一种抗肿瘤药物AZD9291的制备方法与流程
本发明属于医药化工领域,具体涉及一种抗肿瘤药物azd9291的制备方法。
背景技术:
肺癌是世界上最常见的恶性肿瘤之一,已成为我国城市人口恶性肿瘤死亡原因的第1位。非小细胞型肺癌包括鳞状细胞癌(鳞癌)、腺癌、大细胞癌,与小细胞癌相比其癌细胞生长分裂较慢,扩散转移相对较晚。非小细胞肺癌约占所有肺癌的80%,约75%的患者发现时已处于中晚期,5年生存率很低。近年来,针对致癌的基因突变,有许多靶向性新药不断问世。与传统的化学疗法相比,靶向性药物主要影响带有相应突变的癌细胞,毒副性较低,有效率升高。文献研究表明,非小细胞肺癌中有一类是由于表皮细胞生长素受体(egfr)突变所导致。
azd9291(osimertinib),中文名称:迈瑞替尼,化学名称:n-[2-[[2-(二甲基氨基)乙基]甲基氨基]-4-甲氧基-5-[[4-(1-甲基-1h-吲哚-3-基)-2-嘧啶基]氨基]苯基]-2-丙烯酰胺;分子式c28h33n7o2·ch4o3s,结构式如下。它是由英国阿斯利康公司研发,于2015年11月13日在美国上市,商品名tagasso,用于治疗表皮生长因子受体(egfr)t790m突变的晚期非小细胞肺癌患者。
与众多同类针对egfr突变的竞争药相比,azd9291具有一个突出优势,它不仅能抑制致癌的egfr突变,而且能持续抑制抗药性的egfr突变。其他的egfr抑制剂,比如gefitinib(阿斯利康)以及erlotinib(基因泰克genentech和罗氏制药roche),也能够靶向性的抑制具有egfr突变的癌细胞生长,但这些药物都只能短暂见效,癌细胞通常会发展出抗药性的突变。在多于60%的病例中,egfr突变型非小细胞肺癌的抗药性突变发生在egfr序列中的第790位点,由原来的苏氨酸变成甲氨酸(t790m)。这一突变可以有效阻挠一般的egfr抑制剂与egfr作用,从而达到癌细胞抗药的目的。但azd9291不会受egfr突变的干扰,会持续抑制癌细胞生长,因此特别适用于其他egfr突变抑制剂治疗失败的中晚期病人。
目前azd9291的合成路线主要包括以下三种:
合成路线1:wo2013014448专利公开了azd9291的合成方法,反应方程式如下:
该合成路线中采用铁胺还原产生大量铁泥固体废弃物,引入丙烯酰基采用丙烯酰氯或是3-氯丙酰氯,中间体n-(2-二甲基氨基-乙基)-2-甲氧基-n-甲基-n-[4-(1-甲基-1h-吲哚-3-基)-嘧啶-2-基]-5-氨基-苯-1,4-二胺多个位点会发生反应,副产物较多,较难纯化。
合成路线2:cn104817541a专利公开了另外一种azd9291合成方法,反应方程式如下:
该路线引入了保护基团保护氨基,增加了反应步骤,也增加了azd9291工业化生产成本。
合成路线3:cn104910049a公开了另外一种合成方法,反应方程式如下:
该合成路线较长,总收率较低,不适于工业化生产。
技术实现要素:
本发明的目的是为了克服上述现有技术的不足,在专利wo2013014448的基础上,进行了工艺优化,其中中间体2的制备过程中加入活化剂溴化亚铜;中间体3的还原由铁胺还原法改为钯碳催化加氢还原,azd9291的丙烯酰基引入由丙烯酰氯或3-氯丙酰氯改为丙烯酸的混酐溶液,有效的减少了固废的产生,并提高了产品纯度和收率。
本发明的技术方案是:一种抗肿瘤药物azd9291的制备方法,其特征是,
1)3-(2-氯-嘧啶-4-基)-1-甲基吲哚(sm1)与4-氟-2-甲氧基-5-硝基苯胺(sm2)进行反应得到中间体1:即n-(4-氟-2-甲氧基-5-硝基苯基)-4-(1-甲基-1h-吲哚-3-基)-嘧啶-2-胺;
