正电子放射性药物、制备方法及其应用与流程
本发明涉及药物及其制备方法领域,具体涉及一种正电子放射性药物、制备方法及其应用。
背景技术:
转位蛋白TSPO是一种由细胞核编码,内含169个氨基酸(富含色氨酸)的疏水性蛋白,在各种族间高度保守,在组织中主要定位于线粒体外膜,特别是在与甾体合成相关的组织中高度表达,如肾上腺、性腺、唾液腺等,在肾脏及心脏组织中也有部分表达,而在肝脏及脑中表达水平很低。研究表明,TSPO是甾体激素合成的重要组成部分,可促进胆固醇跨膜转运进入磷脂膜,增加孕烯醇酮形成及下游神经类固醇合成,修复损伤神经和促进神经生长等。此外,TSPO还参与许多生理功能,如细胞增殖、免疫应答和线粒体呼吸凋亡等。
在中枢神经系统中,TSPO主要分布于胶质细胞线粒体外膜,在静息小胶质细胞中低水平表达。然而,当发生炎症反应时,大脑中小胶质细胞明显活化增殖,TSPO密度明显增加,并且TSPO密度增高与中枢神经系统炎症反应呈正相关:大脑损伤时,小胶质细胞和星形胶质细胞中TSPO的表达上调,并与大脑损伤的程度直接相关;小胶质细胞的激活增殖并产生致炎性细胞因子如IL-1、IL-6、IL-8、TNF-α和诱导型一氧化氮合酶(iNOS)等,引起脑内炎症反应,随着近年对多种神经系统病变如阿尔茨海默症、多发性硬化、脑梗、进行性肌肉萎缩症、帕金森病和亨廷顿舞蹈病等发病机制的深入探索,越来越多的研究结果表明,小胶质细胞激活介导的脑内慢性炎症反应(即TSPO密度增高)是神经系统病变的病理特征之一。
鉴此,TSPO作为重要的药物靶点,越来越多的得到了研究者们的关注。一方面,靶向TSPO的正电子成像逐渐成为评价脑损伤、诊断脑外伤进展与状态、区分外周炎症与肿瘤的重要工具;同时,该类正电子药物为神经退行性疾病等中枢神经系统疾病的早期诊断提供影像学研究依据。另一方面,各类特异性靶向TSPO的配体在体内实验中已被证实对神经退行性病变和焦虑的动物模型有显著疗效,并具有神经保护、减少脑内炎症、促进神经再生、改善记忆和治疗应激相关障碍等功能。然而,在其治疗型药物的研究与开发中,仍有一些问题是值得探讨的,如在治疗局部神经适应证中TSPO的中长期疗效;考虑到TSPO在外周神经系统中高表达,长期给予TSPO配体药物可能造成的不良反应等。而开发靶向型正电子药物可以对研究TSPO配体的疗效和安全性提供有力支持。
第一个显像TSPO的正电子药物为[11C](R)-PK11195,但由于其透脑率差、信噪比低等问题,无法满足临床的需求。因此,越来越多的新型正电子药物被研发出来,以期实现更优质的成像效果。具有代表性的包括[11C]PBR28,[11C]PBR111,[11C]DAA1106,[11C]MBMP,[11C]ER176,[18F]AB5186,[18F]FEDAA1106,[18F]GE-180和[18F]DPA-714等,其中[18F]DPA-714以其高效便捷的放射标记方法、较高的头脑效率及信噪比等优势,在多种疾病模型上进行了广泛的正电子计算机断层显像(positron emission tomography,PET)研究,包括脑创伤老鼠模型、慢性肝性脑病老鼠模型、亨廷顿病狒狒模型等,临床上已应用于对肌萎缩、中风后遗症以及阿尔兹海默症等疾病的早期诊断和治疗疗效评估。
然而,研究表明,由于[18F]DPA-714分子结构中存在-OCH2CH2[18F]基团,在活体内细胞色素P450作用下,会首先发生脱乙基反应,代谢产生的含有[18F]标记的乙醇片段会被进一步氧化成[18F]乙醛、[18F]乙酸酯及[18F]乙酸,同时还会发生[18F]氟负离子的脱落。由于[18F]乙酸酯片段可以通过血脑屏障,从而产生非特异性结合的正电子成像信号,干扰检测。因此,[18F]DPA-714分子结构的不稳定性严重干扰PET影像的定量化分析。
技术实现要素:
本发明要解决的技术问题是提供一种代谢稳定型靶向TSPO正电子药物[18F]芳基-DPA体系,具体的为[18F]FDPA,此[18F]FDPA的化学及放射化学纯度高、比活度高且可重复性好,符合注射使用正电子药物的质量标准;本发明还提供了[18F]FDPA的无载体放射性氟-18负离子标记方法,此方法步骤少、产率高、操作简便且易于自动化生产;本发明进一步提供了[18F]FDPA的显像应用,其可应用在多种炎症疾病的显像分析上。
本发明第一方面提供了式I的化合物
其中R1和R2各自独立的选自烷基;
任选的,R1和R2各自独立的选自C1-6烷基。
在一优选例中,所述化合物为下列化合物或者下列化合物的立体异构体:
本发明第二方面提供了所述化合物的制备方法,包括如下步骤:
(1)使式A所示化合物氧化后与式所示B化合物接触,以便获得式C所示化合物,以及
(2)使式C所示化合物与氟-18负离子进行接触,以便获得式D所示化合物。
在一优选例中,所述式A所示化合物与所述式B所示化合物的摩尔比为0.8-1.2,优选为1。
在一优选例中,所述式A所示化合物的氧化是采用N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺与单过硫酸氢钾复合盐进行接触。
在一优选例中,所述N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺与单过硫酸氢钾复合盐的重量比为1:4-1:1,优选为1:2。
