一种治疗消化性溃疡的药物化合物及其制备方法与流程
本发明属于医药技术领域,涉及一种治疗消化性溃疡的药物化合物及其制备方法,具体涉及一种富马酸沃诺拉赞一水合物及其制备方法。
背景技术:
富马酸沃诺拉赞,化学名为1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3-基]-n-甲基甲胺富马酸盐,其分子式是:c17h16fn3o2s·c4h4o4,分子量:461.46,化学结构如式i所示。富马酸沃诺拉赞是一种新型的质子泵抑制剂,可用作酸分泌抑制剂、肿瘤病或自身免疫性疾病的治疗药物。奥美拉唑等作为质子泵抑制剂能有效抑制胃酸分泌,但在酸性条件下的不稳定性以及因代谢酶多态性和药物相互作用引起的效果分散,富马酸沃诺拉赞在酸性条件下稳定性优异,以对消化性溃疡、反流性食管炎等显示出更优异的治疗效果。
一个原料药的不同晶型可以有不同的化学和物理特性,包括熔点、化学反应性、表观溶解度、溶解速率、光学和机械性质、蒸气压和密度。这些特性可以直接影响原料药和制剂的处理和/或生产,并且会影响制剂的稳定性、溶解度和生物利用度。当化合物存在多晶型时,由于特定多晶型物具有特异性的热力学性质和稳定性,因此在制备的过程中,了解在各个剂型中应用的化合物的晶型是重要的,以保证生产过程应用相同形态的药物活性化合物。因此,保持药物活性化合物是单一的晶型或是一些晶型的已知混合物是必要的。
专利cn105315258a公开了富马酸沃诺拉赞新的晶型a,b以及适合工业化生产的制备方法,本发明制备晶型制备工艺简单,稳定性好,符合药用要求。晶型a其制备方法为:将富马酸沃诺拉赞游离碱溶解于乙酸乙酯中,加入富马酸甲醇溶液,室温搅拌反应,过滤,滤饼烘干后得到富马酸沃诺拉赞a晶型,晶型a在15.290、20.403、20.704、21.572、25.182、25.559处有特征衍射峰,差热扫描量热图谱在204.8℃有吸收峰。晶型b其制备方法为:将富马酸沃诺拉赞a晶型溶解于c1~c4的烷基醇的水溶液或c4以下的酮类水溶液中,加热搅拌溶解,搅拌放冷;过滤,滤饼烘干后得到富马酸沃诺拉赞b晶型,晶型b在12.253、13.559、15.259、16.889、17.422、20.399、20.764、22.478、25.198、28.077处有特征衍射峰。差热扫描量热图谱在209.0℃有吸收峰。经检测两种晶型均为无水化合物。
cn106478597a公开了一种富马酸沃诺拉赞单晶及其制备方法和用途,本发明富马酸沃诺拉赞易于与其他杂质分离,得到的富马酸沃诺拉赞单晶形态良好,hplc纯度可高达99.5%以上,并且该方法的重现性非常好。此外,利用本发明的方法制备得到的富马酸沃诺拉赞单晶,易于通过单晶的x射线衍射分析确定产品的绝对结构,进而,应用时能够保证含有富马酸沃诺拉赞单晶的药物的准确性。此单晶的制备方法为:向富马酸沃诺拉赞粗品中加入结晶溶剂甲醇和水(0.25-4:1,优选为1:1);以及在预定温度(20-30摄氏度)下,缓慢挥发结晶溶剂,并进行培养5-10天,以便得到结晶产品,所述结晶产品构成所述富马酸沃诺拉赞单晶。本发明单晶在衍射角2θ=11.4,12.3,13.5,15.1,15.3,16.9,18.6,20.4,20.7,22.4和25.1处有特征峰。经检测为无水化合物。
cn106317020a公开了一种富马酸沃诺拉赞晶型α及其制备方法,该晶型纯度高,并具有良好的化学稳定性和晶型稳定性,易于规模化制备,操作简单,成本低,具有广阔的应用前景。晶型α的制备方法为:将富马酸沃诺拉赞粗品加入乙二醇单甲醚中,加热60℃~回流溶解,然后加入纯化水,室温搅拌反应1~2小时,过滤,纯化水洗涤滤饼,晾干,得到富马酸沃诺拉赞晶型α。经差示扫描量热分析(dsc)测定,本发明的富马酸沃诺拉赞晶型α,以2θ角度表示的x-射线粉末衍射(x-rpd)在8.6±0.2°,10.2±0.2°,12.7±0.2°,17.4±0.2°,18.1±0.2°,19.6±0.2°,20.3±0.2°,23.2±0.2°,24.5±0.2°,27.8±0.2°处有衍射峰;其dsc图谱在166~204℃范围内出现吸热峰;优选地,其dsc图谱的吸热峰的峰值出现在194±2℃处。经检测为无水化合物。
