一种基于四苯基甲烷或硅烷的化合物及其制备方法与应用与流程
(一)
技术领域:
本发明涉及一种以四苯基甲烷或硅烷为中心核,不同种类的电致变色基团为外围基团的化合物及其制备方法,以及作为单体在合成电致变色材料中的应用。(二)
背景技术:
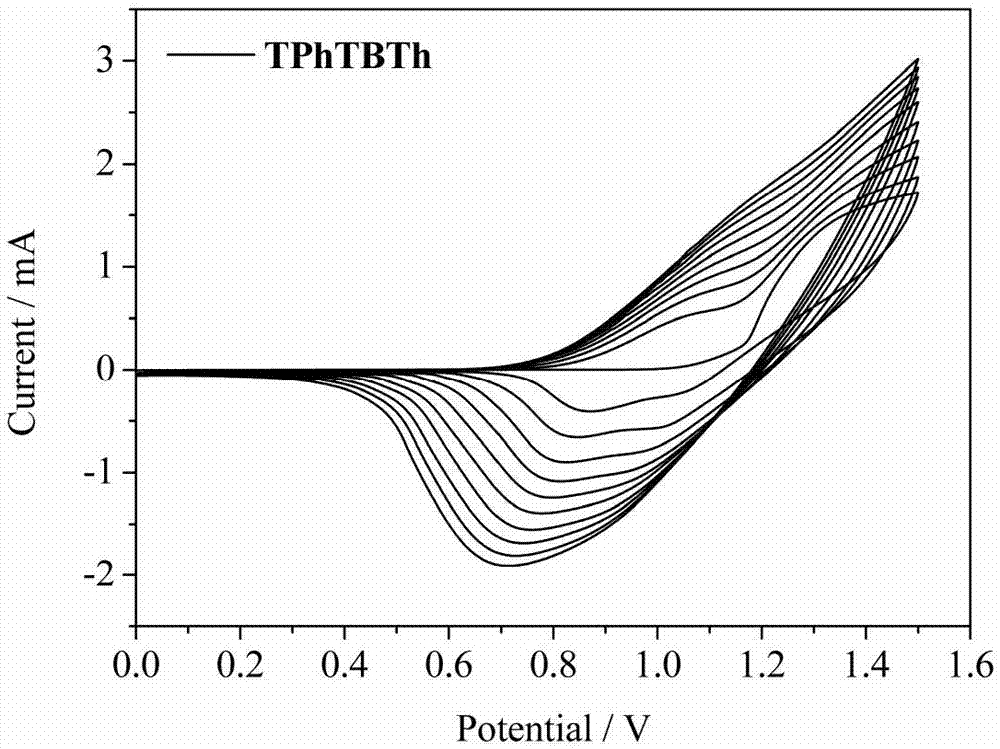
:随着全球能源的急剧消耗和环境的不断恶化。节能环保材料吸引了人们的广泛关注。电致变色材料正是这样一种可以改变人类生产生活方式并且有助于合理利用能源的新型功能材料。该材料具有来源丰富、器件制备工艺简单及工作电压低等优点,且能广泛应用在智能窗、电子纸及显示器等领域。导电聚合物基电致变色材料因结构易修饰、着色效率较高、响应时间较短、光学对比度较高及较丰富的颜色变换而成为电致变色材料中最具有潜力的一类物质。早期的研究工作主要集中在无机电致变色材料,如三氧化钨、二氧化铱等。无机电致变色材料具有良好的光化学稳定性和快速响应速度,但其加工成本较高、颜色种类单调、着色效率低下,限制了其在生产生活中的大规模应用。有机电致变色材料由于可以弥补无机变色材料的不足,已经成为人们研究的重点。相对于有机小分子变色材料的种类较少、分子结构不易修饰,导电聚合物电致变色(pec)材料则由于具有结构种类繁多、变色范围宽广、对比度高、加工性能好、能带可控以及响应速度快等优点而备受人们的关注,被认为是下一代ec材料的发展方向之一;而聚噻吩类导电聚合物,由于结构易修饰、合成相对简单等优点,是目前研究最广泛的一类电致变色材料。目前,关于电致变色导电高分子主要以线型聚合物为主,而关于体型网状电致变色导电高分子少有报道。本发明以四苯基甲烷或硅烷为核,不同种类的电致变色基团如噻吩、3,4-乙撑二氧噻吩为外围基团,设计合成了一类新的基于四苯基甲烷或硅烷电致变色单体,采用电化学的方法成膜及其电致变色方面的应用。(三)技术实现要素:本发明的目的是提供一种以四苯基甲烷或硅烷为中心核、外围修饰不同种类电致变色基团的化合物及其制备方法,以及在电致变色领域中的应用。本发明的技术方案如下:一种基于四苯基甲烷或硅烷的化合物,如式(i)所示:式(i)中,r1=c或si;r2选自如下之一所示的基团:其中“*”表示基团连接位点。本发明提供了所述基于四苯基甲烷或硅烷的化合物的制备方法,所述的制备方法为:(1)噻吩类化合物与三丁基氯化锡反应生成锡化的噻吩类中间体;(2)锡化的噻吩类中间体与四溴-(四苯基)甲烷或硅烷经suzuki反应生成目标产物。所述噻吩类化合物的结构式如下之一所示:与上述噻吩类化合物一一对应的锡化噻吩类中间体的结构式如下之一所示:所述四溴-(四苯基)甲烷或硅烷的结构式如下所示:r1=c或si。具体的,所述步骤(1)的操作过程如下:氮气保护下,将噻吩类化合物溶解于无水无氧的四氢呋喃中,然后于-70~-80℃下滴加正丁基锂的正己烷溶液(2.5mol/l),保温搅拌1~3h,之后加入2-三丁基氯化锡,继续保温搅拌0.5~1.5h,接着自然升至常温(20~30℃,下同)搅拌反应10~24h,之后反应液经后处理,得到锡化的噻吩类中间体;所述噻吩类化合物、正丁基锂、2-三丁基氯化锡的投料物质的量之比为1:1~1.1:1~1.1;所述四氢呋喃的体积用量以噻吩类化合物的质量计为40~100ml/g;所述反应液后处理的方法为:反应结束后,反应液浓缩并进行柱层析纯化,以三氧化二铝为固定相,石油醚为流动相,收集含目标化合物的洗脱液,蒸除溶剂并干燥,得到锡化的噻吩类中间体。