检测异源cfDNA的SNP分子标记及检测方法、用途与流程
本发明属于基因检测
技术领域:
,具体涉及一种检测异源cfdna的snp分子标记及其探针、芯片、试剂盒、方法、检测装置和用途。
背景技术:
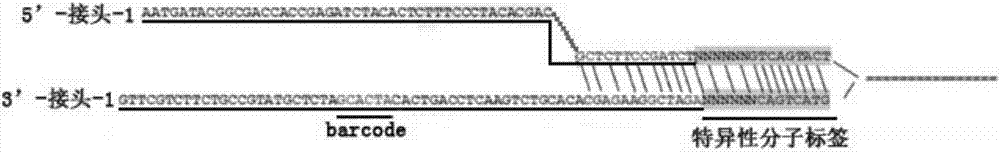
:肾移植是临床治疗终末期肾病的优选手段,而影响肾移植术后患者生存率的最重要原因为排斥反应的发生。移植排斥分为超急性、急性、边缘型急性、亚临床急性或慢性,急性排斥反应(acuterejection,ar)是目前肾移植术后的主要危险并发症,也是导致慢性排斥反应和移植肾失功的最重要的危险因素。早期诊断及时治疗ar可以有效减少移植器官的病理损害程度,延长移植器官的存活时间。通常诊断发生急性排斥反应的指标有:患者临床表现、各项生化指标(丙氨酸氨基转移酶、总胆红素、碱性磷酸酶和γ-谷氨酰转移酶、肌酐或心肌酶谱)和组织活检。前两项检测的灵敏度和特异性低,且检测结果依赖于临床医生的临床经验易产生偏差。穿刺活检一直是肾移植排异反应检测的金标准,但组织活检花费大,属创伤性检查常伴随并发症,包括:疼痛、血尿、肾血肿、移植物血栓症、败血症、休克和移植物衰竭等。近年来诊断肾移植急性排斥反应的发生,一直朝向寻找非侵入性无创检测的方向努力。血清中肌酐水平的增高通常是诊断肾移植后排斥反应的一个有效指标,但肌酐检测具有非特异性,除了肾排斥反应,肾移植患者排异药物中毒引起的急性肾衰竭也会导致肌酐升高,因此,肌酐水平的监测不宜单独作为判断肾移植排斥反应的依据。目前,肾移植患者面临的主要临床问题之一就是缺少用于早期诊断和连续监控移植物排斥风险的高灵敏度、高特异性且非侵入性的检测。游离dna(cellfreedna,cfdna),是指游离于细胞外的部分降解了的机体内源dna,主要来自于细胞的凋亡或坏死。异源或自体的体细胞突变dna都可以在本体的血液中检测到,例如孕妇、肿瘤患者和器官移植患者。由于cfdna的半衰期很短(16min),cfdna作为生物标志物的潜力巨大。dennislo最早在女性肝、肾移植患者的血浆中检测到来自男性供体组织的游离dna,被认为是供体器官在患者体内凋亡或坏死,细胞裂解释放出的cfdna,游离dna可以作为器官移植排异的标志物(参见presenceofdonor-specificdnainplasmaofkidneyandliver-transplantrecipients.lancet.1998may2;351(9112):1329-30.)。正常状态下受体血液中供体来源的cfdna几乎没有或者含量是极低的,通过检测器官移植受体中供体来源的cfdna的比例,可用于判断器官移植后是否发生排斥反应。在中国专利文献cn106544407a中公开了一种确定受体cfdna样本中供体来源cfdna的比例的方法,通过获取器官移植的受体样本的基因组dna,对基因组dna目标区域捕获测序,用于基因分型;同时对移植后受体的血浆cfdna样本进行目标区域的捕获和测序,用以分析供体cfdna占总cfdna的比例。然而上述文献中的技术方案在应用于检测肾移植受体中的供体cfdna比例时还存在如下缺陷:一、肾移植急性排斥反应的早期诊断以及免疫抑制剂使用的剂量,直接关系到肾移植患者的存活率和肾功能的恢复,文献中的snp位点的选择仅以次等位基因频率(maf)为筛选条件,缺少对临床肾移植患者易突变基因,特别是相关药用基因组学检测,因此缺少对肾移植排斥反应的针对性检测,无法为免疫排斥反应的个体提供临床个体化用药依据;二、肾移植排斥反应的病人中,供体来源的cfdna含量低于肺移植和肝移植等器官移植排斥反应病人的外周血中cfdna,使得将异源cfdna信号从测序错误和pcr扩增错误信号中区分出来的难度加大,文献中提供的cfdna建库方法无法实现对cfdna的单分子特异性标记,在对测序数据分析时会引入偏差和错误。技术实现要素:因此,本发明要解决的第一个技术问题在于克服现有技术中缺少对肾移植排斥反应相关基因检测,无法为器官移植患者提供个体化用药指导的缺陷,从而提供一种用于针对肾移植急性排斥反应和药物代谢基因进行高灵敏度和高特异性的早期检测的snp分子标记以及其探针、芯片、试剂盒。本发明要解决的第二个技术问题在于克服现有技术中无法特异性提取cfdna信号以区分偏差和测序错误,从而提供一种单分子标记接头实现对cfdna的单分子标记。