cn106478601a公开了一种富马酸沃诺拉赞新晶型,其制备方法以乙酸乙酯、甲醇、纯化水为混合结晶溶剂,获得了富马酸沃诺拉赞新的晶型。其粉末x-射线衍射图谱中,在衍射角2θ为12.18±0.5、13.43±0.5、22.35±0.5、37.06±0.5、38.25±0.5处有特征峰。本发明所述的新晶型稳定性良好,能有效避免药物杂质的增加,降低了储存带来的高成本,为产品的安全性和确保临床疗效带来了有效保障。经检测为无水化合物。
cn104327051a公开了一种吡咯衍生物的富马酸盐的结晶形式,结晶形式a纯度高,稳定性好,适合制剂工艺过程和长期储存,在工业生产上也具有优越性。其制备方法为:1)将1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3基]-n-甲基甲胺单富马酸盐,或者将1-[5-(2-氟苯基)-1-(吡啶-3-基磺酰基)-1h-吡咯-3基]-n-甲基甲胺和富马酸分别加热溶解于适量的有机溶剂中,冷却、析晶,所述有机溶剂选自碳原子数小于等于3的醇类、酮类、酯类的任意一种或几种的混合溶剂;或者它们与水的混合溶剂;所述的有机溶剂可选自甲醇、乙醇、异丙醇、异丁醇、叔戊醇、丙酮、丁酮、乙酸乙酯、乙酸异丙酯、四氢呋喃、2-甲基四氢呋喃、异丙醚、甲基叔丁基醚、甲基环戊基醚、乙腈、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺。2)过滤结晶并洗涤,干燥。其中在约5.1(17.3),10.1(8.7),11.4(7.7),11.5(7.6),12.2(7.3),13.4(6.6),13.9(6.4),15.2(5.8),16.1(5.5),16.6(5.3),16.9(5.3)17.3(5.1),17.8(5.0),18.5(4.8),19.1(4.7),20.0(4.4),20.3(4.4),20.6(4.3),21.0(4.2),21.5(4.1),22.4(4.0),23.0(3.9),23.5(3.8),24.3(3.7),25.1(3.5),25.5(3.5)26.0(3.4),26.7(3.3),27.6(3.2),27.9(3.2),28.8(3.1),29.1(3.1),和29.9(3.0)处有特征峰。其熔点约为203℃。经检测为无水化合物。
富马酸沃诺拉赞在结晶时,如果采用不同的溶剂和工艺条件,则其分子在各晶型晶胞的排列数目和位置及点阵形式不一样,形成不同的晶体结构,富马酸沃诺拉赞多晶型的变化会改变其性质,质量和药效。因此,富马酸沃诺拉赞的稳定结晶,对于进一步研究该化合物的物理化学性质,研究其药物组合及临床应用,具有十分重要的意义。
富马酸沃诺拉赞属于难溶性药物,在制剂中一般以固体形式应用,其制剂的生物利用度好坏直接影响其疗效,且其对光照较敏感,因此对其晶型的研究具有十分重要的意义。另外,药物中存在的杂质对产品的质量影响较大,因而恰当的除杂方法十分必要。由于富马酸沃诺拉赞中难除杂质较多,而现有技术并未提供高效的富马酸沃诺拉赞纯化方法,难以兼顾收率和纯度。富马酸沃诺拉赞特殊的理化性质给水合物的制备带来了极大的困难。本发明对现有文献技术进行了大量的试验发现,现有技术中制得的富马酸沃诺拉赞均为无水化合物,其存在溶解性差、纯度低等问题,严重影响的人民用药安全问题。
