一种在单细胞水平上检测基因组上核小体排布的方法与流程
本发明涉及一种检测基因组上核小体排布的方法,尤其是检测单细胞甚至单倍体细胞的核小体排布方法。
背景技术:
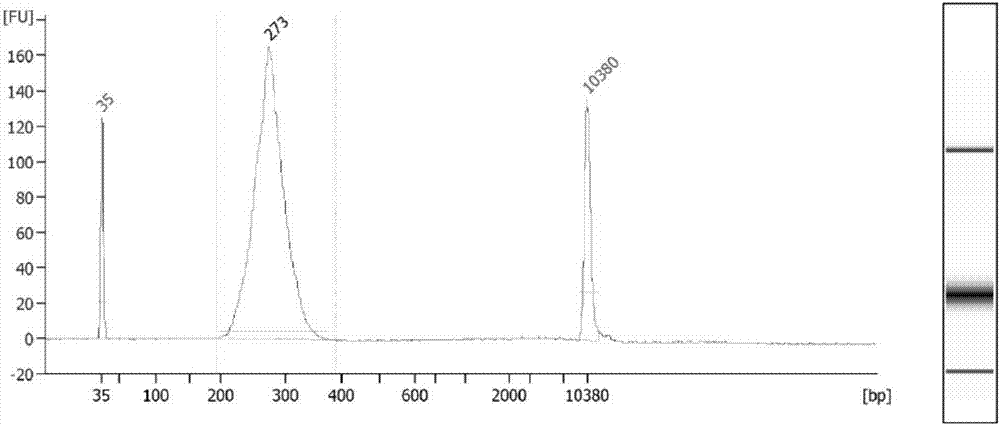
:细胞是生物体功能的基本单位,大量细胞的协调工作保证了生命活动的正常有序进行。但是,在一些疾病中,例如癌症,单个细胞的过度增殖就会引起整个器官功能的紊乱。到目前为止,大部分基因组水平的检测都是以大量细胞为样本进行的,得到的数据是这些样本的平均,这样就很难解释细胞与细胞之间的差异,同样,利用传统方法对一些细胞量很少的样本,例如植入前的胚胎,进行测序是很困难的。单细胞测序技术的实现给基因组学的研究开辟了一个新的领域,使我们可以对复杂的样本进行精细的研究,并且让我们对极少量的样本进行全基因组水平的测序成为可能。在过去的几年里,单细胞测序技术已经能够实现对单细胞基因组,转录组,dna甲基化组等多个水平上的检测,并被广泛应用于发育生物学、神经生物学、微生物学、免疫学以及癌症研究等多个生物学领域。真核生物的dna是缠绕在组蛋白八聚体形成的核小体上的,每个核小体上大约有147bp的dna缠绕,dna形成的双螺旋结构每10个碱基会与组蛋白形成一次直接的接触,使得dna双螺旋的大沟交替地朝向或者远离组蛋白核心八聚体。组蛋白沿dna序列的移动会导致暴露在外面的dna序列的不同,这些裸露在外的dna序列直接决定了dna结合蛋白对该序列的结合能力,因此核小体在dna序列上的位置对dna的生物学功能起了至关重要的作用,了解核小体在基因组上的确切排布对我们深入了解真核生物的基因组是如何调控这些生物学功能的是十分重要的。检测全基因组水平的核小体排布最常用的方法就是依赖于微球核酸酶(mnase)的高通量测序方法(mnase-seq),在这种方法中,微球核酸酶会优先切割裸露的dna以及两个核小体之间的连接dna,与组蛋白结合形成核小体的dna序列会被保护起来,不能够被mnase消化,这些被保护的dna片段经过高通量测序后就可以指示核小体在基因组上的排布情况。传统的mnase-seq包括两个主要的步骤,一个是收集足够多的dna样本,并且用微球核酸酶切割,得到与核小体结合的dna片段,另一个就是对这些得到的dna片段进行高通量测序,通过比对这些dna片段在基因组上的位置来确定核小体在基因组上的排布情况。到目前为止,人以及多种模式生物的核小体排布情况都能够通过mnase-seq检测出来。最近的研究表明核小体的位置会影响转录因子的相互作用,并且影响不同细胞基因的表达状态和表达水平,导致细胞群体基因表达的异质性。传统的mnase-seq需要百万级的细胞来进行一次检测,得到的结果是这些细胞的平均状态,不同细胞之间的核小体排布差异就被忽略,而这些差异正是导致细胞异质性的原因之一,另外,很多情况下,特别是体内的细胞,想要得到传统的mnase-seq所需的百万级的细胞量是很困难的,因此开发适用于少量细胞甚至单细胞的mnase-seq技术对于解决这些问题就至关重要。技术实现要素:针对现有技术的上述不足,根据本发明的实施例,希望提供一种在单细胞水平上检测基因组上核小体排布的方法,适用于少量细胞甚至单细胞的核小体排布检测。根据实施例,本发明提供的一种在单细胞水平上检测基因组上核小体排布的方法,包括如下步骤:(1)裂解细胞,并用微球核酸酶处理基因组上的dna片段;(2)将步骤(1)中酶切后的产物用蛋白酶处理,去掉dna片段缠绕的组蛋白;(3)将步骤(2)中得到的dna片段进行测序文库构建,并进行高通量测序;(4)对高通量测序的结果进行比对分析,并对测序结果的质量进行评估,包括在基因组上的覆盖度、片段长度分布、在转录起始位点和ctcf结合位点的核小体排布等。