本发明要解决的第三个技术问题在于克服现有技术中缺少对肾移植排斥反应相关基因检测的方法和装置,从而提供一种检测异源cfdna的方法和装置。本发明要解决的第四个技术问题在于提供所述的检测异源cfdna的snp分子标记、所述的snp分子标记的探针、所述的异源cfdna的检测芯片或所述的肾移植排斥反应的检测试剂盒在制备用于检测肾移植排斥反应的制剂中的用途。本发明提供了一种检测异源cfdna的snp分子标记,所述snp分子标记选自如表1中所示的snp位点。所述的snp分子标记,所述snp位点包括药物代谢基因的snp位点。所述的snp分子标记,所述药物代谢基因为cyp3a4、cyp3a5和cyp2d6。优选的,所述药物代谢基因的snp位点如表2中所示的snp位点。本发明提供了一种检测所述snp分子标记的探针,所述探针是基于所述snp位点设计而成的。所述的探针,所述探针包括上游探针和下游探针,所述上游探针的序列是以所述snp位点为坐标起点,向5’方向延伸80bp的碱基序列为序列信息设计的;所述下游探针的序列是以所述snp位点为坐标起点,向3’方向延伸80bp碱基序列为序列信息设计的。本发明提供了一种异源cfdna的检测芯片,所述检测芯片上述的探针。本发明提供了一种单分子标记接头,所述单分子标记接头是由正向序列和反向序列部分配对后的y型接头;所述正向序列由5’端至3’端依次包括5’-接头和特异性分子标签a;所述反向序列由3’端至5’端依次包括3’-接头和特异性分子标签b。所述的单分子标记接头,所述5’-接头序列的碱基序列如seqidno.1或seqidno.2所示,所述3’-接头的碱基序列如seqidno.3或seqidno.4所示,所述特异性分子标签a的碱基序列如seqidno.5所示,所述特异性分子标签b的碱基序列如seqidno.10所示,其中n代表随机碱基。所述的单分子标记接头,所述正向序列的3’末端连接有碱基t,所述反向序列的5’末端连接有磷酸基团。所述的单分子标记接头,所述正向序列如seqidno.6或seqidno.7所示,所述反向序列如seqidno.8或seqidno.9所示。本发明提供了一种异源cfdna的检测方法,包括如下步骤:(1)捕获基因组dna的目标区域,所述基因组dna的目标区域包括所述的snp分子标记;(2)将步骤(1)所得的目标区域进行高通量测序,所得测序结果用于基因组dna的snp分型;(3)筛选步骤(2)中snp分型后基因型为纯合子的snp位点,作为有效snp位点;(4)捕获cfdna的目标区域,所述cfdna的目标区域包括所述的snp分子标记;(5)将步骤(4)所得的目标区域进行高通量测序,保留与步骤(3)中所述的有效snp位点相同坐标位置的snp位点;(6)检测cfdna在有效snp位点处区别于步骤(3)中基因型为纯合子snp信号,即异源snp信号;(7)计算步骤(6)中异源snp信号占cfdna在有效snp位点处总测序信号的比例,即得到异源cfdna的定量结果。所述的异源cfdna的检测方法,所述捕获基因组dna的目标区域包括如下步骤:(1)提取血细胞中的基因组dna;(2)将步骤(1)中基因组dna打断为dna片段,所述dna片段末端修复后连接所述的单分子标记接头,得到基因组dna连接产物;(3)pcr扩增步骤(2)中基因组dna连接产物,得到基因组dna文库;(4)利用权利要求6所述的检测芯片捕获得到步骤(3)中所述基因组dna文库的目标区域。所述的异源cfdna的检测方法,其特征在于,所述捕获cfdna的目标区域包括如下步骤:(1)提取血浆中的cfdna;(2)将步骤(1)中cfdna末端修复后连接所述的单分子标记接头,得到cfdna连接产物;(3)pcr扩增步骤(2)中cfdna连接产物,得到cfdna文库;(4)利用所述的检测芯片捕获得到步骤(3)中cfdna文库的目标区域。本发明提供了一种异源cfdna的检测装置,其特征在于,所述装置包括:(1)样本获取单元,用以获取基因组dna和cfdna;(2)异源cfdna定量单元,利用所述的异源cfdna的方法以获取异源cfdna定量结果;(3)质控单元,对上述样本获取单元和异源cfdna定量单元进行监测调控。所述的异源cfdna的检测装置,其特征在于,所述装置为肾移植排斥反应的检测装置。本发明提供了一种肾移植排斥反应的检测试剂盒,所述试剂盒包括探针、检测芯片和单分子标记接头。