本发明经过大量的试验研究,采用新的结晶方法,不仅得到了一种新的富马酸沃诺拉赞晶型而且经测定含有一分子的水,本发明提供的富马酸沃诺拉赞一水合物纯度高、稳定性好,经实验惊喜地发现本发明得到的富马酸沃诺拉赞一水合物溶解性显著提高。本发明还公开了富马酸沃诺拉赞一水合物的制备方法,该制备方法简单易操作,反应条件温和,适合大规模生产。本发明的富马酸沃诺拉赞一水合物制成的组合物片剂溶出度及稳定性显著提高,非常适合临床应用。
技术实现要素:
本发明旨在提供一种富马酸沃诺拉赞一水合物及其制备方法,所要解决的技术问题是提高富马酸沃诺拉赞在水中的溶解性和纯度,从而提高其制剂的溶出度和生物利用度,以克服现有技术缺陷。
为了实现本发明的目的,采用的技术方案为:
一种治疗消化性溃疡的药物化合物,该化合物为富马酸沃诺拉赞一水合物,其分子式为:c17h16fn3o2s·c4h4o4·h2o,结构式如下(式ii),其以2θ±0.2°衍射角表示的x-射线粉末衍射图谱在3.21°、5.62°、6.53°、9.12°、14.43°、15.53°、18.61°、21.72°、24.61°和27.71°处显示有特征衍射峰。
本发明提供的富马酸沃诺拉赞一水合物使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示。
本发明还提供了一种富马酸沃诺拉赞一水合物的制备方法,具体步骤为:
(1)取富马酸沃诺拉赞粗品,加入四甲基乙二胺/水的混合溶液,搅拌溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加预冷的四甲基乙二胺,继续降温析晶;
(3)保温搅拌至析晶完全,养晶,抽滤,水洗,干燥,得白色结晶性粉末。
优选地,步骤(1)所述的四甲基乙二胺/水混合溶液中,四甲基乙二胺与水的体积比为25:1.0~1.2;步骤(1)所述富马酸沃诺拉赞粗品与混合溶液的质量体积比为1g:5ml~7ml;所述的搅拌速度为30-50转/分钟。
进一步优选地,步骤(1)所述的四甲基乙二胺/水混合溶液中,四甲基乙二胺与水的体积比为25:1.1;步骤(1)所述富马酸沃诺拉赞粗品与混合溶液的质量体积比为1g:6ml;所述的搅拌速度为40转/分钟。
优选地,步骤(2)所述四甲基乙二胺与步骤(1)混合溶液的体积比0.5~1:1;步骤(2)所述滴加速度为1.0~2.0ml/min,降温幅度为每10分钟1℃~3℃;步骤(2)所述降温析晶为降温至-10℃~-8℃析晶。
进一步优选地,步骤(2)所述四甲基乙二胺与步骤(1)混合溶液的体积比0.8:1;步骤(2)所述滴加速度为1.5ml/min,降温幅度为每10分钟2℃;步骤(2)所述降温析晶为降温至-9℃析晶。
优选地,步骤(3)所述养晶时间为1h~3h;步骤(3)所述干燥温度为40℃~50℃。
进一步优选地,步骤(3)所述养晶时间为2h;步骤(3)所述干燥温度为45℃。
本发明中,所述的富马酸沃诺拉赞粗品可以为待进一步纯化的富马酸沃诺拉赞固体混合物富马酸沃诺拉赞粗品也可以为市售原料或通过现有技术方法制备得到,所得的晶型结果均在误差范围内,均为本发明新晶型。
本发明还提供了一种含有本发明富马酸沃诺拉赞化合物的药物组合物,该药物组合物为含有富马酸沃诺拉赞一水合物的片剂。
晶体的形成机理很复杂,一个新晶体的获得也具有很大的偶然性,有时不同的溶剂、在不同的结晶条件下会产生相同的晶体结构。某些特定晶型也并非一定会获得更加有利的理化性质。药物的稳定性、吸湿性、溶解性、生物活性、毒性等性质会因晶型的不同而产生巨大的差异。