在一优选例中,所述N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺是先与三氟乙酸和氯仿接触,然后与单过硫酸氢钾复合盐进行接触。
在一优选例中,所述三氟乙酸与氯仿的体积比为2:1-4:1,优选为3:1。
在一优选例中,所述N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺与三氟乙酸的重量比为0.06-0.1,优选为0.08。
在一优选例中,所述式A所示化合物氧化后是先与乙醇先接触,然后再与式B所示化合物接触。
在一优选例中,所述式A所示化合物氧化后与乙醇接触,这时所述式A所示化合物的氧化产物的浓度为10-100mg/mL,优选为50mg/mL。
在一优选例中,所述式B所示化合物是采用乙酸酐与2-金刚烷酮进行接触制备而成。
在一优选例中,所述乙酸酐与2-金刚烷酮的摩尔比为0.8-1.2,优选1.1。
在一优选例中,所述乙酸酐是先与酸性物质先接触,然后再与2-金刚烷酮进行接触。
在一优选例中,所述酸性物质为丙二酸和浓硫酸。
在一优选例中,所述乙酸酐与丙二酸的体积比为0.8-1.3,优选为1.04。
在一优选例中,所述乙酸酐与浓硫酸的体积比为150-250,优选为200。
在一优选例中,所述乙酸酐先与酸性物质先接触时反应条件是升温至50-70℃反应10-20分钟后降至室温。
在一优选例中,所述乙酸酐先与酸性物质先接触时反应条件是升温至60℃反应15分钟后降至室温。
在一优选例中,所述式B所示化合物是与碳酸钠先接触,然后再与式A所示化合物接触。
在一优选例中,所述式B所示化合物与碳酸钠的摩尔比为1:1-1:5,优选1:2。
在一优选例中,所述氟-18负离子是采用QMA固相萃取柱捕获。
在一优选例中,所述QMA固相萃取柱上的氟-18负离子是采用含有季铵盐的洗脱液进行洗脱。
在一优选例中,所述式C所示化合物与季铵盐的重量比0.5:20-20:8,优选为1:12。
在一优选例中,所述季铵盐为四丁基甲磺酸铵。
在一优选例中,所述采用含有季铵盐的洗脱液的pH为7-8。
在一优选例中,所述含有季铵盐的洗脱液是由四丁基甲磺酸铵溶于乙腈和水制备而成。
在一优选例中,所述含有季铵盐的洗脱液中乙腈和水的体积比为0.5-1.5,优选为1。
在一优选例中,所述含有季铵盐的洗脱液中每8-20mg四丁基甲磺酸铵所对应的乙腈和水的使用量为0.5-1.5mL,优选为1mL。
在一优选例中,所述氟-18负离子采用洗脱液洗脱后在100-120℃进行加热并采用5-15mL/min流速的干燥氮气鼓吹,待溶剂完全被吹干,然后向其中加入其体积是0.5-1.5倍洗脱液体积的无水乙腈,继续在100-120℃进行加热,并采用5-15mL/min流速的干燥氮气鼓吹,待溶剂完全被吹干,重复2-4次。
在一优选例中,所述氟-18负离子采用洗脱液洗脱后在110℃进行加热并采用10mL/min流速的干燥氮气鼓吹,待溶剂完全被吹干,然后向其中加入其体积是1倍洗脱液体积的无水乙腈,继续在110℃进行加热,并采用10mL/min流速的干燥氮气鼓吹,待溶剂完全被吹干,重复3次。
在一优选例中,所述式C所示化合物是与乙腈先接触,然后与氟-18负离子进行接触。
在一优选例中,每mg的式C所示化合物对应使用的乙腈的量为0.2-2mL,优选为0.6mL。
在一优选例中,所述式C所示化合物与氟-18负离子进行接触时的反应条件是:100-140℃下反应8-16min,优选为120℃下反应12min。
在一优选例中,所述式C所示化合物与氟-18负离子在反应完成后是采用乙腈和水组成的终止液进行终止反应。
在一优选例中,所述终止液中乙腈和水的体积比为0.5-1.5,优选为1。
在一优选例中,每0.5-20mg的式C所示化合物对应使用的终止液的量为0.5mL。
在一优选例中,每1.8mg的式C所示化合物对应使用的终止液的量为0.5mL。
本发明第三方面提供了一种化合物,具有式C所示的结构:
本发明第四方面提供了所述的化合物的制备方法,其方法为:使式A所示化合物氧化后与式所示B化合物接触,以便获得式C所示化合物。
在一优选例中,所述式A所示化合物与所述式B所示化合物的摩尔比为0.8-1.2,优选为1。
在一优选例中,所述式A所示化合物的氧化是采用N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺与单过硫酸氢钾复合盐进行接触。
在一优选例中,所述N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺与单过硫酸氢钾复合盐的重量比为1:4-1:1,优选为1:2。
在一优选例中,所述N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺是先与三氟乙酸和氯仿接触,然后与单过硫酸氢钾复合盐进行接触。
在一优选例中,所述三氟乙酸与氯仿的体积比为2:1-4:1,优选为3:1。
在一优选例中,所述N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺与三氟乙酸的重量比为0.06-0.1,优选为0.08。