本发明中,所述的细胞包括所有真核生物的细胞。相对于现有技术,本发明创建了一种新的mnase-seq的方法,通过尽量减少dna纯化次数来减少损失,提高文库构建效率,进而可以利用极少量的细胞甚至单个细胞在全基因组水平上检测细胞的核小体排布。利用本发明的方法检测的数据可以覆盖基因组上大部分的区域,该方法为检测单细胞的核小体排布提供了一种十分有效的方法。该方法适用于对单细胞到几百个细胞的检测。附图说明图1是用少量或单个细胞进行文库构建时,文库经两轮扩增后的琼脂糖凝胶电泳图。图2是将构建好的单细胞或者少量细胞文库使用agilent2100生物芯片分析系统进行dna片段的分析图(图中展示的是构建成功的文库的片段分析结果)。图3是用少量细胞或单细胞进行mnase-seq检测,所得到的数据能覆盖基因组的百分比示图。图4是测序数据的片段长度分布图。图5是ctcf结合位点两侧的核小体排布情况图。图6是转录起始位点(tss)前后的核小体排布情况图。图7表示用显微操作仪收集单原核的示意图。图8是用但个原核进行文库构建时,文库经两轮扩增后的琼脂糖凝胶电泳图。图9是将构建好的单原核文库使用agilent2100生物芯片分析系统进行dna片段的分析图(图中展示的是构建成功的文库的片段分析结果)。图10是用单个单倍体的原核进行mnase-seq检测,所得到的数据能覆盖基因组的百分比示图。图11是单原核测序数据的片段长度分布图。图12是单原核检测的ctcf结合位点两侧的核小体排布情况图。图13是单原核检测的转录起始位点(tss)前后的核小体排布情况图。具体实施方式下面结合附图和具体实施例,进一步阐述本发明。这些实施例应理解为仅用于说明本发明而不用于限制本发明的保护范围。在阅读了本发明记载的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等效变化和修改同样落入本发明权利要求所限定的范围。以下实施例中所使用的技术,包括pcr扩增与检测、dna纯化等分子生物学技术,以及细胞培养、检测技术等,除非特别说明,均为本领域内的技术人员已知的常规技术;所使用的仪器设备、试剂和细胞系等,除非是本说明书特别注明,均为一般本领域的研究和技术人员可以通过公共途径获得的。实施例1:少量或单个小鼠胚胎干细胞mnase-seq检测一、实验目的:利用本发明方法检测单个或者少量几个胚胎干细胞的核小体排布情况,构建能用于illumina上机检测的dna文库。二、实验方法:1、细胞获取1)小鼠胚胎干细胞系r1购买自americantypeculturecollection(atcc),并在实验室培养及传代。2)用口吸管挑取一个小鼠胚胎干细胞的克隆,放在杜氏磷酸缓冲液(dpbs)中洗一遍。3)将克隆放在胰酶中,37℃放置5分钟。4)用直径10μm左右的口吸管反复吹吸克隆,直到克隆内的细胞全部散成单个细胞。5)将细胞放到含有0.5%牛血清白蛋白(bsa)的磷酸缓冲液(pbs)中洗三遍。2、细胞裂解1)配置裂解缓冲液(10mmtris-hcl(ph8.5),5mm氯化镁,0.6%乙基苯基聚乙二醇(np40))。2)在一个0.2ml的低吸附管中放入0.65μl裂解缓冲液,取步骤1中一个或几个胚胎干细胞放到裂解缓冲液中,在冰上放置5分钟,使细胞膜被充分裂解。3)将裂解好的细胞放到4℃离心机中,7000rpm离心1分钟。3、微球核酸酶(micrococcalnuclease)反应1)配置微球核酸酶反应体系(1xmnasemasterbuffer,2mm二硫苏糖醇(dtt),5%聚乙二醇(peg6000),30umnase)(mnase及mnasemasterbuffer购自neb公司,货号m0247)。