本发明经过大量的试验筛选,通过选择不同的溶剂溶解及不同的溶剂析晶,得到了本发明的制备方法,通过对搅拌速度、溶剂用量、温度及养晶时间的控制,意外的得到了一种富马酸沃诺拉赞新晶型,经检测为一水合物。本发明提供的富马酸沃诺拉赞纯度高、稳定性好,经实验惊喜地发现本发明得到的富马酸沃诺拉赞一水合物溶解性显著提高。本发明还公开了富马酸沃诺拉赞一水合物的制备方法,该制备方法简单易操作,反应条件温和,适合大规模生产。本发明的富马酸沃诺拉赞一水合物制成的组合物片剂溶出度及稳定性显著提高,非常适合临床应用。
研究表明,在x-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。本发明所提供的富马酸沃诺拉赞结晶其x-射线粉末衍射图谱与现有技术具有明显不同的峰的相对位置,可见其是一种与现有技术不同的新晶型,经检测为一水合物。
本发明所提供的富马酸沃诺拉赞一水合物结晶证实含有1个结晶水,其性状为白色结晶性粉末,在常温干燥条件下不会发生结晶水的流失。且其粉末x-射线衍射图谱与现有技术具有明显不同的峰的相对位置,可见其是一种与现有技术不同的新晶型。
下面通过对本发明提供的富马酸沃诺拉赞一水合物晶型进行研究来解释和说明本发明技术方案:
1、元素分析c17h16fn3o2s·c4h4o4·h2o
仪器:varioelcube元素分析仪;测量元素为c、h、o、n、s;
dioenxdx-500型离子色谱仪;测量元素为f。
元素分析(%)理论值为:h(4.62),c(52.60),n(8.76),o(23.36),s(6.69)、f(3.96)。
元素分析(%)测定值为:h(4.63),c(52.58),n(8.78),o(23.35),s(6.70)、f(3.96)。
与元素分析的理论值基本相符。
2、晶型检测
取本发明制备得到的富马酸沃诺拉赞结晶,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的x-射线粉末衍射图在3.21°、5.62°、6.53°、9.12°、14.43°、15.53°、18.61°、21.72°、24.61°和27.71°处显示有特征峰。
3、差热分析及热重分析
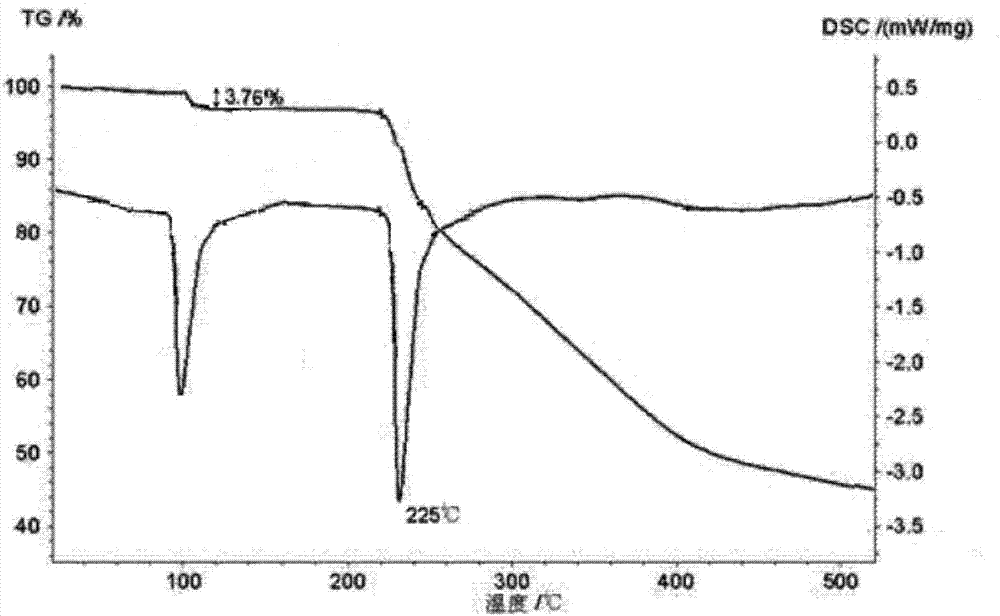
对本发明制备的富马酸沃诺拉赞晶体进行差热和热重分析,结果如附图2所示;结果表明,本品在100℃左右快速失去约1个水分子的重量,并在95℃之前无明显重量变化,证实其失去的水分子为结晶水分子,而非游离水分子;本品在约225℃处有吸热峰,从侧面证明了其为一种不同的晶型。
4、水分分析
采用卡式水分测定仪测定,本发明的富马酸沃诺拉赞的含水量为3.76%,与理论值3.75%相符。
5、纯度检测
经hplc纯度检测,本发明制备得到的富马酸沃诺拉赞结晶的纯度可达到99.98~99.99%。
与现有技术相比,本发明具有如下优点:
(1)本发明所提供的富马酸沃诺拉赞是一种不同于现有技术的新晶型,经检测为一水合物;本发明所提供的富马酸沃诺拉赞一水合物的制备方法简单易操作,反应条件温和,适合大规模生产。
(2)本发明提供的富马酸沃诺拉赞一水合物纯度高、稳定性好,经实验惊喜地发现本发明得到的富马酸沃诺拉赞一水合物溶解性显著提高。本发明的富马酸沃诺拉赞一水合物制成的组合物片剂溶出度及稳定性显著提高,非常适合临床应用。
附图说明
下面结合附图对本发明作进一步的说明:
图1为本发明实施例1制备的富马酸沃诺拉赞一水合物的x-射线粉末衍射图谱。
图2为本发明实施例1制备的富马酸沃诺拉赞一水合物的tg-dsc图谱。
具体实施例
以下用实施例对本发明的技术方案进行详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。
实施例1:富马酸沃诺拉一水合物的制备