本发明提供了一种检测异源cfdna的snp分子标记、检测snp分子标记的探针、异源cfdna的检测芯片或肾移植排斥反应的检测试剂盒在制备用于检测肾移植排斥反应的制剂中的用途。本发明技术方案,具有如下优点:1.本发明所述的检测异源cfdna的snp分子标记,包括表1所示的5764个snp位点,覆盖范围广,snp位点的次等位基因频率(maf)接近0.5,基因多态性高,通过snp分型可以准确区分人群中不同个体,通过上述的检测异源cfdna的snp分子标记可以用于检测进行肾脏移植后的患者中异源cfdna的水平,能够实现针对肾移植急性排斥反应和药物代谢基因进行高灵敏度和高特异性的早期诊断,进而可以为器官移植患者提供个体化用药指导。2.本发明所述的检测异源cfdna的snp分子标记,snp位点包括药物代谢基因的snp位点,如cyp3a4、cyp3a5和cyp2d6;cyp3a4、cyp3a5和cyp2d6的基因多态性与肾移植患者的急性排斥反应密切相关,同时是造成临床个体用药差异的重要因素,能够为患者免疫抑制剂的选择和使用剂量提供参考信息。3.本发明所述的snp分子标记的探针,可以对每个snp位点双向覆盖检测,检测准确度高。4.本发明所述的异源cfdna的检测芯片,包括上述检测探针,检测范围广,芯片应用于高通量测序能够实现对肾移植排斥反应的早期检测,以及个体药物代谢差异检测。5.本发明所述的单分子标记接头,通过接头中的特异性分子标签对dna分子进行特异性标记,在测序分析时通过提取特异性分子标签,可以用来识别pcr产物和原始模板,避免pcr产物重复计数,有效去除重复数据,以及测序过程和pcr过程中随机引入的错误,降低测序背景噪音,提高了检测的准确性;特异性分子标签添加在单分子标记接头的末端(y型接头),连接y型接头和dna片段;特异性分子标签是包含6个随机碱基的互补序列,可以提高y型接头的解链温度,进而提高了y型接头的稳定性。6.本发明所述的异源cfdna的检测方法,包括cfdna文库的构建方法,在cfdna的两端添加单分子标记接头对cfdna进行标记,测序分析时能够对样本cfdna准确筛选,相对增加了有效数据量,实现对起始量低的cfdna的检测。7本发明所述的异源cfdna的检测装置和/或检测试剂盒通过对样本基因组dna的捕获测序,得到样本基因组的snp分型信息;对cfdna的捕获测序,鉴定出样本中不同基因型的异源cfdna,以及异源cfdna的定量结果;将其应用于肾移植排斥反应诊断,可实现针对肾移植的高灵敏度和高特异性的无创早期检测,并可对肾移植患者进行持续性的监测,为分析和判断肾移植排斥反应提供了可靠信息;由于检测基因涉及药物代谢基因,可为患者提供胚系突变检测信息,从而为肾移植排斥患者的早期治疗、个体精准化用药指导以及预后动态监测等提供了可靠的分析依据。附图说明为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。图1为本发明实施例1所示的中国人群数据库与hapmap数据库频率信息的比对结果;图2为本发明实施例3所示的单分子标记接头-1;图3为本发明实施例3所示的单分子标记接头-2;图4为本发明实施例3所示的特异性分子标签矫正测序数据错误率的对比效果,其中横坐标代表定位于染色体上不同位置的序列,纵坐标代表每一序列测序数据的错误率;每一位置处的序列从左向右依次显示序列原始测序数据的错误率、比对序列位置去重、矫正后的错误率,以及使用特异性分子标签矫正后的错误率;图5为本发明实施例7中基因分型结果随测序深度的变化图;图6为本发明实施例9中绘制的标准样本回归曲线;图7为本发明实施例10中检测健康人和两例肾移植排斥患者的结果图。具体实施方式以下通过具体实施例来说明本发明的实施方式,除非另外说明,本发明中所公开的实验方法均采用本
技术领域:
