一类新型双极性聚合物主体材料的设计合成及其性能研究的制作方法
本发明属于有机光电材料技术领域。具体涉及一类新型双极性聚合物主体材料的设计合成及其性能研究,可将该类材料作为主体材料与客体磷光材料掺杂应用于有机发光二极管等有机电子领域。
背景技术:
有机电致磷光器件(PHOLEDs)由于引入了重金属配合物发光体,重金属之间存在着自旋-自旋耦合相互作用,可以同时捕获单线态和三线态激子,理论上能获得将近100%的内量子效率,在大规模器件以及柔性器件上有着潜在的应用价值。PHOLEDs中的重金属配合物磷光材料通常不能单独作为发光层,而是将其掺杂在合适的主体材料中,形成主客体发光体系,以削弱金属配合物浓度过大引起的猝灭效应和三线态-三线态(T1-T1)湮灭效应,从而提高器件的效率。故设计合成高性能的主体材料对PHOLEDs来说也显得尤为重要。小分子PHOLEDs在制备过程中采用复杂的蒸镀技术,繁琐的制备过程和昂贵的成本极大地限制了产品的进一步发展和商业化;而聚合物电致磷光器件(PPLEDs)在制备过程中则采用简单低成本的溶液加工法,例如旋涂和喷墨打印等方法,有利于大面积柔性显示的制造。因此,发展性能优良的聚合物主体材料对于PPLEDs,是一个比较关键的问题。
聚乙烯基咔唑(PVK)作为一类典型的、性能较好的非共轭型聚合物,由于含有较高的ET和好的空穴传输能力,被广泛报道作为主体材料应用于PPLEDs中。据So等报道,基于PVK和FIrpic的双层蓝光PhPLEDs的能量效率和发光效率能够达到14lm/W和22cd/A。然而PVK的电子传输性能和热稳定性能均不是很理想,在作为聚合物主体材料制备高性能、长寿命的PPLEDs上有一定的局限性;且相关非共轭类结构的聚合物主体材料的报道仍然很少。将吸电子能力较强的功能基团引入PVK,获得电子和空穴的传输能力较平衡的双极性堆积聚合物主体材料,将能在蓝色磷光PPLEDs方向取得更好的进展。故我们有必要在此领域进行更加深入细致的研究工作。有鉴于此,我们拟通过功能化的方法,将一些具有特殊位阻、电子结构的吸电子功能基团引入到模型堆积聚合物中,设计合成一系列新型双极性堆积聚合物主体材料。重点研究不同吸电子基团对模型堆积聚合物的ET、热力学稳定性、电荷传输性能、光物理性质和电化学性质的调控。探讨双极性堆积聚合物的分子结构、光电性质、薄膜形貌与器件性能之间的关系。在此基础上,筛选出一系列高性能的聚合物主体材料,为发展蓝色PPLEDs提供原理和材料的支撑。在材料优化的基础上,结合器件优化过程,研究器件中的基本物理机制,揭示器件的内在工作原理,从而指导新型器件的设计与应用。详细请参阅文献(1)Shiyang Shao,Junqiao Ding,Tengling Ye,Zhiyuan Xie,Lixiang Wang,Xiabin Jing,and Fosong Wang Adv.Mater.2011,23,3570-3574.文献(2)Ho-Hsiu Chou,Chien-Hong Cheng Adv.Mater.2010,22,2468-2471.文献(3)Mounggon Kim,Jun Yeob Lee Adv.Funct.Mater.2014,24,4164-4169.