具体的,所述步骤(2)的操作过程如下:氮气保护下,将四-(4-溴苯基)甲烷或硅烷、四三苯基膦钯溶于甲苯/四氢呋喃体积比1:0.5~1的混合溶剂中,然后加入锡化的噻吩类中间体,在避光、120~140℃条件下反应24~50h,之后反应液经后处理,得到目标产物;所述四-(4-溴苯基)甲烷或硅烷、四三苯基膦钯、锡化的噻吩类中间体的投料物质的量之比为1:0.01~0.05:4~8;所述甲苯/四氢呋喃混合溶剂的体积用量以四-(4-溴苯基)甲烷或硅烷的质量计为30~100ml/g;所述反应液后处理的方法为:反应结束后,反应液经二氯甲烷萃取,萃取液浓缩后进行柱层析纯化,以硅胶为固定相,石油醚/二氯甲烷(体积比,1~4/1)为流动相,收集含目标化合物的洗脱液,蒸除溶剂并干燥,得到目标产物。本发明所述的基于四苯基甲烷或硅烷的化合物可经电化学聚合成膜,具体如下:将式(i)所示的基于四苯基甲烷或硅烷的化合物溶解于二氯甲烷中,加入四丁基高氯酸铵(tbap)作为电解质,扫速为100mv/s,经0~1.5v循环伏安法电化学聚合成膜;所述二氯甲烷的体积用量以式(i)所示的基于四苯基甲烷或硅烷的化合物的质量计为1~2ml/mg;所述四丁基高氯酸铵的用量以二氯甲烷的体积计为0.1mol/l。本发明的有益效果在于:本发明所述的基于四苯基甲烷或硅烷的化合物由于具有四面体型的空间结构,经电化学聚合成膜,所得到的薄膜表现出较大的比表面积,同时表现出良好的电致变色等电化学性能。所述薄膜对比度为30%~80%,响应时间为0.5~6s之间,均表现出合理的电化学稳定活性,实验结果表明这类材料是一类潜在的电致变色材料。(四)附图说明图1:本发明所述化合物的合成路线;图2:本发明实施例11薄膜的cv曲线图;图3:本发明实施例11中薄膜在不同电压下的紫外可见吸收光谱图;图4:本发明实施例11中薄膜的电致变色性能。(五)具体实施方式下面以具体实施例对本发明的技术方案做进一步说明,但本发明的保护范围不限于此:实施例12-联噻吩锡三丁基的合成联噻吩2.5g(15mmol),溶解于100ml重蒸过的四氢呋喃溶液中,置于250ml锥形圆底长颈烧瓶中,抽真空氮气保护三次之后,在-78℃氮气保护下缓慢滴加6ml的正丁基锂的环己烷溶液(2.5mol/l),保持低温搅拌两小时后缓慢滴加三丁基氯化锡5.5ml(16.5mmol),在-78℃下搅拌1小时后恢复至室温搅拌24h,反应结束后,浓缩反应混合物,然后用al2o3填料层析过柱,用石油醚洗脱,收集含目标化合物的洗脱液,旋干得到浅绿色产物6.15g,收率90%。确认物质的表征结构如下:1hnmr(500mhz,cdcl3)δ7.33–7.30(m,1h),7.21–7.18(m,2h),7.08(t,j=2.4hz,1h),7.04–7.00(m,1h),1.63–1.56(m,6h),1.40–1.35(m,6h),1.17–1.11(m,6h),0.95–0.91(m,9h)。c20h22s2sn的质谱表征结构如下:455.31g/mol.实施例22-噻吩锡三丁基的合成合成方法同实施例1,不同之处在于,将底物联噻吩替换为噻吩1.26g(15mmol),最终得到产物5.15g,收率92%。c16h30ssn的质谱表征结构如下:373.01。实施例33-噻吩锡三丁基的合成合成方法同实施例1,不同之处在于,将底物联噻吩替换为3-溴噻吩2.445g(15mmol),最终得到产物4.75g,收率85%。c16h30ssn的质谱表征结构如下:373.91。实施例42-三联噻吩锡三丁基的合成合成方法同实施例1,不同之处在于,将底物联噻吩替换为三联噻吩3.725g(15mmol),最终得到产物4.03g,收率50%。