(1)取富马酸沃诺拉赞粗品100g,加入四甲基乙二胺/水(体积比为25:1.0)的混合溶液(700ml),搅拌(40转/分钟)溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加(1.0ml/min)预冷的四甲基乙二胺700ml,继续降温(降温幅度为每10分钟3℃)至-10℃析晶;
(3)保温搅拌至析晶完全,养晶2h,抽滤,水洗,40℃干燥,得白色结晶性粉末。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图见图1。
实施例2:富马酸沃诺拉一水合物的制备
(1)取富马酸沃诺拉赞粗品100g,加入四甲基乙二胺/水(体积比为25:1.2)的混合溶液(500ml),搅拌(50转/分钟)溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加(1.5ml/min)预冷的四甲基乙二胺250ml,继续降温(降温幅度为每10分钟2℃)至-9℃析晶;
(3)保温搅拌至析晶完全,养晶3h,抽滤,水洗,50℃干燥,得白色结晶性粉末。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。
实施例3:富马酸沃诺拉一水合物的制备
(1)取富马酸沃诺拉赞粗品100g,加入四甲基乙二胺/水(体积比为25:1.1)的混合溶液(600ml),搅拌(30转/分钟)溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加(2.0ml/min)预冷的四甲基乙二胺480ml,继续降温(降温幅度为每10分钟3℃)至-8℃析晶;
(3)保温搅拌至析晶完全,养晶1h,抽滤,水洗,45℃干燥,得白色结晶性粉末。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。
实施例4:富马酸沃诺拉一水合物的制备
(1)取富马酸沃诺拉赞粗品100g,加入四甲基乙二胺/水(体积比为25:1.0)的混合溶液(500ml),搅拌(40转/分钟)溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加(2.0ml/min)预冷的四甲基乙二胺500ml,继续降温(降温幅度为每10分钟1℃)至-10℃析晶;
(3)保温搅拌至析晶完全,养晶2h,抽滤,水洗,50℃干燥,得白色结晶性粉末。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。
实施例5:富马酸沃诺拉一水合物的制备
(1)取富马酸沃诺拉赞粗品100g,加入四甲基乙二胺/水(体积比为25:1.2)的混合溶液(600ml),搅拌(40转/分钟)溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加(1.0ml/min)预冷的四甲基乙二胺300ml,继续降温(降温幅度为每10分钟2℃)至-8℃析晶;
(3)保温搅拌至析晶完全,养晶3h,抽滤,水洗,40℃干燥,得白色结晶性粉末。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。
实施例6:富马酸沃诺拉一水合物的制备
(1)取富马酸沃诺拉赞粗品100g,加入四甲基乙二胺/水(体积比为25:1.1)的混合溶液(700ml),搅拌(50转/分钟)溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加(1.5ml/min)预冷的四甲基乙二胺560ml,继续降温(降温幅度为每10分钟3℃)至-9℃析晶;
(3)保温搅拌至析晶完全,养晶2h,抽滤,水洗,45℃干燥,得白色结晶性粉末。
所制得的白色结晶性粉末使用cu-kα射线测量得到的x-射线粉末衍射谱图与实施例1相似。