在一优选例中,所述式A所示化合物氧化后是先与乙醇先接触,然后再与式B所示化合物接触。
在一优选例中,所述式A所示化合物氧化后与乙醇接触,这时所述式A所示化合物的氧化产物的浓度为10-100mg/mL,优选为50mg/mL。
在一优选例中,所述式B所示化合物是采用乙酸酐与2-金刚烷酮进行接触制备而成。
在一优选例中,所述乙酸酐与2-金刚烷酮的摩尔比为0.8-1.2,优选1.1。
在一优选例中,所述乙酸酐是先与酸性物质先接触,然后再与2-金刚烷酮进行接触。
在一优选例中,所述酸性物质为丙二酸和浓硫酸。
在一优选例中,所述乙酸酐与丙二酸的体积比为0.8-1.3,优选为1.04。
在一优选例中,所述乙酸酐与浓硫酸的体积比为150-250,优选为200。
在一优选例中,所述乙酸酐先与酸性物质先接触时反应条件是升温至50-70℃反应10-20分钟后降至室温。
在一优选例中,所述乙酸酐先与酸性物质先接触时反应条件是升温至60℃反应15分钟后降至室温。
在一优选例中,所述式B所示化合物是与碳酸钠先接触,然后再与式A所示化合物接触;
在一优选例中,所述式B所示化合物与碳酸钠的摩尔比为1:1-1:5,优选1:2。
本发明第五方面提供了一种组合物,包含上述三种化合物之一。
本发明第六方面提供了上述三种化合物或者组合物在制备正电子放射性药物中的应用。
在一优选例中,所述正电子放射性药物为放射性氟-18标记的PET显像剂。
在一优选例中,所述正电子放射性药物为机体动态显像剂和断层显像剂。
在一优选例中,所述正电子放射性药物为用于评价外周炎症相关疾病的显像剂。
在一优选例中,所述外周炎症相关疾病为肿瘤、肌肉炎症、毛囊炎、扁桃体炎、肺炎、肝炎或肾炎。
在一优选例中,所述正电子放射性药物为神经系统疾病的早期诊断的显像剂。
在一优选例中,所述神经系统疾病选自脑中风、脑外伤、脑瘤、慢性肝性脑病、癫痫、帕金森病、阿尔茨海默病、亨廷顿病和肌萎缩侧索硬化症。
在一优选例中,所述脑中风选自脑梗塞、脑缺血、脑卒中、脑栓塞和脑出血。
本发明使用的术语具有以下定义,除非另有描述:
本发明涉及的“单过硫酸氢钾复合盐”又名“过一硫酸氢钾复合盐”、“过硫酸氢钾复合盐”,它是一种无机过氧化物,过硫酸氢钾一般与硫酸氢钾、硫酸钾结合成三合盐的形式存在,因此称之为单过硫酸氢钾复合盐,简称:PMPS或KMPS。最早由美国杜邦发明,商品名为OXONE。
本发明涉及的“mg”指的是毫克,“mL”指的是毫升。
本发明涉及的“QMA固相萃取柱”为Waters Sep-Pak light QMA固相萃取柱。
本发明涉及的“NEt2”代表例如
本发明涉及的化合物代表其余依次类推。
本发明化学式中所涉及的“I”代表碘原子。
本发明涉及的裸环辅基SPIAd的中文名称为(1R,3R,5R,7R)-螺[2,2’-[1,3]金刚烷]-4’,6’-二酮。
本发明涉及的“50%CH3CN+50%0.1M甲酸铵水溶液”指的是CH3CN和0.1M甲酸铵水溶液各以占比50%的体积比例混合,其余类似表述依次类推。
本发明具有以下优势:
(1)本发明提供的无载体放射性氟-18负离子标记目标化合物[18F]FDPA是设计将放射性氟-18同位素直接连接在芳环上,相比于现有技术,应用在PET影像时,可以避免发生潜在的代谢反应而产生非特异性结合的正电子成像信号,这样就极大提高了PET影像的定量化分析的质量。
(2)本发明提供的无载体放射性氟-18负离子标记目标化合物[18F]FDPA的制备方法:以裸环三价碘叶立德作为标记前体,以无载体放射性氟-18作为放射源,设计并利用了非常温和的碱性反应条件,一步得到目标化合物[18F]FDPA;其反应条件温和,操作简便,实现自动化标记,非衰减校正产率达到30%以上,纯净度高于98%,比活度高于296GBq/μmol(4Ci/μmol)。
(3)本发明将无载体放射性氟-18负离子标记目标化合物[18F]FDPA应用于炎症动物模型(包括阿尔兹海默症老鼠模型和脑缺血老鼠模型)中的PET影像学评价,结果证明[18F]FDPA作为示踪剂具有很好的专一性、靶向型和稳定性,为后续进行临床炎症相关疾病的诊断、新型TSPO配体药物研发和剂量使用等提供了极大的科学支持。
附图说明
图1为本发明利用通用电气公司Tracerlab FXFN自动化合成系统合成[18F]FDPA的示意图。
图2为本发明的实施例3采用通用电气公司Tracerlab FXFN自动化合成系统制备[18F]FDPA的高效液相色谱检测结果示意图,由上至下分别为:HPLC柱压力示意图(其中横坐标为时间,单位为分钟;纵坐标为柱压力,单位为MPa);HPLC放射量检测器的检测信号图谱(其中横坐标为时间,单位为分钟;纵坐标为放射量强度大小);HPLC紫外检测器的检测信号图谱(其中横坐标为时间,单位为分钟;纵坐标为紫外吸收大小)。
图3为本发明实施例4中采用实施例2手动制备和实施例3自动制备的[18F]FDPA与标准品共注射的高效液相色谱检测结果。从上至下分别为HPLC放射量检测器的检测信号图谱和HPLC紫外检测器的检测信号图谱。其中横坐标为时间,单位为分钟;上图纵坐标代表放射量检测器检测信号,为[18F]FDPA放射线信;下图纵坐标代表紫外检测器检测信号,为FDPA的紫外吸收峰。
