一类含氮杂环结构的三萜皂苷类衍生物及其合成方法和应用与流程
本发明涉及有机合成
技术领域:
,尤其涉及一类含氮杂环结构的三萜皂苷类衍生物的合成方法和应用。
背景技术:
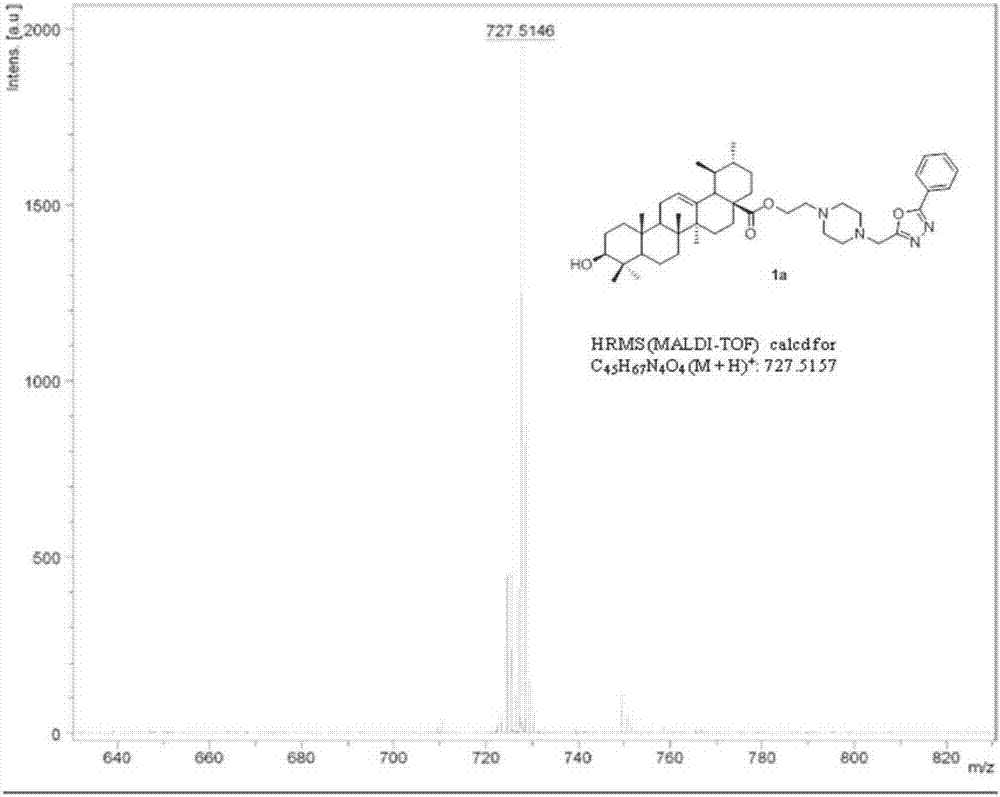
:长期以来,炎症一直是动物和人类生产生活的第一大杀手,许多因素如细菌感染、器官移植等均可诱发机体自身产生炎症反应而严重威胁人类的健康。为治疗炎症反应,人们开始使用的糖皮质激素类固醇类抗炎药物,可有效控制感染性炎症和非感染性炎症,并可有效消除炎症引起的功能障碍。但随着人类科学技术的不断进步和发展,科学家们发现长期使用糖皮质激素类固醇消炎药物可引起机体的肾上腺皮质功能障碍和器官性骨质疏松等并发症。而非甾体类抗炎药(nsaids)最早从含水杨酸的植物中提取,是一类具有解热、镇痛、抗炎作用的药物,其抗炎的共同作用机制是抑制机体环氧化酶(cox)活性,从而进一步抑制机体局部前列腺素(pg)的产生,而发挥抗炎药理作用。后来研究发现长期使用水杨酸药物对身体的胃有很大的影响。而在1897年,霍夫曼合成了高纯度的阿司匹林,随后人们又陆续发现了异丁卡因、布洛芬等众多非甾体抗炎药物。但大量临床试验证明这类非甾体抗炎药均对肠胃有严重的副作用。因此,如何研发出一类新型的药效好、活性更高和低毒副作用的药物来克服目前抗炎药物存在的如上所述的缺点成为全社会关注的问题。所以新型抗炎药物的设计研发具有十分重要的意义。技术实现要素:为了解决上述问题,本发明提供了一类新型的抗炎化合物及其合成方法。本发明的技术方案如下:一类含氮杂环结构的三萜皂苷类衍生物,其结构通式如下:其中,n为0,1,2,3,4;r1分别是氢原子、低级烷基、卤素、氨基、低级链烯基、羟基、烷氧基、硝基、羧基、羧基烷基中的任意一种。该类化合物合成方法简单,合理,生产成本低。式(f)化合物的合成通法如下:a):meoh,h2so4,reflux,5hb):nh2nh2h2o,etoh.reflux,10hc):pocl3,105-110℃reflux,5-6hd):k2co3,kl;acetone,reflux,10h.e):k2co3,klacetone,reflux10h.如式(f)的含氮杂环结构的三萜皂苷类衍生物的合成方法,包括以下步骤:(1)在圆底烧瓶中加入化学物a,用适量甲醇溶解,160-180℃条件下回流搅拌反应,向圆底烧瓶中缓慢加入浓硫酸,反应4-6个小时停止后减压蒸出溶剂得到化合物b;(2)将化合物b与水合肼在乙醇溶液中反应,反应停止后经过乙醇与水重结晶得到白色块状物c;(3)将化合物c与等量的氯乙酸反应,加入适量的三氯氧磷作催化剂,105-110℃条件下回流搅,反应5-6小时;将反应液倒入适量冰水中,过滤,水洗,干燥,得化合物d;(4)将化合物d与过量的无水哌嗪反应,丙酮作溶剂,加入碳酸钾、碘化钾作催化剂,回流反应8-12个小时,tlc监测至反应完全;停止反应后减压蒸除溶剂,残余物加入适量的水溶解后,经二氯甲烷萃取,水洗,无水硫酸镁干燥,旋干溶剂后得到白色固体化合物;柱层析分离杂质,得到中间体化合物e;(5)将中间体化合物e与过量的1,2-二溴乙烷以及熊果酸反应,在适量催化剂碘化钾和碳酸钾的催化下,用丙酮溶液溶解;55-60℃条件下回流搅拌反应8-12h;254nm波长下薄层色谱法点板监测反应进程;反应结束后减压蒸馏出多余溶剂,二氯甲烷与水少量多次萃取,合并有机层,用无水硫酸镁干燥后,抽滤并旋干有机溶剂,得到白色固体;柱层析分离杂质,得到目标化合物f。