技术实现要素:
本发明的目的在于通过往非共轭型的聚乙烯基咔唑(PVK)的咔唑活性位点上引入了不同结构的吸电子功能基团来构建新型双极性聚合物主体材料,利用该材料在有机发光二极管器件领域的应用获得高效的器件性能。
本发明中的新型双极性聚合物主体材料,其结构特点主要是往非共轭型的聚乙烯基咔唑(PVK)的咔唑活性位点上引入了不同结构的吸电子功能基团。其结构特征如下:
其中:R为吸电子基团,化合物I和II中R的连接位点是咔唑单元1、2、3和4号位的其中之一;x、y和n为1~1000中任意数字。
R具体为下列结构中的一种:
本发明中所述新型双极性聚合物主体材料的制备方法,通过R取代的乙烯基咔唑单体进行自由基均聚或者共聚反应制备获得,具体如下:
其中,x、y和n为1~1000中任意数字。
自由基聚合反应是在温度85℃条件下,以偶氮二异丁腈AIBN为催化剂,对R取代的乙烯基咔唑单体进行聚合反应。实验中使用的反应瓶需在重铬酸钾洗液中浸泡并洗净,接着用饱和的水蒸汽除去内壁的盐层,最后在火焰中烘烤并用氮气保护冷却,达到除去吸附在瓶壁上的氧的目的。在氮气保护下,配制R取代的乙烯基咔唑单体或者配置乙烯基咔唑和R取代的乙烯基咔唑单体的混合物浓度为0.1~1.0mol/L,AIBN浓度为1.0~10.0×10-3mol/L的甲苯或四氢呋喃溶液,鼓氮气搅匀。在反应瓶中分别加入上述配置溶液,液氮冷却,抽真空,解冻再充氮气,随后封瓶置入恒温85℃油浴锅中,反应72h结束后使用甲醇淬灭反应,然后甲醇沉降得到聚合物粗产物,粗产物用丙酮抽提,得到聚合物材料。
本发明中所述一类新型聚合物双极性主体材料的制备方法,其特征在于所述R取代的乙烯基咔唑单体是由R取代的咔唑单体分别经过亲核取代和消除反应制备获得,R取代的咔唑单体可由溴代咔唑为原料通过两种不同路线制备获得,具体如下:
步骤a 将溴代咔唑与R-H、醋酸钯、1,1’-双二苯基膦二茂铁(dppf)、三乙胺混合后加入二甲基亚砜溶解后,在避光及氮气保护状态下回流反应12h。反应结束后用二氯甲烷萃取出有机相。使用无水硫酸镁干燥有机相,过滤后旋蒸掉有机溶液,使用柱层析分离法获得R取代的咔唑单体;
步骤a′ 将原料溴代咔唑与联硼酸频那醇酯、醋酸钯、1,1-双(二苯基膦)二茂铁和乙酸钾加入干燥的二甲基亚砜中,在氮气的保护下,边搅拌边加热到90℃,反应12h,后静置冷却至室温,用水和二氯甲烷萃取,再用无水硫酸镁干燥,过滤,并除去有机溶剂,得到终产物粗品;并过柱纯化,得到咔唑的硼酸酯;
步骤b′ 将咔唑的硼酸酯与R-X(X为卤素)和四(三苯基膦)钯加入到甲苯、乙醇和2M碳酸钾水溶液的混合溶液中,在氮气的保护下,边搅拌变加热到90℃,反应12h,后静置冷却至室温,用水和二氯甲烷萃取,再用无水硫酸镁干燥,过滤,并除去有机溶剂,得到产物粗品;并过柱纯化,得到R取代的咔唑单体;
步骤c 将上述R取代的咔唑单体与碳酸钾、氢氧化钾和四丁基溴化铵加入到1,2-二氯乙烷中,在50℃条件下反应5h,后静置冷却至室温,用水和二氯甲烷萃取,再用无水硫酸镁干燥,过滤,并除去有机溶剂,得到产物粗品;并过柱纯化,得到R取代的氯乙基咔唑单体;
步骤d 将上述得到的R取代的氯乙基咔唑单体与氢氧化钾、对苯二酚加入到异丙醇和甲苯的混合溶液中,在85℃条件下反应3h,后静置冷却至室温,旋干溶剂,用水和二氯甲烷萃取,再用无水硫酸镁干燥,过滤,并除去有机溶剂,得到产物粗品;并过柱纯化,得到最终单体。
本发明中所述新型双极性聚合物主体材料的应用,其特征在于该材料与磷光客体发光材料掺杂后作为发光层应用到有机电致发光器件中。有机电致磷光器件中的结构为:透明阳极/空穴传输层/聚合物主体材料和磷光客体发光材料/电子传输层/金属阴极,发光层的主体材料如权利1所述的化合物,客体材料为金属配合物磷光材料。