图4为本发明实施例6中利用[18F]FDPA进行的脑梗大鼠模型中的PET影像学评价的试验结果示意图。其中,图4A为仅注射[18F]FDPA的成像图,图4B为预先1min注射PK11195,之后再注射[18F]FDPA的成像图,图4C为本发明实施例6中仅注射[18F]FDPA的时间-放射量曲线,图4D为本发明实施例6中预先1min注射PK11195,之后再注射[18F]FDPA的时间-放射量曲线,图4C和图4D中,横坐标为以分钟为单位的时间,纵坐标为对[18F]FDPA的摄取量。
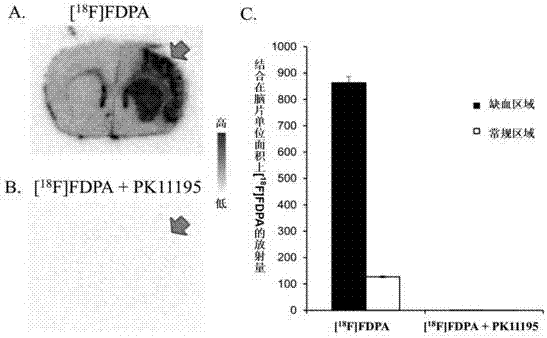
图5为本发明实施例7中利用[18F]FDPA进行的脑梗大鼠模型的体外自显影试验结果示意图。图5A为仅用[18F]FDPA涂染缺血脑片的成像图;图5B为用[18F]FDPA和PK11195混合液涂染缺血脑片的成像图;图5C为图5A和图5B的定量化柱状图,其中纵坐标表示结合在脑片单位面积上[18F]FDPA的放射量,黑色实心区域表示缺血区域,即为图5A的右侧部分,白色空心区域表示常规区域,即为图5A的左侧部分。图5B显示是几乎空白(原因是加入的PK11195把靶点全部占据了,[18F]FDPA根本没有任何结合),图5C右侧部分数据也就是图5B的定量数据,即数据值极低。
图6为本发明实施例8利用[18F]FDPA进行的12月大的正常老鼠和阿尔兹海默症老鼠模型的体内PET影像及体外自显影试验结果示意图。其中,图6A是12个月大正常老鼠的体内动态扫描0-30min的PET成像合图,分为轴向图、冠状图和纵向图;图6B是12个月大阿尔兹海默症老鼠的体内动态扫描0-30min的PET成像合图,分为轴向图、冠状图和纵向图;位于图6A图和6B之间的柱状是量化轴,颜色越深表示放射量越大,最大为2.0SUV;图6C是12个月大正常老鼠和阿尔兹海默症老鼠相同脑区的体外自显影图,量化轴表示颜色越深放射量摄取越多;图6D是[18F]FDPA在12月大正常老鼠和阿尔兹海默症老鼠脑内时间-放射量曲线图,横坐标为以分钟为单位的时间,纵坐标为对[18F]FDPA的摄取量。
具体实施方式
除非特殊说明,本发明所用术语具有本发明所属领域中的一般含义。
下面参考具体实施例,对本发明进行说明,需要说明的是,这些实施例仅仅是说明性的,而不能理解为对本发明的限制。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品;所用试剂在未注明其他要求时,均指符合相应国家标准的分析纯试剂。
实施例1
本实施例提供了一种化合物[18F]FDPA,具有如下结构:
化学名称:N,N-二乙基-2-(2-(4-(氟18-氟代苯基))-5,7二甲基吡唑[1,5-a]嘧啶)-3-基)乙酰胺
分子式:C20H2318FN4O
分子量:353.43
此化合物可以作为靶向转位蛋白TSPO的正电子药物。
实施例2
本实施例提供了实施例1中的[18F]FDPA的制备方法(手动制备),包括如下步骤:
步骤一:制备裸环辅基SPIAd
向4.8mL乙酸酐(成都化夏试剂,CAS108-24-7,货号H1133817-500mL)中加入5.0g丙二酸(CAS:141-82-2,货号:A11526-100g),随后加入24μL浓硫酸,反应体系升温至60℃反应15分钟后降至室温。
在48mmol 2-金刚烷酮(Alfa Aesar,CAS 700-58-3,货号:A14275-100g)中缓慢加入上述反应体系,约半小时到一小时之内加完即可,然后加压蒸干所有溶剂,得到裸环辅基SPIAd初产品,将裸环辅基SPIAd初产品溶解在300mL无水乙醚中,用水返洗三次,每次200mL,收集有机相,用无水硫酸镁干燥,过滤浓缩,之后所得产品用乙醚和正己烷(v:v=1:3,10mL)进行重结晶,得到纯净产物,称为裸环辅基SPIAd,产率52%。1H NMR(500MHz,CDCl3)δ3.60(s,2H),2.25–2.08(m,6H),1.91(s,2H),1.83–1.71(m,6H).13C NMR(126MHz,CDCl3)δ162.94,109.56,37.72,36.77,36.50,33.51,26.12ppm.HRMS(ESI/[M-H]-)calcd.for C13H15O4:235.0976,found 235.0979。
反应过程如下:
步骤二:制备裸环三价碘叶立德前体
(1)制备乙醇反应体系
将50mg(即0.11mmol)N,N-二乙基2-(2-(4-碘苯基)-5–二甲基吡唑[1,5-a]嘧啶-3-基)乙酰胺(结构如式1所示,该化合物的合成方法参考文献:Damont,A.;Médran-Navarrete,V.;Cacheux,F.;Kuhnast,B.;Pottier,G.;Bernards,N.;Marguet,F.;Puech,F.;Boisgard,R.;Dollé,F.Novel Pyrazolo[1,5-a]pyrimidines as Translocator Protein 18kDa(TSPO)Ligands:Synthesis,in Vitro Biological Evaluation,[18F]-Labeling,and in Vivo Neuroinflammation PET Images.J.Med.Chem.,2015,58,7449–7464.)溶解在由0.39mL三氟乙酸和0.13mL氯仿组成的混合溶液中,再向其中加入100mg(即0.165mmol)单过硫酸氢钾复合盐(Sigma-Aldrich,CAS号70693-62-8,货号与规格228036-100G),室温下搅拌50分钟后,加压蒸发掉所有溶剂得到初产品,将此初产品置于真空泵上抽干约30分钟,随后加入3.0mL乙醇,得到乙醇反应体系。