2)将上述反应溶液2.5μl加入步骤2中离心后的细胞裂解液中,轻轻混匀,在室温反应10min。3)加入0.65μl100mm乙二胺四乙酸(edta)溶液终止微球核酸酶反应。4)加入0.65μl2%聚乙二醇辛基苯基醚(tritonx-100),进一步裂解细胞核膜。4、蛋白酶消化1)向上述反应体系中加入0.3μl蛋白酶(购自qiagen公司,货号19155)。2)在pcr仪中50℃反应2小时用蛋白酶降解dna缠绕的组蛋白,75℃反应30分钟使蛋白酶失去活性。5、测序文库构建(文库构建试剂盒购自kapa公司,货号kk8505)1)向上述反应体系中加入12.5μl超纯水,将反应体系扩大到17μl。2)加入2.3μlendrepair&a-tailingbuffer,1μlendrepair&a-tailingenzymemix,混匀。3)在pcr仪中,20℃反应30分钟,65℃30分钟使酶失活。4)加入0.5μladapterstock(购自illumina),3.5μlpcr-gradewater,10μlligationbuffer,3μldnaligase,混匀。5)在pcr仪中20℃孵育60分钟。6)加入37μlxpreagent(购自beckman,货号a63881),进行dna纯化。用10μlelutionbuffer洗脱dna。7)向洗脱下的dna中加入12.5μl2xkapahifihotstartreadymix,2.5μl10xkapalibraryamplicationprimermix,混匀,反应体系为25μl。8)将上述样品进行第一轮pcr扩增,设置pcr程序如下表1:表19)向扩增后的样品中按照1:1加入xpreagent,进行dna纯化,用11μlelutionbuffer洗脱dna。10)取出1μl洗脱下来的dna用qubitdsdnahsassaykit试剂盒(购自invitrogen,货号q32851)进行浓度检测。11)向洗脱下的dna中加入12.5μl2xkapahifihotstartreadymix,2.5μl10xkapalibraryamplicationprimermix,混匀,反应体系为25μl。12)将上述样品进行第二轮pcr扩增,设置pcr程序如下表2:表213)第二轮扩增的循环数根据下表3确定。表3第一轮扩增后的dna浓度(ng/μl)第二轮扩增的cycle’数<0.19-100.1-0.37-80.3-0.56-70.5-1.05-61.0-3.04-53.0-5.03-45.0-10.02-3>10.00-214)将扩增后的样品加入到2%的琼脂糖凝胶中,恒压120伏特,跑电泳30分钟。15)凝胶电泳的结果如图1所示,将单个核小体的条带切割下来,用qiagen公司的qiaquickgelextractionkit(货号28706)进行dna片段的回收。16)回收的dna片段用qubit检测浓度,用agilent2100生物芯片分析系统进行dna片段的分析,分析结果如图2所示。17)建构建好的文库送到公司进行高通量测序,测序使用的机器为illuminaxten,测序模式为双端150bp。三、实验结果:1)如图3所示,每个反应使用1个细胞、5个细胞或100个细胞进行检测,检测数据在基因组上的覆盖度都能达到全基因组的80%左右,并没有明显的降低,这说明利用本发明方法进行单细胞的核小体排布检测是十分有效的。2)如图4所示检测的数据的片段长度大多集中在单个核小体的区域,没有大量过长或者过短的片段。