量的ch-酸性化合物。用于此目的的合适实例是醇,例如甲醇、乙醇、丙醇等,或者羧酸,例如乙酸。本发明还提供了至少一种根据本发明的含有开环聚环烯烃的组合物或至少一种通过根据本发明的方法获得的组合物在包装材料中的用途,其中包装材料优选用于食品和饮料。实施例a.聚合物聚合物1:使用evonik,germany的vestenamer®8020(开环聚环辛烯)作为含有开环聚环烯烃的产物混合物(颗粒的平均尺寸为约3mm×3mm×4mm)。聚合物2:通过再粒化工艺由聚合物1制备聚合物2。将聚合物1经由主料斗供入双螺杆挤出机werner&pfleidererzsk30中。机筒温度为125℃。应用250rpm的螺杆速度,并选择聚合物的通量为6kg/h。用温度计测量的模头处的有效熔体温度为186℃。在离开模头处的挤出机前板之后,熔体线料在水浴中冷却,其后在空气中冷却。然后用造粒机(切割机)将聚合物线料造粒。切割机以57m/min的送线速度(strandspeed)操作。实施再粒化工艺,直到获得100kg的量的聚合物2(颗粒的平均尺寸为约1mm×1mm×1mm)。分子量的测定通过凝胶渗透色谱法测定聚合物的分子量(方法:参见说明书)。mn以g/mol计mw以g/mol计mp以g/mol计多分散性聚合物1870015610011400018.0聚合物2910016430011690018.1mn=数均分子量mw=重均分子量mp=峰值分子量。双键的反式含量通过氘代氯仿(cdcl3)中的1hnmr测定两种聚合物的双键的反式含量。聚合物1和聚合物2的反式含量均为80%。平均颗粒重量聚合物1:颗粒的平均重量为54.0g/1000。聚合物2:颗粒的平均重量为2.1g/1000。(方法:参见说明书)。萃取之前的低聚物单体/mg/kg二聚体/mg/kg三聚体/mg/kg四聚体/mg/kg聚合物142352095766487聚合物218162999696604b.萃取向根据图1(工艺i)或图2(工艺ii)的萃取系统的高压釜中装入待后处理的含有开环聚环辛烯的组合物(萃取材料;聚合物1或聚合物2)。在相同的设备中进行阶段b1和阶段b2。阶段b1通过高压泵将二氧化碳设定成高于临界压力。手动关闭热交换器。温度是20℃(低于临界条件)。二氧化碳处于物质的液态并流过高压釜中的组合物。在每种情况下均使用20kg的co2。阶段b2–工艺i该工艺用分离器进行(工艺i)。向高压釜中装入开环聚环辛烯。从储存容器中取出co2,并用高压泵使其达到超临界萃取压力。温度(通过热交换器增加)高于co2的临界温度。随后,通过相同的高压泵使超临界co2与萃取材料一起以轴向方式连续地流过高压釜,并且在如此进行时溶解co2可溶性单体或低聚物。超临界二氧化碳被连续地引导流过萃取材料。然后在非超临界条件(压力<73.8巴,温度<31℃)下使负载的co2膨胀到分离器中。这使气体冷却下来产生湿蒸气。形成富含萃取物的液化气相(liquidgasphase)和基本上不含萃取物的气相。为了分离,在45巴下连续地蒸发液态二氧化碳,然后以等压方式使其达到27℃的分离温度。溶解在液态co2中的物质在容器底部连续地分离出来。气态co2从分离器的顶部空间中以未负载的形式连续地排出,在-12℃和45巴下的冷凝器中液化并且被送回至高压泵的储存容器。如果co2可溶性物质已被完全萃取(为此目的所需的co2的量由经验确定),则萃取完成。经验确定通过以若干个步骤进行萃取并测定所获得的单体和低聚物的量来实现。当分离器中基本不再获得单体或低聚物时,测定结束。然后将含有净化的开环聚环辛烯的高压釜减压。可以从高压釜中取出开环聚环辛烯用于低聚物测定。添加助溶剂首先,将750g或1500g的丙酮(5重量%或10重量%)或750g的己烷(5重量%)混合到co2中直至s/f=15。随后,用co2将高压釜的容器容积置换两次。此后,在没有助溶剂的情况下以s/f=35加入co2。通过在分离器中初始装入1000g的水来除去助溶剂,以便将助溶剂从气相中洗涤出来。阶段b2-工艺ii与工艺i不同,不使用分离器。然而,在高压釜的下游设置含有300g的活性炭作为吸附剂的容器。该工艺以等压和等温的方式进行。s/f值是相应较高的。在实施阶段b2后的萃取结果#起始重量/g阶段b1工艺s/f萃取压力/巴萃取温度/℃助溶剂吸附剂备注1*1500无i6026035无无聚合物1;颗粒烧结2*2000无i5026035无无颗粒烧结31000有i5042040无无无烧结41000有i50260405%丙酮无无烧结51000有i502604010%丙酮无无烧结61000有i5026040无无无烧结72200有i5026043无无无烧结81000有i50260405%己烷无无烧结9800有ii40026040无活性炭无烧结101000有ii200250–27040无活性炭无烧结*非本发明。除非另有说明,否则使用聚合物2。无论是在尺寸为3mm×3mm×4mm(聚合物1)的情况(实施例1)中还是在尺寸为1mm×1mm×1mm(聚合物2)的情况(实施例2)中,不具有第一萃取阶段b1的实验导致所使用的开环聚环辛烯颗粒烧结。相比之下,如果一开始时使用液态co2,则不会观察到颗粒烧结。评估根据说明书中的指示以双重测定法测定单体和低聚物。#单体/mg/kg二聚体/mg/kg三聚体/mg/kg四聚体/mg/kg聚合物21816299969660431<10015617974n.d.<10022313895n.d.1991010288362<10052524727n.d.<10081021998n.d.<100<150191393<100<1501276103<100<1501763n.d.=未测定。与未经后处理的产物混合物相比,通过所述萃取方法可以显著地减少低聚物。如果萃取压力增加(相比于实施例6中的260巴,实施例3中为420巴),可以进一步降低低聚物含量。使用助溶剂(实施例4、实施例5采用丙酮,实施例8采用己烷)对结果产生完全积极的影响,但助溶剂的比例不能太高:使用5%丙酮的实施例4显示出比使用10%丙酮的实施例5更好的萃取结果。萃取温度从35℃或40℃增加至43℃没有显示出任何显著的效果。如同工艺i(实施例3至实施例8)一样,工艺ii(实施例9、实施例10)适于除去低聚物。相比于工艺i,测得的低聚物含量更低。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1