一种基于转录组测序开发剑麻SSR引物的方法与流程
本发明涉及一种基于转录组测序开发剑麻ssr标记引物的方法,属于分子生物学技术领域。
背景技术:
剑麻属龙舌兰属多年生草本植物,原产于墨西哥等热带、亚热带地区,我国保存的种质资源严重缺乏,仅有100余份。现有的种质资源主要是通过麻园选育、杂交和引种获得,遗传背景模糊,命名极不规范,给种质资源的保存和应用以及育种工作带来了极大的不便。因此需要一种快速有效的方法对现有的种质资源进行遗传多样性分析和资源鉴定。
微卫星dna(microsatellitedna)又称简单重复序列(simplesequencerepeat,ssr)是由1-6个核苷酸为重复单位串联而成的长达几十个核苷酸的重复序列。其侧翼通常都是保守性高的单拷贝序列,因此可根据侧翼序列设计引物进行pcr扩增,由于重复片段大小或重复次数的差异,显示微卫星位点的多态性。与其它分子标记相比,ssr标记具有多态性高、共显性遗传、技术简单、重复性好,特异性强以及操作便利等优点,广泛应用于遗传多样性分析、种质资源鉴定以及遗传图谱构建等领域。但对于ssr标记的应用前提是,必须首先要从该物种中获取重复序列两侧的序列信息,并设计引物,而后才能被应用。
ssr标记可分为基因组ssr(gssr)和表达序列标签ssr(est-ssr),与gssr标记相比,通过表达序列标签(expressedsequencetag,est)序列开发ssr标记更经济,效率更高,而且在不同属内的通用性更好。随着分子生物学和测序技术的快速发展,est序列数据急剧增加,加上生物信息学的飞速发展使得大批量数据处理成为可能,因此给ssr标记的开发提供了大量的序列信息资源和技术支持。并且est-ssr标记是基于某一时期的表达标签序列,能直接与功能基因相关,在分子标记辅助育种如重要性状相关标记关联分析、分离和新基因的鉴定等方面均有极高的应用价值。
目前剑麻的全基因组信息未知,ssr标记缺乏,剑麻ssr标记引物未见报道。随着高通量测序技术和生物信息学的飞速发展,对于无参考基因组的转录组分析,技术非常成熟。因此,利用某一发育时期或逆境胁迫下的剑麻为材料,开展剑麻转录组研究成为可能,这不仅可以挖掘剑麻自身的功能基因,同时对解决剑麻种质资源背景模糊,ssr标记缺乏等实际问题具有十分重要的现实意义。
技术实现要素:
本发明的目的是针对现有技术的不足,提供一种基于转录组测序开发剑麻ssr标记引物的方法,通过转录组测序的方法,获得剑麻某一特定时期的转录组序列,然后通过生物信息学分析软件开发剑麻ssr标记,为后续利用ssr标记引物进行剑麻遗传多样性研究、种质资源的鉴定提供可靠的技术手段。
为实现上述目的,本方法采用的技术方案是:
一种基于转录组测序开发剑麻ssr标记引物的方法,包括如下步骤:
(1)totalrna的提取与转录组文库的构建
采用trizol裂解法提取剑麻叶片总rna,用带有oligo(dt)的磁珠富集mrna,反转录并合成双链cdna,经qiaquickpcrpurificationkit纯化后,在cdna末端添加腺嘌呤核苷进行末端修复并连接测序接头,然后用琼脂糖凝胶电泳回收目标片段,最后对回收片段进行pcr扩增,扩增产物即为转录组文库;
(2)转录组测序及序列质量分析
用illumina2500测序平台进行转录组测序,所得原始序列以fastaq格式保存;由于原始序列含有低质量的序列,在进行数据分析之前先对原始数据进行质量分析和过滤。
(3)转录本组装与ssr分析
利用trinity软件将所得有效序列拼接成一个完整的转录组,作为后续分析的参考序列,取每条基因中最长的转录本作为unigene,采用misa1.0软件对每个unigene进行简单序列重复(ssr)分析;
(4)ssr标记引物设计、扩增与检测
采用引物设计软件primer3进行ssr引物设计,从所设计的ssr引物中随机选取100对引物,首先以热麻1号基因组dna为模板,采用touch-downpcr程序进行扩增,扩增产物经3%琼脂糖凝胶电泳,初步筛选有稳定产物的ssr引物,然后以6份剑麻种质dna为模板,采用touch-downpcr程序,对有稳定产物的ssr引物进行pcr扩增,扩增产物经3%琼脂糖凝胶电泳,进一步筛选多态性的ssr标记引物,检测ssr标记引物的有效性。
步骤(1)所述的接头序列为seqidno.1-seqidno.2。
步骤(2)所述的低质量序列是指不确定碱基n比例大于10%的序列和低质量碱基含量大于50%的序列;低质量碱基指q≤5的碱基。