下面通过实验例进一步说明本发明:
实验例1:溶解度测定
试验品:本发明实施例1-6所制备的样品;
对照品1:参照专利cn104327051a实施例1制备的富马酸沃诺拉赞结晶形式a。
对照品2:参照专利cn105315258a实施例1制备的富马酸沃诺拉赞晶型a。
对照品3:参照专利cn105315258a实施例2制备的富马酸沃诺拉赞晶型b。
对照品4:参照专利cn106317020a实施例1制备的富马酸沃诺拉赞晶型α。
对照品5:参照专利cn106478597a实施例2制备的富马酸沃诺拉赞单晶。
对照品6:参照专利cn106478601a实施例制备的富马酸沃诺拉赞晶型。
对照品7:参照专利cn105085484a实施例1制备的富马酸沃诺拉赞。
对照品8:参照专利cn105130955a实施例一制备的富马酸沃诺拉赞晶体。
对照品9:参照专利cn105294653a实施例13制备的富马酸沃诺拉赞晶体。
对照品10:参照专利cn106366071a实施例10制备的富马酸沃诺拉赞晶体。
对照品11:参照专利cn104860926a实施例1制备的富马酸沃诺拉赞晶体。
对照品12:参照专利101300229a实施例8制备的富马酸沃诺拉赞晶体。
参照中国药典2015年版二部凡例测定其溶解性,方法:取本品适量,分别加入水,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,即得,结果见表1。
表1本发明的晶型和对照品在水中溶解性试验结果
将上述实施例1-6溶解的水溶液样品在25℃恒温搅拌72小时,取样5ml。样品经0.45μm微孔滤膜过滤,弃去初滤液,取续滤液20μl测定药物含量即为水中溶解度(mg/ml)。结果见表2:
表2本发明晶型与现有技术晶型在水中的溶解度对比
从上表可以看出,25℃下,本发明富马酸沃诺拉赞新晶型的在水中的溶解度与现有技术相比,有显著提高,取得了意料不到的技术效果。
实验例2:溶剂筛选试验
采用本发明的制备方法操作,具体如下:
(1)取富马酸沃诺拉赞粗品100g,加入溶剂a/水(体积比为25:1.0~1.2)的混合溶液500ml-700ml,搅拌(30-50转/分钟)溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加(1.0~2.0ml/min)预冷的溶剂b(溶剂b与步骤(1)混合溶液的体积比0.5~1:1),继续降温(降温幅度为每10分钟1℃~3℃)至-10℃~-8℃析晶;
(3)保温搅拌至析晶完全,养晶1h~3h,抽滤,水洗,40℃~50℃干燥,得白色结晶性粉末。
表3溶剂筛选实验结果
实验例3:结晶试验条件筛选
(1)取富马酸沃诺拉赞粗品100g,加入四甲基乙二胺/水的混合溶液,搅拌溶解1-2min,活性炭脱色,抽滤;
(2)将步骤(1)滤液降至-2℃,滴加预冷的四甲基乙二,继续降温析晶;
(3)保温搅拌至析晶完全,养晶,抽滤,水洗,干燥,得白色结晶性粉末。
表4-1结晶试验条件筛选结果
表4-2结晶试验条件筛选结果
实验例4:稳定性试验
实验例通过加速试验,考察本发明提供的富马酸沃诺拉赞一水合物结晶的稳定性。
取实施例1-3制备的样品,于温度40±2℃、相对湿度75±5%的条件下放置6个月,分别于0、1、2、3、6个月末取样测定性状、有关物质、含量,结果见表5。
表5:加速试验结果(温度40±2℃,相对湿度75±5%)
从表5看出,本发明富马酸沃诺拉赞结晶在温度40±2℃、相对湿度75±5%的条件下放置6个月,有关物质含量没有明显升高,各指标均无明显变化,说明本品稳定性好,同时本品在加速实验中,含水量稳定(一分子水),从另一方面证明了该化合物中的水是结晶水不是吸附水。
- 还没有人留言评论。精彩留言会获得点赞!