本发明的有益效果为:
(1)本发明提供了一类新型双极性聚合物主体材料的设计、合成及其应用,具备合适的三线态能级和平衡的电荷传输能力,为高效聚合物电致磷光器件的制备提供了条件;
(2)本发明所述的双极性聚合物主体材料合成方法简单,适合于广泛应用;
(3)本发明所述的双极性聚合物主体材料,具有良好的热力学稳定性和成膜性;
(4)本发明所述的双极性聚合物主体材料,可以与磷光客体材料材料进行有效掺杂,采用溶液旋涂法制备发光器件,有利于大面积柔性显示的制造。
附图说明
附图用来提供对本发明的进一步理解,并构成说明书的一部分,与本发明的实例一起用于解释本发明,并不构成对本发明的限制。实施例中优选实施方式在附图中。
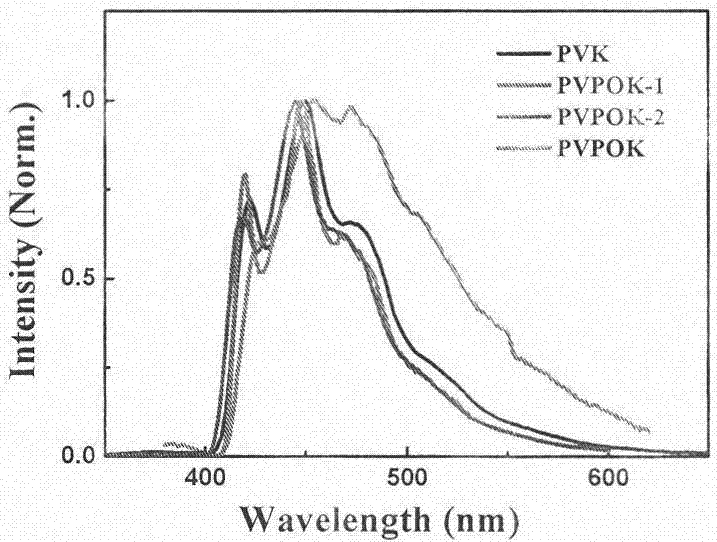
图1聚合物PVPOK、PVPOK-1和PVPOK-2薄膜的紫外-可见吸收和荧光光谱性质对比图;
图2聚合物PVPOK、PVPOK-1和PVPOK-2的低温磷光光谱对比图;
图3聚合物PVK与PVPOK-1、PVPOK-2和PVPOK的循环伏安曲线图;
图4聚合物PVPOK的TGA曲线和DSC曲线图;
图5电致磷光器件结构图;
图6聚合物PVK与PVPOK-1、PVPOK-2器件的电致发光光谱图;
图7聚合物PVK与PVPOK-1、PVPOK-2蓝色磷光器件性质图。
具体实施方式
下面通过具体实施方式来进一步描述本发明,但本发明并不局限于这些实施例。
实例1 PVPOK的合成;
中间体(2)的合成:将化合物2(1.83g,7.4mmol)与二苯基磷氧(1g,4.5mmol)、醋酸钯(0.056g,0.25mmol)、1,1’-双二苯基膦二茂铁(dppf)(0.25g,0.45mmol)、三乙胺(0.8mL,5.9mmol)混合后加入二甲基亚砜(15mL)溶解后,在避光及氮气保护状态下回流反应12h。反应结束后用二氯甲烷萃取出有机相。使用无水硫酸镁干燥有机相,过滤后旋蒸掉有机溶液,使用柱层析分离法获得产物化合物2。
中间体(3)的合成:在50℃下,将化合物2(3.67g,10mmol),氢氧化钾(2.25g,40mol),碳酸钾(13.82g,100mmol),四丁基溴化铵(0.32mg,1mmol)溶于1,2-二氯乙烷(100mL)中在单口烧瓶中反应3~5h。反应结束后除去溶液中固体,使用二氯甲烷萃取,无水硫酸镁干燥有机相,旋干有机溶剂后用柱层析分离法获得化合物4。
中间体(4)的合成:化合物3(4.3g,10mmol)与氢氧化钾(4.5g,80mmol)、对苯二酚(0.11g,1mmol)、甲苯(10mL)及异丙醇(100mL)在85℃下回流3h。反应结束后旋干甲苯和异丙醇,并用二氯甲烷萃取,无水硫酸镁干燥,过滤,旋干后使用柱层析分离法获得化合物4。
最终产物PVPOK的合成:反应管中加入单体化合物4(0.2g,0.5mmol)溶解在经金属钠干燥并蒸馏过的四氢呋喃(1mL)中,加入单体重量比为1%的乙烯基聚合引发剂偶氮二异丁腈(2.0mg),在85℃下搅拌回流3天。反应结束后使用甲醇淬灭反应,然后石油醚沉降得到聚合物粗产物,粗产物用丙酮抽提,得到聚合物材料PVPOK。
实例2 共聚物PVPOK-1与PVPOK-2的合成;
聚合物PVPOK-1与PVPOK-2的合成:反应管中加入单体化合物5(0.1g,0.25mmol/0.2g,0.5mmol)和9-乙烯基咔唑(0.196g,1mmol/0.098g,0.5mmol)以摩尔比为8∶2或5∶5的比例混合,分别对应加入单体总重量比为1%的聚合引发剂偶氮二异丁腈(2.96mg/2.98mg),保持反应管内无水无氧环境,加入经过除水除氧处理的四氢呋喃(2.5mL/2mL),待单体完全溶剂与溶液中后,避光条件下60℃引发聚合反应。聚合引发完成后,将体系置于85℃下反应3天。反应结束后使用甲醇淬灭反应,然后石油醚沉降得到聚合物粗产物,粗产物用丙酮抽提,得到聚合物材料得到共聚物PVPOK-1和PVPOK-2。