步骤(4)所述的ssr标记筛选参数为:单核苷酸的重复次数大于或等于10次,二核苷酸重复次数大于或等于6次,三核苷酸、四核苷酸、五核苷酸、六核苷酸的重复次数大于或等于5次。
步骤(4)所述的ssr标记引物设计参数为:引物长度18-25bp,退火温度tm56-65℃,预期产物长度为100-300bp。
步骤(4)所述的touch-downpcr扩增程序为:首先94℃15s,60℃15s,72℃30s,16个循环,每个循环退火温度降低0.7℃;然后进入下一个扩增阶段:94℃15s,50℃15s,72℃30s,15个循环,最后72℃延伸60min,扩增产物4℃保存备用。
步骤(4)所述的有多态性的18对ssr标记引物组序列为seqidno.3-seqidno.38。
步骤(4)所述的6份剑麻种质为热麻1号、h.11648、番麻、普通剑麻、桂幅4号和广西76416。
本发明的有益效果为:
(1)通过转录组测序的方法,获得剑麻某一特定时期的转录组序列,然后通过生物信息学分析软件开发剑麻ssr标记,开发了一种基于转录组测序开发剑麻ssr标记引物的方法,开发效率更高。
(2)本发明为剑麻ssr标记开发提供了一条新的有效途径,填补了目前剑麻ssr标记引物稀缺的空白。
附图说明
图1剑麻ssr标记引物开发流程图;
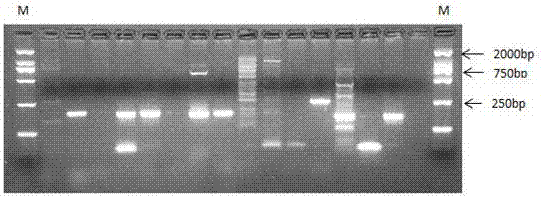
图2琼脂糖凝胶电泳检测部分剑麻ssr标记引物扩增产物;
图3剑麻ssr标记引物多态性筛选。
具体实施方式
下面通过实例对本发明做进一步详细说明,这些实例仅用来说明本发明,并不限制本发明的范围。未加特殊说明,转录组文库构建与测序均按标准流程进行,所有试剂盒操作均按试剂盒说明书进行,所有的试剂均为生物试剂。
本发明所提供的18对ssr标记引物组均来自热麻1号转录组序列,其引物核酸序列分别为seqidno.3-seqidno.38。所用的植物材料为热麻1号,病原菌为烟草疫霉。
一.rna提取与转录组文库构建
取保存于本实验室的烟草疫霉接种于马铃薯培养基(pda)上,28℃培养1周后,接种热麻1号叶片,取不同接种时间的热麻1号叶片进行转录组测序。具体步骤如下:
用灭菌的大头针将叶片正面刺伤,取直径为5mm的菌饼,将菌饼的菌丝生长面贴在伤口的位置,用无菌湿棉花保湿,然后用保鲜膜包裹叶片,置于25-30℃条件下培养,并分别在接种前、接种24小时、36小时、48小时和72小时取叶片,液氮速冻后保存于-80℃备用。
采用trizol裂解法提取热麻1号叶片总rna,具体操作按说明书进行。采用nanodrop-2000分光光度计和bioanalyzer2100生物分析仪对rna质量进行检测。质量合格的rna样品将用于转录组文库构建。文库构建采用illumina公司的文库构建试剂盒进行。首先用带有oligo(dt)的磁珠富集mrna,并将mrna片段化并反转录成第一链cdna,然后合成双链cdna,经qiaquickpcrpurificationkit纯化后,在cdna末端添加腺嘌呤核苷进行末端修复并连接测序接头,然后用琼脂糖凝胶电泳回收目标片段,最后对回收片段进行pcr扩增,扩增产物即为转录组文库,将用于后续的转录组测序。
二.测序与转录组分析
采用illumina2500测序平台进行转录组测序,所得原始序列以fastaq格式保存。由于原始序列含有接头污染和低质量的序列,因此,为了防止这些序列对后续分析产生不利影响,在进行序列分析前,先要去掉测序时的接头序列,不确定碱基(n)比例大于10%的序列以及低质量碱基(q≤5)含量大于50%的序列,所得序列即为有效的转录组序列,并用于后续的序列分析。
利用trinity(版本为v2012-06-08,参数为默认参数)软件将所得有效序列拼接成一个完整的转录组,作为后续分析的参考序列,取每条基因中最长的转录本作为unigene,热麻1号总计获得了103,326个转录本和70,110条unigene序列。转录本和unigene平均长度分别为726bp和645bp。转录本和unigene具体数目分布如表1所示。
表1转录本和unigene拼接长度频数分布
三.ssr位点查找及引物设计