以下以2-苯基-5-((4-乙基哌嗪-1-基)甲基)-1,3,4-恶二唑熊果酸(1a)为例介绍式(f)化合物的合成通法,包括以下步骤:(1)在圆底烧瓶中加入苯甲酸,用适量甲醇溶解,160-180℃条件下回流搅拌反应,向圆底烧瓶中缓慢加入浓硫酸,反应4-6个小时停止后减压蒸出溶剂得到化合物苯甲酸甲酯;(2)将化合物苯甲酸甲酯与水合肼在乙醇溶液中反应,反应停止后经过乙醇与水重结晶得到白色块状物苯甲肼;(3)将化合物苯甲肼与等量的氯乙酸反应,加入适量的三氯氧磷作催化剂,105-110℃条件下回流搅,反应5-6小时;将反应液倒入适量冰水中,过滤,水洗,干燥,得化合物2-(氯甲基)-5-苯基-1,3,4-恶二唑;(4)将2-(氯甲基)-5-苯基-1,3,4-恶二唑与过量的无水哌嗪反应,丙酮作溶剂,加入碳酸钾、碘化钾作催化剂,回流反应8-12个小时,tlc监测至反应完全;停止反应后减压蒸除溶剂,残余物加入适量的水溶解后,经二氯甲烷萃取,水洗,无水硫酸镁干燥,旋干溶剂后得到白色固体化合物;柱层析分离杂质,得到中间体化合物2-苯基-5-(哌嗪-1-亚甲基)-1,3,4-恶二唑;(5)将中间体化合物2-苯基-5-(哌嗪-1-亚甲基)-1,3,4-恶二唑与过量的1,2-二溴乙烷以及熊果酸反应,在适量催化剂碘化钾和碳酸钾的催化下,用丙酮溶液溶解;55-60℃条件下回流搅拌反应8-12h;254nm波长下薄层色谱法点板监测反应进程;反应结束后减压蒸馏出多余溶剂,二氯甲烷与水少量多次萃取,合并有机层,用无水硫酸镁干燥后,抽滤并旋干有机溶剂,得到白色固体;柱层析分离杂质,得到目标化合物2-苯基-5-((4-乙基哌嗪-1-基)甲基)-1,3,4-恶二唑熊果酸(1a)。化合物2-苯基-5-((4-乙基哌嗪-1-基)甲基)-1,3,4-恶二唑熊果酸(1a)的化学式如下:本发明的有益之处在于:本发明的含氮杂环结构的三萜皂苷类衍生物抗炎效果显著,比目前临床上常用的抗炎药物布洛芬和吲哚美辛均具有更好的抗炎抑制效果,而且具有低细胞毒特性。附图说明图1为实施例1中产物的高分辨率质谱图。图2为实施例1中产物的核磁共振谱图,其中图2a为氢谱图,图2b为碳谱图。具体实施方式下面以具体的实施例对本发明的应用原理作详细的描述。实施例1以下以2-苯基-5-((4-乙基哌嗪-1-基)甲基)-1,3,4-恶二唑熊果酸(1a)为例介绍式(f)化合物的合成通法,包括以下步骤:(1)在50ml圆底烧瓶中加入苯甲酸(5mmol),用适量甲醇溶解,170℃条件下回流搅拌反应,向圆底烧瓶中缓慢加入5滴浓硫酸,反应5个小时停止后减压蒸出溶剂得到化合物苯甲酸甲酯。(2)将化合物苯甲酸甲酯与3倍量的水合肼在乙醇溶液20ml中反应,反应停止后经过乙醇与水重结晶得到白色块状物苯甲肼。(3)将化合物苯甲肼与等量的氯乙酸反应,加入适量的三氯氧磷作催化剂,108℃条件下回流搅,反应5.5小时。将反应液倒入适量冰水中,过滤,水洗,干燥,得化合物2-(氯甲基)-5-苯基-1,3,4-恶二唑。(4)将2-(氯甲基)-5-苯基-1,3,4-恶二唑与过量的无水哌嗪反应,丙酮作溶剂,加入碳酸钾、碘化钾作催化剂,回流反应10个小时,tlc监测至反应完全。停止反应后减压蒸除溶剂,残余物加入适量的水溶解后,经二氯甲烷萃取(3×5ml),水洗三次,无水硫酸镁干燥,旋干溶剂后得到白色固体化合物。柱层析(二氯甲烷:甲醇=200:1)分离杂质,得到中间体化合物2-苯基-5-(哌嗪-1-亚甲基)-1,3,4-恶二唑。