c24h34s3sn的质谱表征结构如下:538.08。实施例52-(3,4-乙烯二氧)噻吩锡三丁基的合成合成方法同实施例1,不同之处在于,将底物联噻吩替换为(3,4-乙烯二氧)噻吩2.132g(15mmol),最终得到产物4.20g,收率65%。c18h32o2ssn的质谱表征结构如下:538.08。实施例6化合物四-[4-(联噻吩-2)-苯基]甲烷/硅烷的合成(a)将四-[4-溴-苯基]甲烷0.636g(1mmol)、四三苯基膦钯0.18g加入到50ml两口圆底烧瓶中,抽真空氮气保护下加入甲苯/四氢呋喃混合溶剂20ml(体积比为1:1),搅拌均匀溶解之后,注入2-联噻吩锡三丁基3.642g(8mmol),将温度升至140℃,避光条件下反应24h。反应结束后,二氯甲烷萃取,萃取液旋干拌样,过柱分离,以硅胶为填料,石油醚与二氯甲烷(体积比4:1)为洗脱剂,最终得到浅黄色产物0.39g,产率为40%。确认物质的表征结构如下:1hnmr(500mhz,cdcl3)δ7.67(d,j=8.6hz,2h),7.53(dd,j=5.1,1.0hz,1h),7.48(d,j=3.8hz,1h),7.35(dd,j=3.6,1.1hz,1h),7.32(d,j=3.6hz,2h),7.31(s,1h),7.11(dd,j=5.1,3.6hz,1h).c57h36s8质谱表征为976.06。(b)将(a)中四-[4-溴-苯基]甲烷替换为四-[4-溴-苯基]硅烷0.647g(1mmol),其余条件不变,则得到产物四-[4-(联噻吩-2)-苯基]甲烷0.31g,产率31%。c56h36s3si的质谱表征结构如下:992.04.实施例7化合物四-[4-(噻吩-2)-苯基]甲烷/硅烷的合成合成方法同实施例6,不同之处在于:(a)将2-联噻吩锡三丁基替换为2-噻吩锡三丁基2.984g(8mmol),最终得到四-[4-(噻吩-2)-苯基]甲烷0.23g,产率36%。c41h23s4的质谱表征结构如下:648.11.(b)将(a)中四-[4-溴-苯基]甲烷替换为四-[4-溴-苯基]硅烷0.6520g(1mmol),其余条件不变,则得到产物四-[4-(噻吩-2)-苯基]硅烷0.2g,产率30%。c40h28s4si质谱表征结构如下:664.08.实施例8化合物四-[4-(噻吩-3)-苯基]甲烷/硅烷的合成合成方法同实施例6,不同之处在于:(a)将2-联噻吩锡三丁基替换为3-噻吩锡三丁基2.991g(8mmol),最终得到四-[4-(噻吩-3)-苯基]甲烷0.16g,产率25%。c41h28s4质谱表征结构如下:649.11.(b)将(a)中四-[4-溴-苯基]甲烷替换为四-[4-溴-苯基]硅烷0.6520g(1mmol),其余条件不变,则得到产物四-[4-(噻吩-3)-苯基]硅烷0.13g,产率19%。c40h28s4si质谱表征结构如下:665.09.实施例9化合物四-[4-(三联噻吩-2)-苯基]甲烷/硅烷的合成合成方法同实施例6,不同之处在于:(a)将2-联噻吩锡三丁基替换为2-三联噻吩锡三丁基4.30g(8mmol),最终得到四-[4-(三联噻吩-3)-苯基]甲烷0.2g,产率15%。c73h44s12质谱表征结构如下1304.01.(b)将(a)中四-[4-溴-苯基]甲烷替换为四-[4-溴-苯基]硅烷0.6520g(1mmol),其余条件不变,则得到产物四-[4-(三联噻吩-3)-苯基]硅烷0.24g,产率18%。c72h44s12si质谱表征结构如下:1320.99.