(2)反应制备裸环三价碘叶立德前体的粗产品
取步骤一制备得到的裸环辅基SPIAd 25.3mg(即0.11mmol)溶于0.5mL的10%(m/m)的碳酸钠水溶液中,缓慢加入上述乙醇反应体系,室温下剧烈搅拌下至体系呈透明状,随后向其中补加0.3mL的10%的碳酸钠水溶液中调整pH等于9。反应液室温下剧烈搅拌1小时后,加5mL水稀释体系,随后用二氯甲烷萃取三次,每次5mL。合并有机相,用无水硫酸镁干燥后,过滤旋干有机溶剂,得到裸环三价碘叶立德前体的粗产品。
(3)裸环三价碘叶立德前体的粗产品的纯化
将裸环三价碘叶立德前体的粗产品进行硅胶色谱柱层析,洗脱体系为200mL的乙酸乙酯,之后加大极性至甲醇和乙酸乙酯的混合液(v/v=1:10,200mL),得到产物浓缩旋干为白色固体粉末状,即裸环三价碘叶立德前体(式2),质量26.1mg,产率为34%。表征如下:Mp 142-143℃.1H NMR(300MHz,DMSO)δ7.87-7.81(m,4H),6.90(s,1H),3.89(s,2H),3.50(q,J=7.2Hz,2H),3.31(s,3H),3.24(q,J=7.1Hz,2H),2.69(s,3H),2.34(s,2H),1.93(d,J=13.8Hz,4H),1.79(s,2H),1.65(d,J=11.0Hz,6H),1.19-1.13(m,3H),0.9(t,J=7.2Hz,3H);13C NMR(75MHz,DMSO)δ169.2,163.0,158.4,152.5,147.6,145.2,136.4,132.9,130.5,116.3,109.5,105.5,102.2,58.0,42.1,37.0.35.3,33.7,27.7,26.4,24.7,16.7,14.7,13.5;HRMS(m/z):[M+Na]+calculated for 719.1706,found 719.1703.
反应过程如下:
步骤三:利用裸环三价碘叶立德前体制备[18F]FDPA
(1)利用QMA固相萃取柱捕获氟-18负离子
氟-18负离子的生产是通过18O(p,n)18F,应用小体积[18O]H2O靶,用18MeV的质子束流持续轰击60min。利用Waters Sep-Pak light QMA固相萃取柱(天津德祥茂隆科技有限公司,WAT023525,SEP-PAK LIGHT QMA 50BX。),从[18O]H2O捕获并分离8-10mCi的高纯度氟-18负离子,具体操作步骤参考文献:Damont,A.;Hinnen,F.;Kuhnast,B.;-Peyronneau,M.;James,M.;Luus,C.;Tavitian,B.;Kassiou,M.;Dollé,F.Radiosynthesis of[18F]DPA-714,a selective radioligand for imaging the translocator protein(18kDa)with PET.J.Labelled Comp.Radiopharm.2008,51,286-292。
(2)洗脱QMA固相萃取柱上的氟-18负离子
将13.5mg四丁基甲磺酸铵(Tetrabutylammonium methanesulfonate,TBAOMs,CAS 65411-49-6,上海瀚鸿化学,SR15010135)溶于由0.5mL乙腈和0.5mL水组成的混合溶液中,得到洗脱液;利用1mL注射器吸取洗脱液后将注射器与QMA固相萃取柱连接,以6mL/min的流速缓慢推出洗脱液,使之流经QMA固相萃取柱时充分洗脱其上吸附的8-10mCi的氟-18负离子同位素,收集在V形反应瓶中。
这里需要强调的是,通常洗脱氟-18负离子时采用的碱液为碳酸钾或四乙基碳酸氢铵的乙腈/水混合溶液,pH在9-10左右;但该碱性环境下裸环三价碘叶立德前体会在放射标记条件下(氟-18负离子)迅速分解,无法完成下列步骤(4)的反应,也就无法得到标记产物。经过反复尝试,最后选择弱的pH为7-8的四丁基甲磺酸铵(tetrabutylammonium methanesulfonate,TBAOMs)洗脱时,下面步骤(4)的反应才能发生,这是通过长期摸索且不断尝试才发现的,是本发明非常突出的特点。此外,pH在7-8的其它碱例如四乙基高氯酸铵(tetraethylammonium perchlorate,TEAOCl4)、草酸钾或四乙基三氟甲磺酸铵(Tetraethylammonium trifluoromethanesulfonate,TEAOTf)也可实现部分转化,但经多次条件筛选结果发现采用TBAOMs时标记产率是最高的。
(3)氟-18负离子的共沸干燥
