大豆苗期促侧根生长基因Gmr937及应用的制作方法
技术领域
本发明属分子生物学领域,具体涉及大豆苗期促侧根生长基因Gmr937及应用。
背景技术:
根系是作物吸收水分和营养的重要器官,强壮的根系对促进地上部分的光合作用、提高作物的产量、提高作物的抗逆性等方面都有重要意义。从分子水平上了解作物根系的基因功能,对提高作物产量及植物在抗旱研究中的相关研究提供理论依据。由于土壤限制根系的可观察性,田间条件下研究植物根系困难,所以有关作物根系的研究不如地上部深入。
目前对侧根生长相关基因的研究主要集中在侧根的生长发育和抗逆等方面。王峰等[3]对水稻侧根发育相关基因Os02g0198200的研究表明,该基因对水稻侧根的生长发育有促进作用,对水稻的生长发育,高产抗逆有重大意义;任永哲[4]在拟南芥根系发育的分子机制研究中,对RML1基因进行了功能分析,发现转该基因植株侧根发育水平显著提高,而该基因与谷胱甘肽合成有关,说明谷胱甘肽对侧根发育起到了促进作用。王鼎慧在对GmHAP3-17基因抗旱分析中发现在自然干旱条件下,转GmHAP3-17基因植株与野生大豆表现出根系更发达,扎根更深,侧根更多等明显现象,说明该基因对大豆植株具有正调控的抗旱功能,该基因成为培育抗旱转基因作物的基因资源。
技术实现要素:
本发明的目的是为了植物具有强壮的根系,促进地上部分的光合作用、产量及抗逆性,而提供一种大豆苗期促侧根生长基因Gmr937及应用。
大豆基因Gmr937,它的碱基序列如序列表SEQ ID NO.1所示;
所述的基因为人工合成。
大豆基因Gmr937在植物苗期促侧根生长的应用。
大豆基因Gmr937在培育耐旱转基因植物品种的应用。
所述的植物为大豆。
本发明提供了大豆苗期促侧根生长基因Gmr937,它是采用EastepTM试剂盒提取M18幼苗根系总RNA,反转录cDNA,克隆大豆苗期促根系发育相关Gmr937基因。分别构建植物过表达载体和RNAi表达载体,采用农杆菌介导法将重组质粒转入大豆JN18。经PCR检测得到T1代过表达载体阳性植株21株,RANi表达单拷贝整合到受体大豆JN18基因组中。随机各选取3株阳性植株进行荧光定量PCR测定,阳性植株13株。以35s启动子为探针进行Southern杂交检测,结果表明转化的目标基因以结果表明Gmr937基因在大豆幼苗叶片中的表达量最高,是对照的12倍,在茎的表达量最低,是对照的7倍;Gmr937干扰植株在大豆苗期根、叶片中的表达量是对照的0.7倍,在茎中的表达量是对照的0.85倍。测量适宜条件下培育的对照植株及转基因植株侧根总长度,结果表明超表达转基因植株的侧根总长度为90cm,比对照增加了10cm;RNAi干扰转基因植株侧根总长度为70cm,比对照组减少了8cm,说明Gmr937基因对大豆侧根的发育有促进作用。对干旱处理7d后的转基因大豆和CK对照组的抗旱生理生化指标进行测定,结果表明: Gmr937 基因在大豆中超表达可以提高大豆的耐旱性。
附图说明
图 1超表达载体结构图;
图2 RNA干扰载体结构图;
图 3克隆载体目的基因PCR验证,M: DNA Marker DL2000; P: 阳性对照; N: 阴性对照;A1,2:扩增目的片段PCR产物;B1-4克隆载体PCR产物;
图4 超表达载体的PCR和双酶切验证,M:DNA Marker DL2000; 1超表达载体PCR产物 , 2:超表达载体质粒双酶切产物;
图5干扰载体PCR和双酶切验证,M: DNA Marker DL2000; 1:N937正义PCR产物;2:N937反义片段PCR产物;3:干扰载体双酶切产物;4内含子PCR产物;
图6 T0代转化苗PCR检测;M: DNA Marker DL2000; P: 阳性对照; N: 阴性对照; CK: 未转基因植株; A:35s启动子检测B:Bar基因检测C:NOS终止子检测 1 ~3: 转基因植株PCR产物Note: A: PCR detection for 35S promoter; B: PCR detection for bar gene; C: PCR detection for NOS gene; M: DNA Marker DL2000; P: Positive control; N: Negative control; CK: Non - transformed plant; 1 ~ 3: Transgenic plants;