并且用单细胞进行检测的数据质量没有明显的下降。3)如图5所示,用单细胞或少量细胞进行检测在ctcf的结合位点两侧能够看到有规律的核小体排布,这说明检测数据的质量是很好的。4)如图6所示,无论用单细胞还是用少量细胞进行检测,都能看到在转录其实位点上游核小体的缺失区域以及下游核小体的有规律的排布。因此,从上述实验结果可以看出,利用本发明方法进行单细胞的核小体检测,得到的实验数据质量与多个细胞相比没有明显的差异,说明本发明方法能够很好的适应单细胞核小体检测的需要,本发明方法使得对很少的细胞进行核小体检测成为可能,这样极大地降低了原材料的获取难度,并且在稀有材料以及对细胞异质性的研究中将得到广泛的应用。实施例2:原核的mnase-seq检测一、实验目的:利用本发明方法对受精初期的原核的核小体排布情况进行检测,构建能用于illumina上机检测的dna文库。以此来检测本发明方法能否适用于对于单倍体基因组的核小体排布检测。二、实验方法:1、原核的获取1)实验用spf级c57bl/6小鼠采购自北京维通利华实验动物技术有限公司,并在同济大学实验动物中心饲养。2)取8-10周龄的母鼠,腹腔注射5u的孕马血清促性腺激素(pmsg),48小时后腹腔注射6u的人绒毛膜促性腺激素(hcg)。将注射激素后的母鼠与公鼠按照1:1的比例合笼。3)合笼后的第二天早上检查小鼠交配情况,保留阴道见栓的母鼠。4)处死母鼠后取出受精卵,用hoechest33342染料染色5分钟,如图7所示在显微操作仪上将受精卵的两个原核分别取出。5)将取出的原核放到含有0.5%牛血清白蛋白(bsa)的磷酸盐缓冲液(pbs)中洗三遍。2、原核的裂解1)配置裂解缓冲液(10mmtris-hclph8.5),5mm氯化镁,0.6%乙基苯基聚乙二醇(np40))2)在一个0.2ml的低吸附管中放入0.65μl裂解缓冲液,用口吸管吸取步骤1中单个原核放到裂解缓冲液中,在冰上放置5分钟,使细胞膜被充分裂解。3)将裂解好的细胞放到4℃离心机中,7000rpm离心1分钟。3、微球核酸酶(micrococcalnuclease)反应1)配置微球核酸酶反应体系(1xmnasemasterbuffer,2mm二硫苏糖醇(dtt),5%聚乙二醇(peg6000),30umnase)(mnase及mnasemasterbuffer购自neb公司,货号m0247)。2)将上述反应溶液2.5μl加入步骤2中离心后的细胞裂解液中,轻轻混匀,在室温反应10min。3)加入0.65μl100mm乙二胺四乙酸(edta)溶液终止微球核酸酶反应。4)加入0.65μl2%聚乙二醇辛基苯基醚(tritonx-100),进一步裂解细胞核膜。4、蛋白酶消化1)向上述反应体系中加入0.3μl蛋白酶(购自qiagen公司,货号19155)。2)在pcr仪中50℃反应2小时用蛋白酶降解dna缠绕的组蛋白,75℃反应30分钟使蛋白酶失去活性。5、测序文库构建(文库构建试剂盒购自kapa公司,货号kk8505)1)向上述反应体系中加入12.5μl超纯水,将反应体系扩大到17μl。2)加入2.3μlendrepair&a-tailingbuffer,1μlendrepair&a-tailingenzymemix,混匀。3)在pcr仪中,20℃反应30分钟,65℃30分钟使酶失活。4)加入0.5μladapterstock(购自illumina),3.5μlpcr-gradewater,10μlligationbuffer,3μldnaligase,混匀。5)在pcr仪中20℃孵育60分钟。6)加入37μlxpreagent(购自beckman,货号a63881),进行dna纯化。