将V形反应瓶置于110℃进行加热,并同时伴有10mL/min流速的干燥氮气鼓吹,进行5分钟后V形反应瓶中的溶剂完全被吹干,之后向其中加入1mL无水乙腈,继续在110℃加热条件下,并同时伴有10mL/min流速的干燥氮气进行鼓吹,进行5分钟后至溶剂完全被吹干,此过程重复3次,最后一次将V形反应瓶从加热器中取出,氮气鼓吹至体系温度降至室温。
(4)氟-18负离子与裸环三价碘叶立德前体的反应生成[18F]FDPA
将步骤二制备得到的1.8mg的裸环三价碘叶立德前体(式2)溶于1mL乙腈中,随后加入本步骤中(3)的V形反应瓶中,体系封口,置于加热器中在120℃下反应12分钟,然后将V形反应瓶从加热器中取出置于冰中冷却30秒后开盖,加入0.5mL由乙腈与水(体积比为1:1)组成的混合溶液终止反应。
反应过程如下:
(5)[18F]FDPA的分离纯化
将所有反应溶液(含有式3)注射入半制备高效液相色谱HPLC进行分离纯化。
高效液相色谱法分离纯化条件如下:
色谱仪器:美国沃特世高效液相色谱仪
半制备色谱柱:CAPCELL PAK C18,250x 10mm
流动相:45%乙腈+55%水,混合完毕后加入总体积0.1%的三乙胺。
三者混合后,超声1min去除空气气泡,即得流动相。
流速:5.0mL/min
柱温:23℃
检测器:沃特世2487双波长吸收检测器(Waters 2487DualλAbsorbance Detector)由紫外检测器(λ=254nm)和放射量检测器共同检测。
收集产物保留时间:14.7分钟
收集分离得到的产物,蒸干所有溶剂,重新溶于3毫升无菌生理盐水中,并通过0.22微米针头式滤器,得到可供注射用的[18F]FDPA放射量为2.8±0.6GBq(75.6±16.2mCi)。全部反应时间为75分钟,衰减校正产率为49±12%,比活度大于260GBq/μmol(7Ci/μmol)。
产物鉴定:
(1)经测试,[18F]FDPA的半衰期为110.1±0.2分钟,符合[18F]同位素的半衰期数值,说明其同位素纯度高。
(2)通过与非放射性化合物FDPA的共注射HPLC判断,产物结构为式3所示化合物(对应于权利要求的式D所示化合物)。具体谱图数据见实施例4。
实施例3
本实施例提供了[18F]FDPA合成系统,如图1所示,包括:
氟-18负离子产生装置;
氟-18负离子洗脱装置;
加料装置;
反应装置;
HPLC纯化装置;
萃取、收集装置;
以及阀门v1、阀门v3、阀门v5、阀门v6、阀门v7、阀门v8、阀门v10、阀门v13、阀门v18、阀门v20、阀门v24和阀门v25。
所述氟-18负离子产生装置包括GE回旋加速器和氦气产生器,所述GE回旋加速器经过氦气产生器通过阀门v10与洗脱装置的QMA固相萃取柱连接,
所述氟-18负离子洗脱装置包括洗脱液产生器和QMA固相萃取柱,所述洗脱液产生器为小瓶1,小瓶1依次通过阀门v1和v10与QMA固相萃取柱上下连接,所述QMA固相萃取柱分别通过阀门v10和阀门v13与反应装置的反应瓶12上下连接。
所述加料装置包括小瓶3、小瓶5和小瓶6,所述小瓶3通过阀门v3与反应装置的反应瓶12上下连接,所述小瓶5通过阀门v5与反应装置的反应瓶12上下连接,所述小瓶6通过阀门v6与反应装置的反应瓶12上下连接
所述反应装置包括反应瓶12。
所述HPLC纯化装置包括瓶14和半制备HPLC设备
所述萃取、收集装置包括大瓶15、C18固相萃取柱16、小瓶7、小瓶8、收集瓶17和针头式滤器,所述小瓶7通过阀门v7与C18固相萃取柱16上下连接,所述小瓶8通过阀门v8与C18固相萃取柱16上下连接。
本实施例接着利用上述[18F]FDPA合成系统进行[18F]FDPA的制备(自动化制备),包括如下步骤:
(1)利用QMA固相萃取柱捕获氟-18负离子
使用GE回旋加速器通过18O(p,n)18F核反应产生的放射性氟-18负离子通过阀门v10进入反应模块中,随后通过氦气产生器产生的氦气压力被吸附于Waters QMA固相萃取柱上。
(2)洗脱QMA固相萃取柱上的氟-18负离子
将20.0mg四丁基甲磺酸铵(TBAOMs)溶于由0.5mL乙腈和0.5mL水组成的混合溶液中,预先注入小瓶1中,反应开始后通过真空泵将小瓶1中的TBAOMs溶液通过阀门v10、QMA固相萃取柱、阀门v11抽入反应瓶12中,即将放射性氟-18负离子从QMA固相萃取柱上洗脱至反应瓶12中。
(3)氟-18负离子的共沸干燥
反应瓶12处开始85℃进行加热、鼓氮气过程,持续3分钟后,在氦气压力下将预先置于小瓶5中的1mL干燥乙腈溶液注入反应瓶12,85℃下鼓氮气8分钟,之后体系升至110℃,鼓氮气同时进行真空抽气,持续4分钟,确保反应瓶12中的溶剂全部蒸干。之后反应体系在空气气流下降温至40℃待加料。
(4)氟-18负离子与裸环三价碘叶立德前体的反应生成[18F]FDPA
预先将2mg裸环三价碘叶立德前体(式2,制备方法见实施例2)溶于1mL无水乙腈中,加入小瓶3中,在氦气压力下将小瓶3的该溶液通过阀门v3注入反应瓶12中,随后关闭反应瓶12周围的阀门v13、阀门v20和阀门v24,反应体系升温至120℃反应12分钟。反应完毕后打开阀门v24和阀门v25,体系降温至40℃,然后将预先置于小瓶6中的由1.5mL乙腈和1.5mL水组成的混合溶液加入反应体系停止反应。