图7 转基因植株T1代植株PCR检测,M: DNA Marker DL2000; P: 阳性对照; N: 阴性对照; CK: 未转基因植株; 1 ~ 4: 转基因植株A35sPCR检测;B:Bar基因检测;C:NOS终止子检测; Note: A: PCR detection for 35S promoter; B: PCR detection for bar gene; C: PCR detection for NOS gene; M: DNA Marker DL2000; P: Positive control; N: Negative control; CK: Non - transformed plant; 1 ~ 4: Transgenic plants;
图 8 T1 转基因植株 Southern 杂交检测,M: λ Hind Ⅲ DNA Marker; P: 阳性对照; CK: 未转化植株; A:超表达植株;B:干扰植株1 - 3: 转化植株 Note: M: λ Hind Ⅲ DNA Marker; P: Positive control; CK: Non - transformed plant; 1 - 3: Transgenic plants;
图9大豆苗期Gmr937基因在 T1 转化 植株根、茎、叶组织中的相对表达量;
图10大豆苗期干旱处理24h后目的基因在根、茎、叶组织中的相对表达量;
图11干旱处理后转基因植株和CK对比图。
具体实施方式
实施例1Gmr937基因克隆及表达载体的构建
Gmr937基因的克隆植物材料
1)植物材料 栽培大豆(Glycine max)吉农 18、M18植物材料,由吉林农业大学植物生物技术中心提供。
载体与菌种 大肠杆菌( Escherichia coli)菌株DH5α ,农杆菌菌株EHA105,克隆载体pMD18-T,基础植物表达载体pCAMBIA3301,由吉林农业大学植物生物技术中心提供;
生物试剂 Easteptm通用型总RNA提取试剂盒,DNA GEL Extraction kit凝胶回收试剂盒,pMD18-vector载体试剂盒,限制性内切酶(BglⅡ,BsteⅡ),无缝克隆试剂盒和反转录试剂盒购自中美泰和(北京)公司。
2)Gmr937基因的克隆
利用 Primer 5 软件设计Gmr937基因全长序列的引物N937A1/937NAS1见表1,通过EastepTM试剂盒提取M18幼苗根系总RNA,反转录成cDNA,克隆Gmr937基因。利用反转录所得的cDNA为模板,用特异性引物N937A1/937NAS1的最适温度上下10℃梯度PCR。反应体系: DNA 模板 2 μl, 10×buffer 2.5 µl, 2 mmol·L-1 dNTP 1.5 µl, 25 mmol·L-1 MgSO4 2.0 µl, 10 µmol·L-1引物( F/R)各 1 µl,高保真酶 Kod Plus( 5 U·µl-1) 0.3 µl,加 ddH2O 至 25 µl。反应条件: 预变性94 ℃ 5 min,变性 94 ℃ 45 s ,退火 58℃ 45 s , 延伸72℃ 1.5 min , 35 个循环; 后延伸72 ℃ 5 min ℃ 。取 10 µl 扩增产物在 1.5 %的琼脂糖凝胶分离回收,将回收产物连接到pMD18T载体上形成PMD-18T-Gmr937克隆载体,挑去单克隆测序。
测序结果与NCBI数据库比对,利用Expasy在线分析Gmr937序列的氨基酸序列,进行氨基酸序列的同源性比较及功能的预测。如表1引物信息;
提取M18幼苗根部总RNA为模板,反转录成cDNA,用所设计的特异性引物N937A1/N937AS1扩增Gmr937目的片段,大小为1000bp见图3A,将扩增片段连接到PMD-18T上,对目的片段进行PCR鉴定,得到与预期大小一致的1000bp大小的Gmr937基因片段,见图3B。
3)Gmr937载体的构建
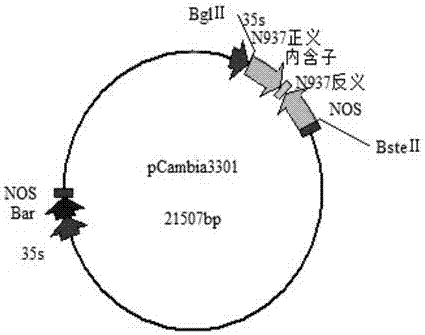