以热麻1号70,110条unigene为材料,利用misa1.0软件对unigene序列进行ssr位点查找,查找含有1、2、3、4、5和6碱基重复的ssr位点,且重复次数依次不小于10、6、5、5、5和5次。总计查找到了13,175个ssr位点,ssr密度分布出现频率最高的依次为单核苷酸重复、二核苷酸重复和三核苷酸重复,分别为5001个、4339个和3676个。使用primer3.0软件对ssr候选位点进行引物设计。引物设计参数为,引物长度18-26bp,gc含量40%-60%,退火温度tm值55-65℃(上下游引物的tm值相差不能大于5℃);pcr目标产物在100-300bp;尽量避免产生引物二聚体,发夹结构、错配等。总计设计了11,946对ssr引物。其中长度为20bp的引物最多,为10,270对,占总引物的85.97%。
四.ssr标记引物的有效性验证
从10,270对引物中随机选取100对引物进行pcr扩增,筛选有稳定产物条带的ssr标记引物,然后以6份剑麻种质基因组dna为模板,对有稳定产物的ssr标记引物进行扩增,筛选有多态性的ssr标记引物,检测ssr标记引物的有效性。引物由生工生物工程(上海)有限公司合成,引物序列见核苷酸序列表(序列分别如seqidno.3-42)。
序列说明:sedidno.3-38为热麻1号ssr标记引物序列,其中sedidno.3和4为一对ssr引物,sedidno.5和6为一对ssr引物,依次类推,18对引物的退火温度和扩增产物大小见表2。
表218对剑麻ssr引物的退火温度和扩增产物大小
1.dna提取
采用天泽公司的柱式植物dnaout试剂盒提取热麻1号、h.11648、番麻、普通剑麻、桂幅4号和广西76416等6份剑麻种质的基因组dna。取叶片1克,经液氮速冻后快速研磨成粉末,先加入750µl65℃预热的裂解液,充分混匀后65℃预热5-10分钟,室温13,000rpm离心5min,取上清液500µl分别进行抽提、漂洗后过柱,最后用100µl洗脱液洗脱2次,洗脱液即为提取的dna,4℃保存备用。
2.pcr反应体系的建立
20µlpcr反应体系中各组份的浓度及使用量:2×novataq-pluspcrforestmix(江苏愚公生命科技有限公司):2µl;引物(10μmol/l)f:0.2µl,r:0.2µl;dna:50ng(1µl);灭菌ddh2o:8.6µl。novataq-pluspcrforestmix购自江苏愚公生命科技有限公司。
touch-downpcr扩增程序:94℃(15s),60℃(15s)(△℃=-0.7,即每增加一个循环,退火温度降低0.7℃),72℃(30s)(16个循环),然后进入下一个扩增阶段,94℃(15s),50℃(15s),72℃(30s)(15个循环),最后72℃(60min),扩增产物4℃保存备用。
3.pcr产物的检测
采用3%琼脂糖凝胶(100ml1xtbe缓冲液中加入3g琼脂糖)电泳检测pcr产物。将20µlpcr产物全部上样,先150v电泳10min,然后120v电泳40min,电泳结果于凝胶成像系统拍照保存。
本发明采用touch-downpcr程序,从100对热麻1号ssr标记引物中,用琼脂糖凝胶电泳筛选出了66对有稳定产物条带的ssr标记引物(附图2),然后用6份剑麻种质,从66对ssr标记引物中,筛选出了18对有多态性的ssr标记引物(附图3),证实了通过转录组测序开发剑麻ssr标记引物是切实可行的。
序列表
<110>中国热带农业科学院南亚热带作物研究所
<120>一种基于转录组测序开发剑麻ssr引物的方法
<141>2017-07-14
<160>38
<170>siposequencelisting1.0
<210>1
<211>58
<212>dna
<213>人工序列
<400>1
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58
<210>2
<211>57
<212>dna
<213>人工序列
<400>2
gatcggaagagcacacgtctgaactccagtcacatctcgtatgccgtcttctgcttg57
<210>3
<211>20
<212>dna
<213>人工序列
<400>3
tcgcgtgcaccaacaatttc20
<210>4
<211>20
<212>dna
<213>人工序列
<400>4
gtagcggatgtaggagacgc20
<210>5
<211>20
<212>dna
<213>人工序列
<400>5
tgcttcgactcctgcttctg20
<210>6
<211>20
<212>dna
<213>人工序列
<400>6
agtggtggccgtggaaatag20