常规技术,下文中所称的供体和受体,是相对的两个个体,是基于移植,例如基于器官或组织移植时的供给一方和接受一方来说的。实施例1snp位点筛选选择dbsnp(databasesnp,https://www.ncbi.nlm.nih.gov/snp/)数据库和中国人群数据库(保存于苏州人人基因科技有限公司),中国人群数据库是基于3000例中国人的全基因组样本信息构建的遗传多态性位点目录,如图1所示,中国人群数据库与hapmap数据库(http://www.hapmap.org)存在部分差异性频率信息,代表了中国人群中的差异性遗传位点,使中国人群数据库能够更精准的呈现中国人群的遗传多态性位点信息。从上述两种数据库中筛选次等位基因频率(maf)接近于0.5的位点,得到表1所示的5754个目标snp位点。所述药物代谢基因的snp位点,如cyp3a4、cyp3a5和cyp2d6基因,cyp3a4、cyp3a5和cyp2d6基因的snp位点如表2所示,上述基因单核苷酸多态性影响肾移植急性排斥反应和多种免疫抑制剂(如他克莫司或环孢素)血药浓度/剂量比(c/d);通过检测表2中所示的位点,可以针对性检测肾移植后的急性排斥反应,为肾移植的早期治疗、个体化精准用药指导以及预后动态监测等提供了关键的突变位点信息,辅助医生选择合适的药物和药物剂量,提高肾移植后的存活率和肾功能的恢复程度。表1目标snp位点表2药物代谢基因snp位点rs55951658rs55901263rs4646438rs28371759rs4986913rs776746rs56411402rs3892097rs5030655rs1065852实施例2探针设计和芯片合成1.探针设计根据表1以及表2所示的snp位点在基因组上的坐标位置,以snp位点为坐标起点,向5’端延伸80bp碱基的碱基序列为序列信息设计得到上游探针,上游探针不包括snp位点序列;以snp位点为坐标起点,向3’端延伸80bp碱基的碱基序列为序列信息设计得到下游探针,下游探针不包括snp位点序列;以rs1036431位点示例,截取snp上下游各80bp序列分别得到上游探针(ggcttttgagcgcagttaacttttgggctgaaggaaaatggacatgcaagcaggagggagaatcctagttagactcctcg)和下游探针(ttttcacaagtcattgtcgatgtcactgtcagtgtagtgggccttcaagctatagggagccactcctaatgagcaacatg)。通过所述上游探针和下游探针实现对每个snp位点的双向覆盖检测,探针检测准确度高。2.芯片合成步骤1中针对表1以及表2所示的共5764个snp位点共设计合成11528条探针,探针合成后(由生工生物工程(上海)股份有限公司合成)等摩尔体积混合,得到用于检测表1以及表2所示snp位点的检测芯片。实施例3单分子标记接头的制备单分子标记接头(由生工生物工程(上海)股份有限公司合成)包括接头和特异性分子标签,其中接头包括正向序列中的5’-接头和反向序列中的3’-接头;如图2和图3所示,5’-接头选自选自seqidno.1所示的5’-接头-1或seqidno.2所示的5’-接头-2,3’-接头序列选自seqidno.3所示的3’-接头-1或seqidno.4所示的3’-接头-2;5’-接头-2是5’-接头-1的3’末端的部分序列,3’-接头-2是3’-接头-1的5’末端的部分序列;如图2所示,由5’-接头-1、3’-接头-1和特异性分子标签组成y型的单分子标记接头-1,如图3所示,5’-接头-2、3’-接头-2和特异性分子标签组成y型的单分子标记接头-2;特异性分子标签是由正向序列中的特异性分子标签a和反向序列中的特异性分子标签b互补配对形成的双链dna分子,其中特异性分子标签a的碱基序列如seqidno.5所示,特异性分子标签b的碱基序列如seqidno.10所示;单分子标记接头的正向序列3’末端连接有碱基t,反向序列的5’末端有磷酸基团,在文库构建过程中,单分子标记接头通过3’末端的碱基t与游离dna片段相连,单分子标记接头5’末端的磷酸基团保护碱基t不被核酸外切酶剪切,保障单分子标记接头与dna片段的连接效率。所述正向序列如seqidno.6或seqidno.7所示,所述反向序列如seqidno.8或seqidno.9所示。3’-接头-1序列中具有barcord序列为atcacg,barcord在高通量测序过程用于区分来自不同样本的序列;特异性分子标签中包括polyn序列,其中n代表随机碱基,特异性分子标签中包含6个随机碱基,形成46种唯一标签;每个dna端的3’端和5’端分别连接一个单分子标记接头-1,通过前后12bp的随机碱基进一步对同一样本中的不同dna片段进行特异性标记,获得测序数据后,通过读取特异性标签序列,有效去除了在测序过程和pcr过程引入的错误,使测序背景噪音由1%下降到1‰;另一方面,特异性分子标签可以用于识别原始dna片段和pcr后的扩增产物,有效区分pcr重复片段,提高了测序的精确度。