利用无缝克隆软件CE Design V1.03设计引物,超表达引物N937cbdA′/N937cbdAS′见表1。将上述克隆载体PMD-18T-Gmr937与载体pCambia3301质粒进行双酶切(BglⅡ,BsteⅡ)纯化回收大片段的线性片段和Gmr937目的片段,按照无缝克隆试剂盒说明书进行连接,得到pCambia3301-Gmr937表达载体,将构建好的表达载体转化到大肠杆菌中保存,提取保存菌液质粒,经PCR和双酶切验证,所得阳性菌株送长春库美生物技术有限公司测序,按照测序结果筛选阳性菌株。pCambia3301-Gmr937表达载体结构见图1。
使用BglⅡ,BsteⅡ双酶切植物过表达载体得到大小1000bp的基因片段与目的基因片段大小一致,对构建好的超表达载体质粒进行目的片段的PCR检测和双酶切检测见图4,经PCR和双酶切鉴定结果表明目的基因的超表达载体构建成功。
4)RNAi表达载体的构建
利用RNAi干扰引物N937gr1A′/N937gr1AS′、N937gr2A′/N937gr2AS′对克隆载体PCR得到目的基因Gmr937的正反义片段,利用引物N937nhz1A′/N937nhzAS′对大豆基因组进行PCR,克隆功能性间隔序列用于形成茎环结构。按照无缝克隆试剂盒说明进行连接,转化获得以抗除草剂Bar基因为筛选标记的RNAi表达载体pCambia3301-RNAi- Gmr937。RNA干扰载体结构如图2。
使用BglⅡ,BsteⅡ双酶切植物干扰载体得到大小2200bp大小的基因片段与目的基因的正反义片段和内含子片段的加和一致,对干扰载体进行正反义片段和内含子序列的PCR检验和双酶切验证见图5,鉴定结果表明目的基因的正反义片段和内含子序列已经成功连接到3301载体上,植物干扰载体构建成功。
实施例2 Gmr937载体农杆菌转化法获得转基因植株及检测
依据钟影大豆子叶节侵染方法[8]将构建好的植物超表达载体和RNAi表达载体通过农杆菌介导法转入到受体植株JN18中,获得转基因植株。
1)PCR检测
CTAB法提取转基因植株基因组DNA,设计35s启动子引物,NOS终止子引物和bar基因引物, 以Gmr937表达载体质粒和JN18对照组DNA分别作为阳性对照和阴性对照, PCR扩增目的片段,扩增产物在1%琼脂糖凝胶上电泳分析。
采用农杆菌介导法将重建的植物表达载体转化到受体大豆JN18中,对获得的T0代转化植株进行PCR检验,见图6。结果说明35s启动子,Bar基因和NOS终止子已经转入转化植株中。
对T1代植株进行PCR检测,结果如图7,得到35s启动子特异性条带,Bar基因特异性条带和NOS终止子特异性条带,与预期结果相符。100粒T1代种子有34株转化为阳性植株,其中21株为超表达植株,13株为干扰植株。
2)Southern杂交检测
对转化成功的阳性苗进行培育,得到T1代种子,以内切酶BamHⅠ单酶切,以35s启动子序列为探针进行Southern杂交,检测目的基因在T1代植株基因组中的整合情况。
Southern blot 检测 结果为阳性的植株进行 qRT - PCR 检测见图9,结果证明N937基因在转基因大豆植株苗期的根、 茎和叶片中均有表达,且超表达植株目的基因表达量都有明显的提高,在叶片提高最明显,相对表达量是对照的12倍,在根中表达量是对照的10倍;转干扰表达植株目的基因的表达量与CK相比降低,在根、叶片中表达量是对照的0.8倍,在茎中表达量是对照的0.85倍。
干旱处理24小时后,超表达植株目的基因表达量有显著提高,在叶片中表达量是对照组的14倍,在根中表达量是对照组的12倍。转RNAi-Gmr937载体植株根、茎表达量是对照组的0.8倍,在叶片中表达量是对照组的0.7倍,见图10。结果表明,干旱条件下Gmr937基因在超表达转化株中表达量明显上升。
3)qRT-PCR检测
提取T1代转Gmr937超表达植株和转RNAi-Gmr937植株幼苗时期根部、茎部和叶片的RNA,反转录成cDNA。 设计荧光定量PCR的特异性引物QA/QAS见表1 ,以大豆肌动蛋白为内参进行qRT-PCR检测,反应体系:引物QA/QAS各2µl,DNA模板2µl,DD water 4µl ,SYBR Premix Mix 10µl;反应条件:预变性95℃ 5min,变性 95℃ 30s,退火 58℃ 30s,延伸 72℃ 30s。采用Mx3000型实时荧光定量PCR仪,按照SYBR Premix ExTaql试剂盒说明书扩增目的序列,每个样品三次重复,通过2﹣△△CT计算分析目的基因的相对表达量。