实例3 PV(2-PO)K的合成;
中间体(2)的合成:将化合物2(1.83g,7.4mmol)与二苯基磷氧(1g,4.5mmol)、醋酸钯(0.056g,0.25mmol)、1,1’-双二苯基膦二茂铁(dppf)(0.25g,0.45mmol)、三乙胺(0.8mL,5.9mmol)混合后加入二甲基亚砜(15mL)溶解后,在避光及氮气保护状态下回流反应12h。反应结束后用二氯甲烷萃取出有机相。使用无水硫酸镁干燥有机相,过滤后旋蒸掉有机溶液,使用柱层析分离法获得产物化合物2。
中间体(3)的合成:在50℃下,将化合物2(3.67g,10mmol),氢氧化钾(2.25g,40mol),碳酸钾(13.82g,100mmol),四丁基溴化铵(0.32mg,1mmol)溶于1,2-二氯乙烷(100mL)中在单口烧瓶中反应3~5h。反应结束后除去溶液中固体,使用二氯甲烷萃取,无水硫酸镁干燥有机相,旋干有机溶剂后用柱层析分离法获得化合物4。
中间体(4)的合成:化合物3(4.3g,10mmol)与氢氧化钾(4.5g,80mmol)、对苯二酚(0.11g,1mmol)、甲苯(10mL)及异丙醇(100mL)在85℃下回流3h。反应结束后旋干甲苯和异丙醇,并用二氯甲烷萃取,无水硫酸镁干燥,过滤,旋干后使用柱层析分离法获得化合物4。
最终产物PV(2-PO)K的合成:反应管中加入单体化合物4(0.2g,0.5mmol)溶解在经金属钠干燥并蒸馏过的四氢呋喃(1mL)中,加入单体重量比为1%的乙烯基聚合引发剂偶氮二异丁腈(2.0mg),在85℃下搅拌回流3天。反应结束后使用甲醇淬灭反应,然后石油醚沉降得到聚合物粗产物,粗产物用丙酮抽提,得到聚合物材料PV(2-PO)K。
实施例4 PVDPTK的合成;
中间体(2)的合成:将3-溴咔唑(4.92g,20mmol)与联硼酸频那醇酯(6.6g,26mmol)、醋酸钯(0.134g,0.6mmol)、1,1-双(二苯基膦)二茂铁(0.66g,1.2mmol)和乙酸钾(5.88g,60mmol)加入干燥的二甲基亚砜中,在氮气的保护下,边搅拌边加热到90℃,反应12h,后静置冷却至室温,用二氯甲烷萃取,再用无水硫酸镁干燥,过滤,旋干有机溶剂,得到粗产物;并过柱纯化,得到中间产物5。
中间体(3)的合成:将中间产物3(0.586g,2mmol)、2-氯-4,6-二苯基-1,3,5-三嗪(0.536g,2mmol)和四(三苯基膦)钯(0,023g,0.02mmol)加入到2M的碳酸钾水溶液(9mL)、甲苯/乙醇(18mL,v/v=2∶1)的混合溶液中,在氮气的保护下,在110℃条件下反应12h,反应结束后冷却至室温,用二氯甲烷萃取,再用无水硫酸镁干燥,过滤,旋干有机溶剂,得到粗产物;并过柱纯化,得到中间产物6。
中间体(4)的合成:在50℃下,将中间产物3(0.398g,1mmol),氢氧化钾(0.225g,4mmol),碳酸钾(1.382g,10mmol),四丁基溴化铵(0.032g,0.1mmol)溶于1,2-二氯乙烷(10mL)中在单口烧瓶中反应5h。反应结束后除去溶液中固体,二氯甲烷萃取,无水硫酸镁干燥有机相,过滤,旋干有机溶剂后,用柱层析分离法获得中间产物7。
中间体(5)的合成:将中间产物4(4.61g,10mmol)与氢氧化钾(4.49g,80mmol)、对苯二酚(0.11g,1mmol)、甲苯(10mL)及异丙醇(100mL)在85℃下回流3h。反应结束后旋干甲苯和异丙醇,并用二氯甲烷萃取,无水硫酸镁干燥有机相,过滤,旋干有机溶剂后,用柱层析分离法获得单体8。