2)中间体1与n,n,n′-三甲基乙二胺进行反应得到中间体2,即n-(2-二甲基氨基-乙基)-2-甲氧基-n-甲基-n-[4-(1-甲基-1h-吲哚-3-基)-嘧啶-2-基]-5-硝基-苯-1,4-二胺;
3)中间体2经氢化还原反应得到中间体3,即n-(2-二甲基氨基-乙基)-2-甲氧基-n-甲基-n-[4-(1-甲基-1h-吲哚-3-基)-嘧啶-2-基]-5-氨基-苯-1,4-二胺;
4)中间体3再进一步引入丙烯酰基生成azd9291;
其特征是,所述步骤4)为:中间体3与丙烯酸的混酐溶液进行反应得到产物azd9291。
其中,丙烯酸的混酐溶液的制备方法为:将酰氯与无机碱加入到溶剂中,0℃~10℃搅拌状态下加入丙烯酸,得到丙烯酸的混酐溶液。所述酰氯为特戊酰氯、对甲基苯磺酰氯、苯磺酰氯、甲磺酰氯中的至少一种。所述无机碱为碳酸钾、碳酸铯、碳酸锂中的至少一种。所述溶剂为乙腈、甲基叔丁基醚、1,4-二氧六环中的至少一种。所述丙烯酸、酰氯与无机碱的摩尔比为1:1.0~1.2:1.0~1.5。
进一步的,所述步骤2)中还加入溴化亚铜作为催化剂活化中间体1的氟,促进n,n,n′-三甲基乙二胺的亲核取代反应,提高反应的速度和收率;溴化亚铜用量为中间体1重量的1.5-2.5%。
进一步的,所述步骤3)的氢化还原为钯碳催化加氢还原,钯碳的用量为中间体2重量的2%~10%。
本发明步骤1)中反应溶剂优选为2-戊醇、正丁醇、异丁醇和1,4-二氧六环中的一种或几种。优选反应温度为60℃~110℃,反应时间为2h~6h。sm1与sm2摩尔比优选为1.0:1.0~1.5。由于嘧啶环中的2-氯反应活性降低,因此加入酸作为催化剂活化嘧啶环,从而使苯胺的亲核取代反应能够进行,优选酸为甲磺酸、对甲苯磺酸、一水合对甲苯磺酸中的一种或几种,它与sm1的摩尔比为2~2.5:1。
本发明步骤2)中反应溶剂优选为n,n-二甲基甲酰胺、二甲基亚砜、n,n-二甲基乙酰胺中的一种或几种。此步反应为亲核取代反应,反应有氢氟酸生成,还需加入适量的碱作为缚酸剂促进反应的进行,优选为无机碱或有机碱,无机碱选自碳酸钠、碳酸氢钠、碳酸钾中的一种或几种;有机碱选自三乙胺、n,n-二异丙基乙胺中的一种或几种。中间体1、n,n,n′-三甲基乙二胺和碱的摩尔比为1.0:1.0~1.2:1.2~2.0。优选反应温度为90℃~110℃,反应时间为2~8h。
本发明步骤3)中反应溶剂优选为甲醇、乙醇、异丙醇中的一种或几种。优选反应温度为15℃~40℃,常压下反应2h~4h。
本发明步骤4)中反应温度为-10℃~15℃。优选的中间体3与丙烯酸的摩尔比为1:2~2.5。
本发明的最佳实施方案如下:
1)中间体1的制备
将sm1、sm2和一水对甲苯磺酸加入到异丁醇中,升温至回流,反应4~6h,有黄色固体析出,降至室温,过滤、洗涤、干燥得到黄色固体中间体1;
2)中间体2的制备
将中间体1、n,n,n′-三甲基乙二胺和n,n-二异丙基乙胺加入到n,n-二甲基甲酰胺中,升温至95~105℃反应4~6h,降至室温,加入水析出固体,经过滤、水洗、干燥后得中间体2;
更进一步的,上述步骤还加入溴化亚铜,反应时间2~4h,降至室温后过滤除去溴化亚铜;
3)中间体3的制备
氮气保护中,将中间体2和钯碳加入到甲醇中,通入氢气常压反应3~5h,过滤,滤液减压浓缩至干,得中间体3;
4)azd9291的制备
将中间体3在0℃加入到丙烯酸的混酐溶液中进行反应;反应完全后加入水终止反应,经萃取、干燥、浓缩,乙腈重结晶后,得到产物azd9291。
本发明的有益效果:该工艺步骤简单,操作方便,产生的固废少,易于工业化生产,得到的产品纯度较高(高达99.8%),总收率高(高达72%)。
附图说明
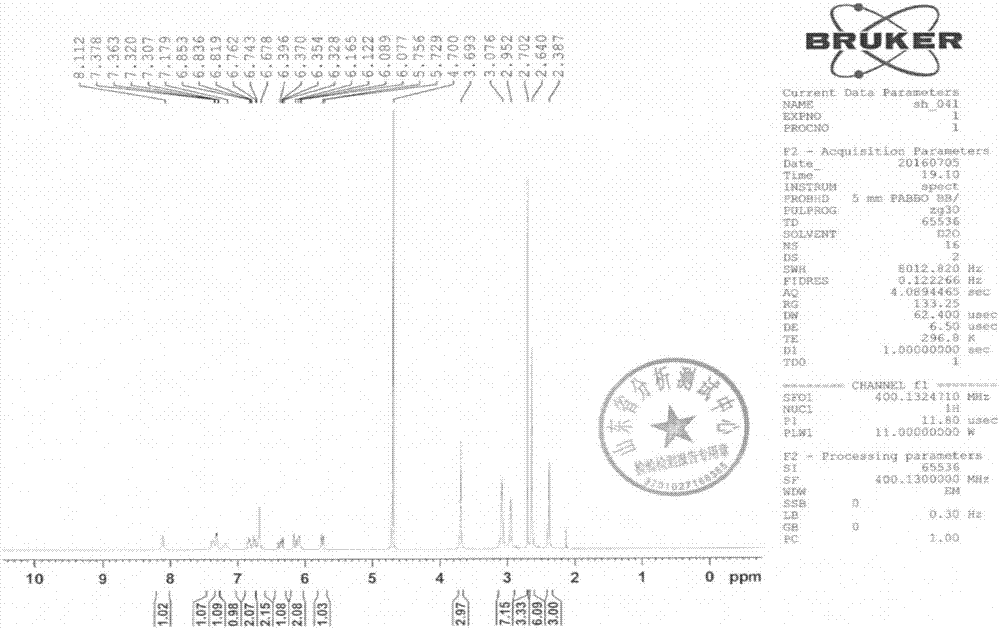