(5)将中间体化合物2-苯基-5-(哌嗪-1-亚甲基)-1,3,4-恶二唑与过量的1,2-二溴乙烷以及熊果酸反应,在适量催化剂碘化钾和碳酸钾的催化下,用20ml的丙酮溶液溶解。57℃条件下回流搅拌反应10h。254nm波长下薄层色谱法点板监测反应进程。反应结束后减压蒸馏出多余溶剂,二氯甲烷与水少量多次萃取,合并有机层,用无水硫酸镁干燥后,抽滤并旋干有机溶剂,得到白色固体。柱层析(二氯甲烷:甲醇=150:1)分离杂质,得到目标化合物2-苯基-5-((4-乙基哌嗪-1-基)甲基)-1,3,4-恶二唑熊果酸(1a)。目标化合物1a的产率、谱图数据如下所示:产率46%,m.p.257-259℃;1hnmr(cdcl3,300mhz,ppm)δ8.13–8.04(m,2h),7.54(q,j=5.8hz,3h),5.24(s,1h),4.17(s,2h),3.92(s,2h),3.22(d,j=6.0hz,1h),2.70(s,8h),2.21(d,j=11.5hz,1h),2.00(dd,j=15.3,11.4hz,1h),1.94–1.86(m,2h),1.79(d,j=13.7hz,1h),1.73–1.44(m,12h),1.38–1.25(m,7h),1.08(s,3h),0.99(d,j=7.1hz,4h),0.94(d,j=10.5hz,6h),0.86(d,j=6.4hz,3h),0.75(t,j=12.4hz,7h).13cnmr(cdcl3,75mhz):δ177.30,165.39,163.30,138.11,131.80,129.03,127.01,125.62,123.79,79.00,77.46,77.04,76.62,61.64,56.43,55.23,53.06,52.87,52.70,51.94,48.00,47.54,42.04,39.57,39.06,38.87,38.76,38.63,36.97,36.69,33.05,30.65,29.70,28.16,27.98,27.24,24.23,23.41,23.41,21.16,18.30,17.09,17.09,15.55,15.55.hrms(maldi-tof)calcdforc45h67n4o4(m+h)+:727.5157,found:727.5146。以下用小鼠耳肿胀法测定炎症抑制率实验化合物的体外抗炎活性实验采用二甲苯致小鼠耳肿胀法测定。初步评价抗炎活性采用腹腔注射给药途径:首先将实验用动物昆明种试验小鼠随机分每组10只(雄性小鼠和雌性小鼠每组各5只),分别设置为测试药品组、阳性对照药吲哚美辛组、布洛芬组以及空白实验组。其次,我们将所有需要测试的目标化合物溶解在二甲基亚砜(dmso)溶液中,用相对应的测试样品分别给测试药品组、阳性对照组和空白实验组的小鼠进行腹腔注射,腹腔注射30分钟后,将20微升的二甲苯试剂均匀地涂抹于实验小鼠右耳的正反面,待试验小鼠涂抹完二甲苯30分钟后将小鼠进行断颈处死法,然后将处死后的实验小鼠的左右两只耳朵分别剪下,随后用同一直径为7毫米的打孔器在实验小鼠左右耳朵的同一位置进行打孔,得到两个同等大小的圆形耳片,稍后用分析天平分别进行称重,记录两只耳朵的重量。数据用统计学软件处理得到初步测定的抗炎抑制率。进一步的抗炎活性评价采用口服给药途径测定,从抗炎抑制率数据中挑选抗炎活性最好的化合物,将其研磨后,用羧甲基纤维素钠溶液进行溶解,随后对实验小鼠进行口服给药,以便进一步验证其不同时间段、不同浓度下的抗炎活性,此时选取吲哚美辛为阳性对照药。