最终产物PVDPTK的合成:反应管中加入单体化合物5(0.2g,0.45mmol)溶解在经金属钠干燥并蒸馏过的四氢呋喃(1mL)中,加入单体重量比为1%的乙烯基聚合引发剂偶氮二异丁腈(2.0mg),在85℃下搅拌回流3天。反应结束后使用甲醇淬灭反应,然后甲醇沉降得到聚合物粗产物,粗产物用丙酮抽提,得到聚合物材料PVDPTK。
实施例5 聚合物材料光物理性质的测定;
a紫外可见吸收光谱:在比色皿中配制聚合物的甲苯溶液,浓度约为1×10-5mol/L,采用岛津(Shimadzu)UV-1750紫外可见光谱仪和日立(Hitachi)F-4600荧光光谱仪进行吸收光谱和发射光谱测定。其中,薄膜制备使用的仪器为中国科学院微电子研究所研发的KW-4A型旋涂仪;
b荧光发射光谱:所有材料的荧光发射光谱(溶液及薄膜)均测试于Hitachi F-4600光谱仪,测试样品配制浓度为1×10-5mol L-1的甲苯溶液,激发波长为紫外可见吸收光谱的最大波长。其中,薄膜制备使用的仪器为中国科学院微电子研究所研发的KW-4A型旋涂仪;
c低温磷光发射光谱:低温磷光发射光谱测试于Hitachi F-4600光谱仪,在液氮冷却条件下,材料温度达到77K,将材料配制成1×10-5mol L-1的甲苯溶液测试。
实施例6 聚合物材料电化学性质的测定;
聚合物主体材料的电化学性质是用电化学循环伏安法(CV)来测定的,实验仪器是辰华CHI660E电化学工作站,该仪器采用的是三电极体系,包括玻碳工作电极、Ag/AgCl参比电极以及铂丝对电极。聚合物电化学测定使用的溶剂一般为干燥的乙腈,电解质为六氟磷四丁基铵(Bu4NPF6),浓度是0.1M;测试环境需要氮气保护。仪器扫描的速率是0.1V/s,基准物为二茂铁(FOC),分别通过测量氧化和还原过程的开始电压计算该材料的HOMO和LUMO能级。
实施例7 聚合物材料热力学稳定性的测定;
差热扫描(DSC)测试:DSC图谱测试于Shimadzu DSC-60A差热议,在氮气保护条件下,样品首先以10℃/min的速率加热到低于分解温度的状态,然后,在液氮条件下降温回到起始温度,第二次再以10℃/min的速率升温扫描。
热重(TGA)测试:TGA图谱测试于Shimadzu DTG-60H热重仪,在氮气保护条件下,升温速率为10℃/min,同时保护气流氮气的流速为20cm3/min,材料重量发生变化直至达到恒重状态。
实施例8 聚合物材料电致蓝色磷光器件的制备与表征;
本发明的新型双极性电致磷光主体材料的应用于发光层,有机电致磷光器件中的结构为:透明阳极/空穴传输层/主体和客体发光材料/电子传输层/阴极,发光层由主体材料和掺杂材料组成,发光层的主体材料如权利1所述的化合物,客体材料为金属配合物磷光材料。
作为主体材料制备的蓝色PhPLEDs器件结构为ITO/PEDOT:PSS(40nm)/Host:10-15wt%FIrpic(100nm)/TPBI(20nm)/LiF(0.8nm)/Al(100nm)。其中ITO是方块电阻为10-20Ω的透明电极;PEDOT:PSS为聚(3,4-二氧乙基噻吩)/聚(对苯乙烯磺酸)作为空穴传输材料,Host:FIrpic为实施例中双极性聚合物主体材料掺杂10-15%的铱(III)二((4,6-二氟苯)-吡啶-N,C2′)吡啶甲酸酯,采用溶液旋涂技术,薄膜厚度为100nm;再蒸镀LiF缓冲层,最后,蒸镀Al阴极。
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进,这些改进也视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!