实施例10化合物四-[4-[(3,4-乙烯二氧)噻吩-2]-苯基]甲烷/硅烷的合成合成方法同实施例6,不同之处在于:(a)将2-联噻吩锡三丁基替换为2-(3,4-乙烯二氧)噻吩锡三丁基4.304g(8mmol),最终得到四-[4-[(3,4-乙烯二氧)噻吩-2]-苯基]甲烷0.3171g,产率36%。c49h36o8s4质谱表征结构如下:880.13.(b)将(a)中四-[4-溴-苯基]甲烷替换为四-[4-溴-苯基]硅烷0.6520g(1mmol),其余条件不变,则得到产物四-[4-[(3,4-乙烯二氧)噻吩-2]-苯基]硅烷0.31g,产率35%。c48h36o8s4si质谱表征结构如下:896.11.实施例11上述单体的电化学聚合成膜,以四-[4-(联噻吩-2)-苯基]甲烷为例说明电化学聚合的具体过程及电致变色性质。将四-[4-(联噻吩-2)-苯基]甲烷9.7mg(1mm)、tbap(0.1m)为电解质,加入到容量瓶中,二氯甲烷定容10ml,超声3min,充分溶解之后,用于电化学聚合。以ito为工作电极、铂片(0.9*3cm)为对电极、ag/agcl为参比电极,采用循环伏安法0-1.5v成膜,在空白溶液中(0.1mtbap溶解于乙腈)脱掺杂2min,制备得到的膜在烘箱中60℃干燥2小时后,在tbap/乙腈中进行电化学、光学及电致变色性能测试。该薄膜表现出良好的氧化还原性能,在0-1.3v电压阶跃,表现出黄色到蓝绿色之间颜色的改变,1100nm的光学对比度为37%,1100nm下的着色和褪色时间分别为2.88s,0.55s,这些表明这类材料是一类非常潜在的电致变色材料。与噻吩-苯-噻吩类聚合物电致变色材料相比较,这类化合物经电化学聚合形成稳定的交联结构聚合物薄膜,电致变色响应速度得到了明显的改善,提高了将近一秒,同时这类材料表现出很好的电化学稳定性。其他物质的电致变色性能测试数据如下,所有薄膜均是在含有tbap的二氯甲烷溶液中循环伏安4圈制备得到,测试条件为在含有tbap的乙腈溶液中进行。表1上述聚合物的光学对比度及响应时间polymer(p=polymerizaiton)contrastcoloringtime(s)discoloringtime(s)ptphtth\\\ptphtbth37%2.88s0.55sptphttth55%2.98s0.73sptphsitth\\\ptphsitbth52%3.13s0.72sptphsittth56%3.32s0.68sptphtedot42%3.64s1.68sptphsitedot45%3.72s1.74sptpht-3-th\\\ptphsit-3-th\\\(斜线代表物质不能成膜,无法测试电致变色性能)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 含有2,3-二氟-5,6-二甲基苯基的液晶化合物及其制备方法与应用与流程
- 一类四苯基亚甲基二氢卟吩化合物及其制备方法与应用与流程
- 一种1,3-二(二苯基氯硅烷基)-2,2,4,4-四甲基环二硅氮烷的合成方法与流程
- 一种1-[2-(2,4-二甲基苯基硫烷基)苯基]哌嗪氢溴酸盐α型晶体的制备方法与流程
- 一种1‑(4‑氯苯基)‑2‑环丙基‑1‑丙酮的制备方法与流程
- 一种双[三(2‑甲基‑2‑苯基)丙基锡]衣康酸酯配合物及其制备方法与应用与流程
- 甲基苯基硅树脂的制备方法与流程
- 一种苯基二甲基硅氧基笼型倍半硅氧烷硅橡胶交联剂及其制备方法与流程
- 一种甲基氯硅烷单体合成循环反应系统的制作方法与工艺
- E‑3‑(苯基衍生物‑次甲基)‑吡喃并二氢黄酮衍生物及制备及应用的制作方法与工艺