同样将实验小鼠分成测试药品组,阳性对照组和空白实验组,每组昆明种小鼠雌雄各5只,将测试化合物和吲哚美辛分别溶解于1/1000的缩甲基纤维素钠水溶液中,空白组为1/1000的缩甲基纤维素钠水溶液。我们对测试目标化合物组,阳性对照组和空白实验组分别测定了不同时间、不同剂量下的抗炎活性以及,实验处理方法同上,随后称重,数据用统计学软件得到不同时间段、不同剂量下药物的抗炎活性结果。目标化合物的抗炎活性结果如表1所示:剂量(mg/kg)小鼠数量耳肿胀平均值(mg)抑制率(%)二甲基亚砜—813.23±0.81—吲哚美辛组5089.68±0.9426.83布洛芬组5089.90±0.6125.17熊果酸5085.60±1.2757.671a5085.31±0.8359.84测试用细胞选用人体宫颈癌细胞株hela,人肝癌细胞hep3b以及正常人体肝细胞lo2。使用含体积比为10%的牛胎儿血清和100u/ml的双抗(青霉素-链霉素混合物)的细胞培养基高糖培养基来培养测试用的人体宫颈癌细胞株hela,人肝癌细胞hep3b以及正常人体肝细胞lo2。培养过程中用消化液胰蛋白酶(0.25%inpbs;ph7.4)将处于对数生长期的人体宫颈癌细胞株hela,人肝癌细胞hep3b以及正常人体肝细胞lo2消化2分钟,随后加入细胞培养基培养基5ml终止消化,在离心机上进行离心并移取上层液,加入15ml细胞培养基,放置在37℃,5%co2培养箱中孵育24小时后,再更换一次培养基。当细胞培养至长满瓶壁后,计数,制成1×104个·ml-1的细胞悬液,接种于96孔培养板内,每孔100μl。实验分别设置空白对照组和测试药品组,每组8复孔。同样放置到37℃,5%co2孵育24小时后,弃去原培养液。空白对照组加入新鲜细胞培养液,供试品药品组分别加入含有不同浓度目标化合物的培养液,每孔100μl,置37℃,5%co2培养箱孵育48小时。然后向96孔培养板每个小孔加入10μlmtt溶液(5mg/mlinpbs),置37℃,5%co2培养箱孵育4小时,移取上清液,随后每孔分别加入100μldmso。在振荡器上震摇10min后,使用酶标仪在570nm吸光度下测其光密度。最后计算其半抑制浓度ic50值如表所示:aic50为半抑制浓度,数据代表三个独立实验的平均值,数据变动一般在5-10%之间。sgcb为人体肺癌细胞;agsc为人体肺癌细胞;l02d人体正常肝细胞。以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本
技术领域:
的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 苯并双(噻二唑)衍生物、包含其的油墨、以及使用了其的有机电子装置的制造方法
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 一种苯并噻咯类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并噻吩并咔唑类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 靛红杂合喹唑啉类化合物合成的靛红衍生物及其在制备抗肿瘤药物中的应用的制造方法与工艺
- 一种杂环衍生物及使用该杂环衍生物的有机发光器件的制造方法与工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 一种含氮杂环衍生物及使用该含氮杂环衍生物的有机发光器件的制造方法与工艺