图1为本发明实例3制备的azd9291的1hnmr图;
图2为本发明实例3制备的azd9291的13cnmr图;
图3为本发明实例3制备的azd9291的ir图;
图4为本发明实例3制备的azd9291的质谱图;
图5为本发明实例3制备的azd9291的hplc图谱;
图6为对比例2制备的azd9291的hplc图谱。
具体实施方式
下述实施例用于进一步解释本发明,但不限制本发明的范围。
实施例1:化合物中间体1至中间体3的制备
1)中间体1的制备:
将40g(0.164mol)sm1、30.6g(0.164mol)sm2和62.3g(0.328mol)一水对甲苯磺酸加入到200ml异丁醇中,升温至回流,反应6h,有黄色固体析出,降至室温,过滤,异丙醇洗涤,50℃真空干燥得54.8g黄色固体,产率85.0%。
2)中间体2的制备:
方法1:将50g(0.127mol)中间体1、15.0g(0.147mol)n,n,n′-三甲基乙二胺、1g溴化亚铜和19.65g(0.152mol)n,n-二异丙基乙胺加入到350mln,n-二甲基甲酰胺中,升至100℃反应3h,降至室温过滤除去溴化亚铜,加入80ml水,有大量固体析出,过滤,水洗,45℃真空干燥得57.2g橘红色固体,产率94.9%。
方法2:将50g(0.127mol)中间体1、15.0g(0.147mol)n,n,n′-三甲基乙二胺和19.65g(0.152mol)n,n-二异丙基乙胺加入到350mln,n-二甲基甲酰胺中,升至100℃反应5h,降至室温,加入80ml水,有大量固体析出,过滤,水洗,45℃真空干燥得50.7g橘红色固体,产率84.0%。
3)中间体3的制备:
氮气保护中,将28.5(0.060mol)中间体2、2.5g钯碳加入到500ml甲醇中,25℃下通入氢气常压反应4h,过滤,滤液减压浓缩至干,得26g褐色固体,产率97.3%。
实施例2:azd9291的合成
特戊酰氯3.37g(28mmol)、碳酸钾4.13g(30mmol)加入到60ml1,4-二氧六环中,在0℃将丙烯酸2.02g(28mmol)加入到上述溶液中,搅拌1h,制成混酐溶液,实施例1制备的中间体3为6.17g(13.8mmol)在0℃加入到混酐溶液中,tlc监测原料消失。加入水搅拌0.5h,用乙酸乙酯100ml萃取,有机相无水硫酸钠干燥后,过滤,滤液45℃下真空浓缩得到azd9291粗品。用50ml乙腈重结晶,得到棕色固体6.29g;产率91.3%,纯度99.8%。
中间体2的制备以方法1计算,产品的总收率以71.7%。中间体2的制备以方法2计算,产品的总收率以63.2%。
实施例3:azd9291的合成
甲磺酰氯3.2g(28mmol),碳酸锂2.22g(30mmol)加入到100ml乙腈中,在10℃搅拌,将丙烯酸2.02g(28mmol)加入到上述溶液中,搅拌1.5h,将实施例1制备的中间体3为6.17g(13.8mmol)在0℃加入到混酐溶液中,tlc监测原料消失。加入水搅拌1h,乙酸乙酯100ml萃取,有机相无水硫酸钠干燥后,过滤,滤液45℃下真空浓缩得到azd9291粗品。用50ml乙腈重结晶,得到棕色固体6.35g,产率92.2%,纯度99.8%,hplc图谱如图5所示。中间体2的制备以方法1计算,产品的总收率以72.4%。中间体2的制备以方法2计算,产品的总收率以63.8%。
其1hnmr、13c-nmr、ir和esi-ms图谱分别如图1-4所示,具体数据如下所示。
1hnmr(400mhz,d2o)δppm:2.39(s,1h),2.64(s,6h),2.70(s,3h),3.01(d,7h),3.69(s,1h),5.74(d,1h),6.07-6.16(m,2h),6.33-6.39(m,1h),6.68(s,2h),6.74-6.85(m,2h),7.18(br,1h),7.31-7.38(m,2h),8.11(s,1h)。