如图4所示,选择定位于染色体不同位置的共21个dna分子,对每个dna分子的测序数据分别使用比对序列位置去重和矫正错误率以及特异性分子标签矫正错误率,比较原始测序数据错误率、比对序列位置去重和矫正后的错误率和使用特异性分子标签矫正后的错误率,结果发现特异性分子标签矫正可以显著降低原始数据中错误率,且效果优于传统比对比对序列位置去重和矫正错误率的方法。如图3所示的单分子标记接头-2是截断后的单分子标记接头-1,在单分子标记接头-2连接待测dna分子后,利用引物1(5’-aatgatacggcgaccaccga-3’)和引物2(5’-caagcagaagacggcatacga-3’)在对文库进行富集扩增时将在单分子标记接头-2的序列延伸为单分子标记接头-1的序列;单分子标记接头-2的序列相对单分子标记接头-1的序列比较短,有效降低了单分子标记接头的合成成本。实施例4提取基因组dna和cfdna1、样本采集和血浆分离(1)样本采集:抽取供体血液8ml左右,用抗凝血管保存运输;(2)血浆分离:获得新鲜全血样本,1600g,4℃离心10min,分离上清液至新的离心管,将上清液继续16000g,4℃离心10min,离心后的上清液转移至新的离心管,得到分离后的血浆,血浆可在-20℃下保存;(3)血细胞分离:全血样本离心后的下层沉淀即为血细胞。2、使用莱枫基因组dna提取试剂盒(dk603)提取分离后的血细胞,得到基因组dna;用莱枫游离dna提取试剂盒(dk607)抽提分离后血浆,得到cfdna。实施例5文库构建1、分别取1μlcfdna进行quantifluortm-st(promega)定量,另取1μl使用agilent2100检测质量。2、以基因组dna和cfdna作为样本,使用kapaltplibrarypreparationkit分别制备基因组dna文库和cfdna文库,具体步骤如下所示:(1)末端补平a.向标记好的离心管中加入表3所示的末端补平混合液,用移液器吹打混匀;表3末端补平混合液试剂名称体积水8μlkapaendrepairbuffer(10x)7μlkapaendrepairenzymemix5μl总体积20μlb.取50μl步骤2中定量后的样本,加入20μl表3所示的末端补平混合液,得到总体积为70μl的样本反应液,用移液器吹打混匀;c.在pcr仪上运行如下程序:20℃保持30min,10℃静置。(2)补平后纯化重悬浮agencourtampurexpreagent,磁珠室温放置30min。a.样品管中加入120ul磁珠和步骤(1)中末端补平后的样本反应液,充分吹打混匀,室温静置5min;b.将样品管置于磁力架上,待上清澄清后,吸弃上清;c.保持样品管在磁力架上,加入200μl新鲜配制的80%乙醇,室温孵育至少30sec,转动离心管以清洗磁珠,吸弃上清;d.保持样品管在磁力架上,加入200μl新鲜配制的80%乙醇,室温孵育至少30秒,转动离心管以清洗磁珠,吸弃上清;e.将样品管瞬时离心,放置于磁力架上去掉残存的乙醇,室温晾干至磁珠表面无明亮反光后,从磁力架上取下样品管,加入42ulnuclease-freewater重悬磁珠。(3)加a尾a.根据样本的数量,计算所需试剂用量,于标记好的离心管中加入表4所示的混合液,用移液器吹打混匀;表4加a尾混合液试剂名称1个样本8个样本48个样本kapaa-tailingbuffer(10x)5μl40μl240μlkapaa-tailingenzyme3μl24μl144μl总体积8μl64μl384μlb.取42μl步骤(2)中纯化后的样本,加入8μl表4所示的混合液,总体积为70μl,用移液器吹打混匀;c.在pcr仪上运行如下程序:30℃保持30min,10℃静置。(4)加a尾后纯化a.样品管中加入kapapeg/naclsprisolution90μl和步骤(3)中加a尾后的样本反应液,充分吹打混匀,室温静置5min;b.将样品管置于磁力架上,待上清澄清后,吸弃上清;c.保持样品管在磁力架上,加入200μl新鲜配制的80%乙醇,室温孵育至少30sec,转动离心管以清洗磁珠,吸弃上清;d.