分别提取34株阳性植株总DNA,分别以表达载体pCambia3301-Gmr937和RNAi表达载体pCambia3301-RNAi-Gmr937质粒作为阳性对照,以未导入基因的JN18作为阴性对照,以35s为探针,内切酶BamHⅠ酶切大豆全基因组,进行Southern杂交检测,结果如图8所示,未转化的JN18植株没有出现杂交信号,检测的42株植株有6株阳性植出现了明显的杂交信号,结果表明35s基因以单拷贝的形式整合到受体大豆基因组中,整合位点不同。
4)转化植株幼根长度和相关生理生化检测
设置两组处理,第一组在25℃下湿度10%水分正常条件处理的转化植株与CK植株的对照组,第二组在25℃下湿度5%干旱条件处理的转化植株与CK植株的对照组,每组设置三次重复,7d后分别用直接测量法测量各组的侧根总长度,用交流电法测各组叶片组织的相对电导率,用紫外分析法测各组叶片组织的丙二醛含量,用比色法测定各组的过氧化物酶活性,并用DPS软件处理数据,Duncan法进行样本间的差异分析。
设置适宜水分处理和干旱处理2个实验组,分别测定2个处理条件下CK、过 表达和干扰 T1代植株的侧根总长度,结果显示超表达植株在适宜条件下其侧根根系发育与CK对照组相比,侧根系总长度增长了17.5%~23%,干旱处理后超表达植株与CK对照组相比侧根系总长度增长了24.6%~28.3%,干扰植株在适宜条件下侧根发育比CK对照组相比侧根系总长度下降了6.3%~10.2%(表2)。说明该基因对大豆侧根的生长发育有明显的促进作用。干旱处理24h后,转N937基因未有明显变化,CK对照组叶片微微下垂、萎蔫,而转RNAi-N937基因的植株叶片明显萎缩,打卷,如图11。
注/Note: Duncan’s,P = 0. 05 A: Gmr937表达植株 a:Gmr937干扰植株
5)、 转化植株幼根长度和相关生理生化检测
设置两组处理,第一组在25℃下湿度10%水分正常条件处理的转化植株与CK植株的对照组,第二组在25℃下湿度5%干旱条件处理的转化植株与CK植株的对照组,每组设置三次重复,7d后分别用直接测量法测量各组的侧根总长度,用交流电法测各组叶片组织的相对电导率,用紫外分析法测各组叶片组织的丙二醛含量,用比色法测定各组的过氧化物酶活性,并用DPS软件处理数据,Duncan法进行样本间的差异分析。
一般在逆境胁迫下,植物细胞膜的透性会发生改变,从而适应环境,细胞膜受伤害的程度可以通过测定细胞渗透在水溶液中的电导率来间接获得植物受到逆境伤害越大越 容易导致相对电导率增大。由表 3 可知,常温条件下,转 Gmr937 基因植 株叶片与未转化植株叶片相对电导率无显著差异,干旱胁迫处理 7d 后,转化株系 1.5g叶片的相对 电导率与未转化植株叶片的相对电导率间的差异达到了显著水平,转Gmr937基因植株相对电导率明 显低于非转化植株,降低了 13.8~ 15.15% ,说 明转表达基因株系叶片的细胞膜受到逆境的伤害较轻,而转干扰基因植株叶片相对电导率升高了3.27 ~5.31%。
丙二醛是常用的膜脂过氧化指标,可反映细 胞膜质的过氧化程度和对逆境胁迫反应的强弱。由 表 4 可知,适宜条件下,转Gmr937基因植株叶片 与未转化植株叶片丙二醛含量无显著差异,干旱处理 7d后,转 Gmr937基因植株丙二醛含量明显低于 非转化植株,降低了 9. 86% ~ 10. 34% ,二者间的 差异达到了显著水平,说明转基因株系的叶片比未 转基因株系的叶片受到的逆境伤害小。
转RNAi-Gmr937基因的植株丙二醛含量与非转基因植株无显著差异,说明转干扰载体植株叶片受到逆境伤害并未发生明显变化。
过氧化物酶( POD) 可衡量植物抗逆性的 强弱。由表 5可知,适宜条件下,转Gmr937基因植株 叶片与CK植株叶片中 POD 活性无显著差异,干旱处理 7d后,转Gmr937基因植株过氧化物酶 活性 明 显 高 于 非 转 化 植 株,增 加 了 24. 62% ~ 31. 78% ,二者间的差异达到了显著水平,表明了转 基因植株抗氧化系统的活性氧清除能力要高于非转 基因植株。