用10μlelutionbuffer洗脱dna。7)向洗脱下的dna中加入12.5μl2xkapahifihotstartreadymix,2.5μl10xkapalibraryamplicationprimermix,混匀,反应体系为25μl。8)将上述样品进行第一轮pcr扩增,设置pcr程序如下表4:表49)向扩增后的样品中按照1:1加入xpreagent,进行dna纯化,用11μlelutionbuffer洗脱dna。10)取出1μl洗脱下来的dna用qubitdsdnahsassaykit试剂盒(购自invitrogen,货号q32851)进行浓度检测。11)向洗脱下的dna中加入12.5μl2xkapahifihotstartreadymix,2.5μl10xkapalibraryamplicationprimermix,混匀,反应体系为25μl。12)将上述样品进行第二轮pcr扩增,设置pcr程序如下表5:表513)第二轮扩增的循环数根据下表6确定。表614)将扩增后的样品加入到2%的琼脂糖凝胶中,恒压120伏特,跑电泳30分钟。15)凝胶电泳的结果如图8所示,将单个核小体的条带切割下来,用qiagen公司的qiaquickgelextractionkit(货号28706)进行dna片段的回收。16)回收的dna片段用qubit检测浓度,用agilent2100生物芯片分析系统进行dna片段的分析,分析结果如图9所示。17)建构建好的文库送到公司进行高通量测序,测序使用的机器为illuminaxten,测序模式为双端150bp。三、实验结果:1)如图10所示,对单个原核的核小体排布情况进行检测,检测数据在基因组上的覆盖度与单细胞以及多个细胞相比都有明显的降低,但是依然能覆盖全基因组50%左右的区域,这对于单个基因组的检测来说也是很高的,而这种覆盖度的降低也是随机的,可以通过几个重复实验来弥补覆盖度低的问题,这说明利用本发明方法进行单倍体基因组的核小体排布检测是十分有效的。2)如图11所示,检测的数据的片段长度大多集中在单个核小体的区域,没有大量过长或者过短的片段。并且用单原核进行检测的数据质量没有明显的下降。3)如图12所示,用单原核进行检测在ctcf的结合位点两侧能够看到有规律的核小体排布,这说明检测数据的质量是很好的。4)如图13所示,用单个原核进行检测,能看到在转录其实位点上游核小体的缺失区域以及下游核小体的有规律的排布。因此,从上述实验结果可以看出,利用本发明的实验方法进行单原核的核小体检测,得到的实验数据质量是很好的,说明本发明方法能够很好的适应单倍体基因组核小体检测的需要,这一结果进一步拓展了本发明方法的应用领域。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种多肽——核转换蛋白i-9.57和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人核转换蛋白41.69和编码这种多肽的多核苷酸的制作方法
- 曲古抑菌素a衍生物及其制备方法和用途的制作方法
- 一种新的多肽-核转换蛋白i10和编码这种多肽的多核苷酸的制作方法
- 一种新的多肽——人含atp/gtp结合结构域的核转换蛋白13和编码这种多肽的多核苷酸的制作方法
- 一种二核簇结构的三维配位聚合物及其制备方法
- 具有脲结构的新型噻吩二胺衍生物的制作方法
- 喹唑啉二酮衍生物及其制备方法、其药物组合物及用途的制作方法
- 一类茶树组蛋白特异表达序列标签及其生物芯片的制作方法
- 一种新的多肽——人含atp/gtp结合结构域的核转换蛋白10和编码这种多肽的多核苷酸的制作方法