保持样品管在磁力架上,加入200μl新鲜配制的80%乙醇,室温孵育至少30sec,转动离心管以清洗磁珠,吸弃上清;e.将样品管瞬时离心,放置于磁力架上去掉残存的乙醇,室温晾干至磁珠表面无明亮反光后,从磁力架上取下样品管,加入32ulnuclease-freewater重悬磁珠。(5)加单分子标记接头a.根据样本的数量,计算所需试剂用量,于标记好的离心管中加入如下表5所示的接头连接混合液和步骤(4)所得纯化后的样本(beadswithdna),用移液器吹打混匀:表5接头连接混合液试剂名称1个样本单分子标记接头5μl5xkapaligationbuffer10μlkapat4dnaligase5μlbeadswithdna30μl总体积18μlc.连接:20℃保持15min。(6)加接头后磁珠纯化(7)文库扩增a.室温解冻2xkapahifihotstartreadymix,于标记好的离心管中加入表6所示的文库扩增体系,用移液器吹打混匀;表6文库扩增体系b.取23μl步骤(6)中的纯化样本加入27μl表6所示的pcr扩增体系,总体积50μl,用移液器轻轻混匀,瞬时离心2s,在pcr仪上运行如下程序:98℃变性45s,8个循环(98℃变性45s,65℃退火30s,72℃延伸30s),72℃延伸1min,4℃冷却静置。(8)文库鉴定取2μl步骤(7)所得pcr产物于2%琼脂糖凝胶电泳,确定片段分布于250-500bp之间。(9)文库纯化a.加入agencourtampurexpreagent50μl于样品管中,样本管中盛装有步骤(7)中扩增后的文库样本,充分吹打混匀,室温静置5min。b.瞬时离心样品管2s,置于磁力架上5min,吸弃上清。c.保持样品管在磁力架上,加入200μl新鲜配制的80%乙醇,快速转动样品管以清洗磁珠,吸弃上清。d.将样品管瞬时离心,放置于磁力架上去掉残存的乙醇,室温晾干至磁珠表面无明亮反光,加入22μlnucleasefreewater,用移液器吹打混匀。室温静置2min,再次放置于磁力架上,取20μl上清于新的离心管。e.取1μl样本使用quantifluortm-st(promega)精确定量,得到样本文库,将样本文库保存至-20℃或进行下一步的杂交捕获。实施例6文库目标区域杂交捕获、测序1、根据实施例5中纯化后的文库样本浓度吸取总量为500ng的样本至新的1.5ml离心管,加入表7所示试剂,放入浓缩仪浓缩至完全干燥,如不立即进行下一步实验,可室温(15-25℃)放置过夜;表7浓缩体系名称体积cot-1dna5μlxgenuniversalblockingoligop51μlxgenuniversalblockingoligop7(6nt)1μl2、杂交体系配制与文库的变性(1)室温溶解xgen2xhybridizationbuffer,根据文库样本数量,配制表8所示的杂交混合液,用移液器吹打混匀,通过传递窗传递至513室;表8杂交混合液试剂名称1个反应体积xgen2xhybridizationbuffer8.5μlxgenhybridizationbufferenhancer2.7μlnuclease-freewater1.8μl总体积13μl(2)向每管步骤1中浓缩完成后的样本加入13μl表8所示的杂交混合液,室温静置5min;(3)用移液器吹打混匀样本并转移至low-bind0.2mlpcr管,将样本放入pcr仪运行如下程序:95℃保持10min,65℃静置;(4)当95℃运行结束时,保持样本在pcr仪上,立即加入4μlxgenlockdownprobepool,用移液器吹打混匀,避免产生气泡,此时反应总体积为17μl。(5)记录杂交开始时间,根据实验进度,选择4h或16h杂交时间。3、配制washbuffer(1)根据表9将xgen2xbeadwashbuffer、xgen10xwashbufferi、xgen10xwashbufferii、xgen10xwashbufferiii、xgen10xstringentwashbuffer配制成1x工作液,移液器吹打混匀;表9工作液体系(2)准备稀释好的washbufferi和stringentwashbuffer,按表10条件存放,其它试剂于室温存放(保证65℃孵育时间不少于2h);表10存放条件试剂名称1x工作液体积1x工作液储存温度washbufferi100μl65℃washbufferi200μl室温(15-25℃)stringentwashbuffer400μl65℃(3)准备m-270磁珠从4℃冰箱中取出m-270磁珠,确认磁珠已放置于室温30min,涡旋混匀使磁珠重悬浮;①在1.7mllow-bind管中为每个样本准备100μl磁珠;②将low-bind管置于磁力架上,静置至管内液体澄清,吸弃上清;③加入200μl1xbeadwashbuffer,涡旋混匀10sec,静置于磁力架上至管内液体澄清,吸弃上清;④重复步骤③一次;⑤加入100μl1xbeadwashbuffer,移液器吹打混匀。