(5)[18F]FDPA的分离纯化
在瓶14中预先加入2.5mL的HPLC流动相溶剂,反应瓶12中的全部溶液由氦气压力转移至瓶14中,接着将瓶14中的所有溶液通过氦气压力注射入半制备HPLC设备中,随即开始分离纯化,
高效液相色谱法分离纯化条件如下:
色谱仪器:美国沃特世高效液相色谱仪
半制备色谱柱:Luna C18semi-preparative,250×10.00mm,
流动相:50%CH3CN+50%0.1M甲酸铵水溶液
流速:5.0mL/min
柱温:23℃
检测器:沃特世2487双波长吸收检测器(Waters 2487DualλAbsorbance Detector)由紫外检测器(λ=254nm)和放射量检测器共同检测。
(6)萃取、收集
高效液相色谱法检测结果如图2所示,将产物峰对应的部分(保留时间为17.2分钟,见图2中的方框部分),通过阀门v18收集如大瓶15中,大瓶15中预先加入了23mL注射用级别的无菌水(United States Pharmacopeia(USP);Hospira);大瓶15中的溶液在氦气压力作用下通过置于16号位置的C18固相萃取柱(即Waters Sep-pak C18固相萃取小柱,产品编号WAT043395),并用小瓶7中预先加入的10mL无菌水冲洗C18固相萃取柱16以去除可能残余的盐类杂质、HPLC流动相和放射性氟-18负离子。最后在氦气压力下利用小瓶8中预先注入的1.0mL无水乙醇洗脱C18固相萃取柱16上的产物,收集至预先加入了10mL无菌生理盐水的产物收集瓶17中。收集瓶17中的所有溶液在氦气压力作用下通过0.22微米针头式滤器,得到可供注射用的[18F]FDPA。
自动化合成完毕后,测量得到产物[18F]FDPA的非衰减校正产率为27±5%(n=7),比活度大于148GBq/μmol(4Ci/μmol)。
实施例4
将实施例2和实施例3制备得到的[18F]FDPA均进行产物纯度及专一性测试,以确定收集产物是[18F]FDPA。具体方法为采用高效液相色谱法:取0.2mg FDPA(结构如下图)溶解于1mL乙腈中;将实施例2和实施例3制备得到的[18F]FDPA溶液各吸取0.1mL,分别溶于上述乙腈溶液中。得到的溶液震荡混合后,分别注射入HPLC中。
化学名称:N,N-二乙基-2-(2-(4-氟苯基))-5,7二甲基吡唑[1,5-a]嘧啶)-3-基)乙酰胺
简称:FDPA
分子量:354.43
性状:白色固体粉末,无特殊气味,不挥发。
合成方法见参考文献:Selleri,S.;Bruni,F.;Costagli,C.;Costanzo,A.;Guerrini,G.;Ciciani,G.;Costa,B.;Martini,C.2‐Arylpyrazolo[1,5‐a]pyrimidin‐3‐yl Acetamides.New Potent and Selective Peripheral Benzodiazepine Receptor Ligands,Bioorg.Med.Chem.2001,9,2661–2671.
高效液相色谱法检测条件如下:
色谱仪器:美国沃特世高效液相色谱仪
分析色谱柱:Phenomenex Luna C18,250x 4.6mm
流动相:70%CH3CN+30%0.1M甲酸铵水溶液
流速:1.0mL/min
柱温:23℃
检测器:沃特世2487双波长吸收检测器(Waters 2487DualλAbsorbance Detector),由紫外检测器(λ=254nm)和放射量检测器共同检测(紫外检测器串联在放射量检测器之前,即物质随流动相先流经紫外检测器,再流经放射量检测器,所以从精确性上,紫外出峰一般早于放射峰大约0.1-0.2min左右被认为是同时出峰)。
高效液相色谱法检测结果如图3所示。从图可知,出峰信号时间完全一致,从而确定经质量控制测试后,实施例2的手动制备得到的[18F]FDPA及实施例3自动制备得到的[18F]FDPA结构确定无误,化学及放射化学纯度均大于99%,满足临床前PET影像要求。
实施例5
本实施例提供了[18F]FDPA在动物血浆中的稳定性评价,具体方法为:
分别利用实施例2和实施例3制备的[18F]FDPA各分5份,每份10μL,约2.0MBq(54μCi),均与100μL正常血浆(购于美国Abcam公司)混合,在37℃震荡下孵育。分别在20min、40min、60min、90min、120min依次取出混合液,加入等体积的乙腈溶液使蛋白变性,剧烈震荡后离心2min(离心速率为6000转每分钟)沉降蛋白,然后吸取上清液50μL注射入高效液相色谱中,根据放射检测器出峰时间来判断[18F]FDPA在动物血浆中的稳定性。
高效液相色谱仪器:美国沃特世高效液相色谱仪
分析色谱柱:Phenomenex Luna C18,250x 4.6mm
流动相:70%CH3CN+30%0.1M甲酸铵水溶液
流速:1.0mL/min
柱温:23℃
检测器:沃特世2487双波长吸收检测器(Waters 2487DualλAbsorbance Detector),由紫外检测器(λ=254nm)和放射量检测器共同检测。