通过数据分析对比发现转Gmr9337基因植株对干旱胁迫的抗性提高了,而转干扰基因的植株对抗旱性反而降低,进而说明Gmr937基因的功能与植物的抗旱有关。N937基因表达的蛋白部分是蛋白激酶活性蛋白功能原件,而热激蛋白与植物的抗旱和抗高温有关,这与本实验研究结果相一致[18-20]。经干旱处理后的 转Gmr937基因植株在侧根的数量以及侧根的长度上与CK对照植株和Gmr937干扰植株均表现出显著水平,而CK植株侧根系发育较转干扰株系根系发育较好。正常条件下转N937基因的大豆在根系发育的形态学上主根长度与侧根数量较正常植株多,而导入Gmr937干扰基因的株系其根系发育较为迟缓,与莫金钢[21]等研究结果相一致,所以鉴定Gmr937基因对大豆的侧根发育有一定的功能。
在本试验中干旱胁迫下,通过对转化植株及非转化植株进行生理生化指标测定,转化Gmr937阳性植株与对照 组之间均有显著差异,大豆Gmr937 基因的表达与相对电导率、丙二醛含量和 过氧化物酶的变化有关。说明Gmr937基因在受到干旱胁迫处理后引起了细胞膜渗透性、膜质过氧化程度 和抗氧化系统的生理生化变化[22]。结 果证明 了 转Gmr937 基因的过量表达能够提高转基因大豆的耐 旱能力,为该基因的利用奠定了基础。 由于目的基因对植物的生长发育 及非生物胁迫起着重要作用,将有耐受非生物胁迫 功能的植物 Gmr937基因转入经济作物 中,对提高经济作物的耐胁迫能力,加速根部的生长发育及稳定农作物的产 量有着非常重要的意义[23-25]。有关 Gmr937基因在蛋白水平 上的表达有待于进一步研究。
<110> 吉林农业大学
<120> 大豆苗期促侧根生长基因Gmr937及应用
<160> 1
<210> 1
<211> 1000
<212> DNA
<213> 人工
<400> 1
acgattcgag ctcggtaccc gggatcctct agagattttt ttcagcgcca tctccccttg 60
cgtttgggcc tggcccatta gttcccttgc ctccgtaatt tcttgaccca gttcgaccca 120
ttgccaccaa gggatttctc tggcccgatg cggtcctagt tccggattcc atacatgtgt 180
ctccacgtcc cacccatgct ctgtttttgc tagtcatctc cattttgacg gccctcgcta 240
tattcatcaa acgtcctctc gttaacagat tggcaacgtg caagctcttc aaaccatgtg 300
cgaagcaagc gaaatattgt tcgtcttgca atttgggaat ctgagcaatt aaacattcaa 360
accgttgaat gtagtcatca atgctcctcg tctgttgcag cgccgataat tgctcatatg 420
ctgttccctt ccctgcatac ctctgcatca gttcatactt caattcctcc cacgtcaggt 480
tctcattctc aagcagtaac aaattgaaga aatgaacggt cccgttctcc atgctaatct 540
gggcgagcct caccttcacc tcgtcgcttg tttcctgcac ctggaaatag atttcagccc 600
ttgaaatcca gccgacggga tcttcgccag tgaaagaagg gagctccacc ttcttgatcg 660
attgtcggaa ttcgtctagt gcttctgctt ctaagctctt ccaaaccgaa ccagcactga 720
tcggatttgg cctctttcca ccatgctctc cgttcgtttt ggccttgcga cttccgccat 780
gtgatccttc tccctcttct ccatcgttga tgttcatcat caagctcttc gtcaccagtt 840
ctaggatctt cgactggttc tcctttgctt cttttgcttc tgcttgtatc gcctccatca 900
tcgatttcat catctccaaa tcaccttcaa ccttttccat cctcgcgtcc atcgctgatt 960
cgtccgctgg ttttatcgtc gacctgcagg catgcaagcg 1000
- 还没有人留言评论。精彩留言会获得点赞!