⑥将100μl悬浮磁珠转移至新的0.2mllow-bind管,置于磁力架上,静置至管内液体澄清,吸弃上清;4、捕获(1)确认步骤2的杂交反应已满足4h,保持样本和磁珠在pcr仪上,将样品转至准备好的磁珠管内,移液器吹打混匀,避免产生气泡;(2)65℃孵育45min,期间每隔12min将磁珠混匀(保持样本在pcr仪上),避免产生气泡。5、洗涤注意以下步骤需在65℃条件下快速操作:(1)每个样本加入100μl1xwashbufferi(65℃预热),快速混匀;(2)将样品转移至一新的1.7ml管中(65℃预热),快速混匀;(3)样品管置于磁力架上,静置至管内液体澄清,吸弃上清;(4)加入200μl1xstringentwashbuffer(65℃预热),轻柔混匀,65℃水浴5min。样品管置于磁力架上,静置至管内液体澄清,吸弃上清;(5)重复步骤(4)一次;(6)加入200μl常温1xwashbufferi,涡旋混匀2min。样品管置于磁力架上,静置至管内液体澄清,吸弃上清;(7)加入100μl常温1xwashbufferⅱ,涡旋混匀1min。样品管置于磁力架上,静置至管内液体澄清,吸弃上清;(8)加入200μl常温1xwashbufferⅲ,涡旋混匀30sec,样品管置于磁力架上,静置至管内液体澄清,吸弃上清;(9)将样品管从磁力架上取下,加入20μlnuclease-freewater,移液器吹打混匀,确保所有磁珠处于悬浮状态并全部转至0.2mlpcr管。6、pcr富集(1)室温解冻2xkapahifihotstartreadymix,配制表9所示的pcr扩增体系,快速混匀:表9pcr扩增体系试剂名称基因组文库2xkapahifihotstartreadymix25μl10ump5/p7primer2μl总体积27μl(2)每个样本(23ul)加入27μlpcrmix,总体积50μl,用移液器轻轻混匀,瞬时离心2s,在pcr仪上运行如下程序:98℃变性45s,12个循环(98℃变性15s,60℃退火30s,72℃延伸30s),72℃延伸1min,4℃冷却静置。7、纯化重悬浮agencourtampurexpreagent,确认磁珠已放置室温30min,每个样本用70μlbeads纯化,20μl洗脱。8、文库定量a.取1μl步骤7中纯化后的样品用qubitfluorometer3.0定量;b.取2μlpcr产物用于2%琼脂糖凝胶电泳,剩余样本于-20℃保存。9、送测样本数据量为10mreads,插入片段选择178(测序平台为illuminex-ten,或其他高通量测序平台)。实施例7异源cfdna定量分析1、对基因组dna文库和cfdna文库高通量测序后数据callsnp(samtool),提取5764个snp位点信息;2、对基因组dna文库中的5764个snp位点进行分析,进行基因分型,如图5所示,测序深度为50x时,即可获得基因分型;筛选受体患者基因组上为纯合型的snp位点作为有效snp位点;3、根据步骤2中的有效snp位点对cfdna文库进行分析,检测cfdna测序结果的有效位点处异源snp信号占cfdna在有效位点处总测序信号的比例;4、计算方式如下所示:假设受体患者基因组上有一个有效snp位点,基因组上纯合型的位点基因型为aa,检测到异源snp信号为a,则可以得到供体在此位点的基因型为aa或aa,基因分型如表6所示,根据异源snp信号的两种基因型,统计异源cfdna信号,计算出异源cfdna浓度,如下:表6基因分型当供体基因型为aa,异源信号比例=a型reads数/a型reads数;当供体基因型为aa,异源信号比例=2*a型reads数/a型reads数。