结果显示,[18F]FDPA在37℃下动物血清中非常稳定,120分钟之内没有任何分解,这同时也说明了[18F]FDPA用在PET影像时可以避免发生潜在的代谢反应而产生非特异性结合的正电子成像信号。
实施例6
本实施例为利用[18F]FDPA进行了脑梗大鼠模型中的PET影像学评价,具体方法为:
制备脑梗大鼠模型,在手术七天后,通过尾静脉注射入20MBq(540μCi)实施例2的[18F]FDPA,动态扫描90分钟,进行PET影像实验。老鼠模型制作方式及PET扫描操作见参考文献:Tiwari,A.K.;Yui,J.;Fujinaga,M.;Kumata,K.;Shimoda,Y.;Yamasaki,T.;Xie,L.;Hatori,A.;Maeda,J.;Nengaki,N.;Zhang M.-R.Characterization of a novel acetamidobenzoxazolone-based PET ligand for translocator protein(18kDa)imaging of neuroinflammation in the brain.J.Neurochem.2014,129,712-720。
PET影像实验结果如图4所示,结果显示,在脑梗一侧[18F]FDPA的摄取量明显高于正常脑侧;脑梗侧的最大摄取量出现在注射后的第10分钟,为1.20±0.08SUV;正常脑侧最大摄取量仅为0.54±0.01SUV,并且呈较快的消除行为,第2分钟摄取量与第45分钟摄取量比值为2.7;在注射后第10分钟和第90分钟,脑梗侧和正常脑侧的摄取量比值分别为5.41±0.12和4.26±0.34,有明显差异;抑制实验采用预先通过尾静脉注射靶向TSPO的配体PK11195(3毫克每千克),之后1分钟时再注射示踪剂[18F]FDPA,结果显示脑梗侧和正常脑侧对示踪剂的摄取量几乎一致,表明了[18F]FDPA在体内有很好的靶向性、专一性。
进而,利用简化参照组织模型分析方法,对[18F]FDPA在体内与转位蛋白TSPO的非取代性结合能力(BPND)进行了计算。结果表明,[18F]FDPA对TSPO的结合能力高达4.02±1.32(p<0.05),优于现有已经报道的示踪剂[11C]MBMP(2.03±0.24,p<0.05),[18F]FEBMP(2.72±0.27,p<0.05)和[11C]PK11195(1.59±0.33,p<0.05),说明[18F]FDPA将对定量化TSPO能提供更加准确的科学信息。
实施例7
本实施例提供了利用实施例2制备得到的[18F]FDPA进行的脑梗大鼠模型的体外自显影试验,利用[18F]FDPA对脑梗模型大鼠脑片进行培育。老鼠模型脑片制作方式及自显影数据采集操作见参考文献:Tiwari,A.K.;Yui,J.;Fujinaga,M.;Kumata,K.;Shimoda,Y.;Yamasaki,T.;Xie,L.;Hatori,A.;Maeda,J.;Nengaki,N.;Zhang M.-R.Characterization of a novel acetamidobenzoxazolone-based PET ligand for translocator protein(18kDa)imaging of neuroinflammation in the brain.J.Neurochem.2014,129,712-720。
结果如图5所示,结果显示,对比于正常脑侧,脑梗侧摄取信号明显高,脑梗侧与正常脑侧的信号比约为7:1;相似的,将2.0MBq(54μCi)[18F]FDPA与1mg PK11195(上海浩然生物技术有限公司,品牌ALEXIS,货号ALX-550-346-M010,规格10mg)混合后对脑片进行培育,发现信号差异消失。这些结果说明了[18F]FDPA在体外具有很好的靶向型和专一性。
实施例8
本实施例提供了针对实施例2制备得到的[18F]FDPA的阿尔兹海默症老鼠模型评价。具体的,是对12个月大的正常老鼠(B6C3F1/J)和阿尔兹海默症转基因老鼠模型(APP/PS1)(购自北京医科利昊生物科技有限公司,购买网址:http://www.bio-equip.com/sho w1service.asp?serviceid=13039)进行了[18F]FDPA的评价工作。
具体步骤为:将正常老鼠和上述阿尔兹海默症转基因老鼠麻醉后置于小动物PET扫描仪中,分别通过尾静脉注射方式将实施例2制备得到的可供注射用的[18F]FDPA150μL(约45μCi放射量)缓慢注射入两种老鼠体内,注射时间均为10秒,随即开始分别进行动态扫描,时间长度为60分钟,影像采集窗口设置为0.5分钟2个、1分钟5个、12分钟2个、30分钟1个(共10个)。扫描完毕后机器自动进行数据整理,转换成可供分析的PET图像。
结果显示,在APP/PS1老鼠模型中,[18F]FDPA可以迅速通过血脑屏障,在注射后3分钟达到1.50±0.13SUV,对比于正常老鼠对[18F]FDPA的摄取量而言,阿尔兹海默症老鼠大脑摄取量高1.6倍;体外自显影实验表明,转基因老鼠脑片中大脑皮层、丘脑、纹状体各脑区摄取量对小脑摄取量比值远远高于同样年龄的正常老鼠相应区域摄取量,说明[18F]FDPA可以作为阿尔兹海默症疾病的早期诊断试剂。
- 还没有人留言评论。精彩留言会获得点赞!