(100mhz,d2o):32.30,38.50,42.63,49.30,54.64,56.10,104.17,106.84,110.02,111.99,116.02,120.55,121.11,122.07,123.70,124.76,124.93,128.47,130.35,133.99,137.47,139.29,147.13,153.69,156.87,162.27,166.07。
ir(atr,cm-1)m/z:3244.25(vnh),2932.57(vch),1671.97(c=h)。
esi-ms:m/z500.3[m+h]﹢。
下述对比例对路线1、wo2013014448报道的实验方法最后两步进行验证。
对比例1:中间体3的制备
将22g实施例1制备的中间体2,15.5g铁粉和1.7g氯化铵加入到240ml乙醇和80ml水的混合体系中,加热回流2h,降温至40℃,过滤去除不溶物,滤液45℃下真空浓缩至干,过柱纯化,得到17g固体,产率82.5%。
对比实例2:azd9291的合成:
将丙烯酰氯3.45g(38mmol)的二氯甲烷100ml溶液滴加到冰水浴冷却中的对比例1制备的中间体3为17g(38mmol)和n,n-二异丙基乙胺7.2ml(42mmol)的二氯甲烷250ml溶液中,搅拌1.5h。然后用二氯甲烷250ml稀释,用饱和碳酸氢钠溶液2×250ml洗涤,水层用二氯甲烷萃取2次,合并有机相,无水硫酸镁干燥,过滤,滤液40℃真空浓缩。粗品过柱纯化,得到16.01g棕色固体,产率83.0%,纯度99.3%。
对比实例3:azd9291的合成:
在0℃下向对比例1制备的中间体3为10g(22.4mmol)在thf95ml和水9.5ml的混合溶液中滴加3-氯丙酰氯3.28g(25.59mmol),滴完后,升至室温搅拌15min,加入氢氧化钠3.48g(85.28mmol),将体系升温至65℃保温10h。冷却到室温,加入甲醇40ml和水70ml,室温搅拌过夜,过滤,水洗至中性,50℃真空干燥,得到9.50g固体azd9291,产率85.0%,纯度99.0%。
根据本发明的实施例1~3,本发明得到的纯度≥99.7%的azd9291。根据路线1,得到的azd9291的纯度≥99.0%,并有铁泥固废产生。可见采用本发明的路线,产生的固废少,产品纯度和收率明显提高。同时从中间体2的制备也可以看出:采用溴化亚铜活化中间体1促进亲核取代反应进行,提高反应的速度和收率,反应时间缩短2小时,收率提高≥10%。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种抗肿瘤药物奈拉滨化合物的制造方法与工艺
- 四苯基卟啉连二胺芳基钌化合物及其制备方法和用途与制造工艺
- 一种具抗肿瘤活性的不对称双1‑取代5‑N杂靛红席夫碱类化合物的合成方法与制造工艺
- 一种具抗肿瘤活性的不对称双7‑N杂靛红席夫碱类化合物的合成方法与制造工艺
- 一种具抗肿瘤活性的不对称双取代靛红席夫碱类化合物及其合成方法与制造工艺
- 一种抗肿瘤药物奈拉滨组合物的制造方法与工艺
- 含Micranthanone A用于抗肿瘤的药物的制造方法与工艺
- 一种小分子靶向药物脂质体制剂的制备方法与制造工艺
- 一种5?溴?1H?吡咯并[2,3?b]吡啶的合成方法
- 一种作用于EGFR敏感突变激酶EGFR<sup>L858R</sup>,EGFR<sup>(d746-750)</sup>的6-取代氨基嘌呤类化合物及 ...的制作方法