实施例9绘制标准样回归曲线1、取2个正常人血样,一个作为供体,另一个作为受体;2、根据实施例4所述方法提取受体基因组dna、受体cfdna和供体cfdna;3、受体的cfdna中掺入供体cfdna,按1%、2%、4%、6%、8%的供体cfdna浓度混合,同时每个浓度梯度有三个样品的重复;3、根据实施例5所述方法构建基因组dna文库和步骤3中得到的混合cfdna文库;4、根据实施例6所述方法构建杂交捕获基因组dna文库、混合cfdna文库的目标区域后测序;5、根据实施例7所述方法测定混合cfdna文库中供体cfdna的比例,绘制得到图6所示的线性回归图,检测待测样品中的异源信号比例即异源cfdna检测值代入上述线性回归图中,进而可以计算得到待测样品中异源cfdna浓度。实施例10肾移植排斥患者检测选择两例活检后确认有排斥反应的肾移植病人,以健康人作为阴性对照,检测肾移植病人血浆中的cfdna异源信号,如图7所示,在健康的对照样本中,未检出异源cfdna信号,图片中仅显示健康样本的测序噪音;在两例肾移植排斥病人中可以检出明显的cfdna信号,说明异源cfdna的定量方法检测灵敏度和准确度高。显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。sequencelisting<110>苏州人人基因科技有限公司<120>检测异源cfdna的snp分子标记及检测方法、用途<130>sha201700229<160>10<170>patentinversion3.3<210>1<211>58<212>dna<213>人工序列<400>1aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58<210>2<211>33<212>dna<213>人工序列<400>2acactctttccctacacgacgctcttccgatct33<210>3<211>64<212>dna<213>人工序列<400>3agatcggaagagcacacgtctgaactccagtcacatcacgatctcgtatgccgtcttctg60cttg64<210>4<211>33<212>dna<213>人工序列<400>4agatcggaagagcacacgtctgaactccagtca33<210>5<211>15<212>dna<213>人工序列<220><221>misc_feature<222>(1)..(6)<223>nisa,c,g,ort<400>5nnnnnngtcagtact15<210>6<211>73<212>dna<213>人工序列<220><221>misc_feature<222>(59)..(64)<223>nisa,c,g,ort<400>6aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatctnn60nnnngtcagtact73<210>7<211>48<212>dna<213>人工序列<220><221>misc_feature<222>(34)..(39)<223>nisa,c,g,ort<400>7acactctttccctacacgacgctcttccgatctnnnnnngtcagtact48<210>8<211>78<212>dna<213>人工序列<220><221>misc_feature<222>(9)..(14)<223>nisa,c,g,ort<400>8gtactgacnnnnnnagatcggaagagcacacgtctgaactccagtcacatcacgatctcg60tatgccgtcttctgcttg78<210>9<211>47<212>dna<213>人工序列<220><221>misc_feature<222>(9)..(14)<223>nisa,c,g,ort<400>9gtactgacnnnnnnagatcggaagagcacacgtctgaactccagtca47<210>10<211>14<212>dna<213>人工序列<220><221>misc_feature<222>(9)..(14)<223>nisa,c,g,ort<400>10gtactgacnnnnnn14当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1