<210>7
<211>20
<212>dna
<213>人工序列
<400>7
gtgtgtgtgtgtgtgttggg20
<210>8
<211>20
<212>dna
<213>人工序列
<400>8
ggccgaatcctttccactca20
<210>9
<211>20
<212>dna
<213>人工序列
<400>9
cgctcgtcctcttctttcgt20
<210>10
<211>23
<212>dna
<213>人工序列
<400>10
tcccatccaatagtccccca20
<210>11
<211>20
<212>dna
<213>人工序列
<400>11
ggtcatgatgaaggccacca20
<210>12
<211>20
<212>dna
<213>人工序列
<400>12
gcgaacctgcattgctgaat20
<210>13
<211>20
<212>dna
<213>人工序列
<400>13
ccttaggctccctgctgttc20
<210>14
<211>23
<212>dna
<213>人工序列
<400>14
ccacaagagccgctaccatc20
<210>15
<211>20
<212>dna
<213>人工序列
<400>15
aacaaccagagcccaaacca20
<210>16
<211>20
<212>dna
<213>人工序列
<400>16
ggggaggtggtttggtgatc20
<210>17
<211>20
<212>dna
<213>人工序列
<400>17
ggttagggttcttggtgggg20
<210>18
<211>20
<212>dna
<213>人工序列
<400>18
gcttcctgatcttcttgttggc22
<210>19
<211>20
<212>dna
<213>人工序列
<400>19
aaaatccatgaggcggctga20
<210>20
<211>20
<212>dna
<213>人工序列
<400>20
tagtagctaggcccaggcaa20
<210>21
<211>20
<212>dna
<213>人工序列
<400>21
acagcacgagaaatgagctca21
<210>22
<211>20
<212>dna
<213>人工序列
<400>22
ccgatccggcgtaattctct20
<210>23
<211>20
<212>dna
<213>人工序列
<400>23
gccttctcccacggaatcaa20
<210>24
<211>20
<212>dna
<213>人工序列
<400>24
tgtggagtgtgatgggagtg20
<210>25
<211>20
<212>dna
<213>人工序列
<400>25
tggagggtgatggatagggg20
<210>26
<211>20
<212>dna
<213>人工序列
<400>26
gatgaggccatcgttttggt20
<210>27
<211>20
<212>dna
<213>人工序列
<400>27
agagttgccagatgtgtgca20
<210>28
<211>20
<212>dna
<213>人工序列
<400>28
aggtgggattcttgcggatg20
<210>29
<211>20
<212>dna
<213>人工序列
<400>29
tcaaaagcaacgaacagcgg20
<210>30
<211>20
<212>dna
<213>人工序列
<400>30
cgacttcctcatcgatgcga20
<210>31
<211>20
<212>dna
<213>人工序列
<400>31
gcaggccctgtagtttgact20
<210>32
<211>20
<212>dna
<213>人工序列
<400>32
ttcgtgcccagtttctcctc20
<210>33
<211>20
<212>dna
<213>人工序列
<400>33
atcttcaggtttccgctgca20
<210>34
<211>20
<212>dna
<213>人工序列
<400>34
ccgagagagagcgagagaga20
<210>35
<211>20
<212>dna
<213>人工序列
<400>35
accgcattcatcggtctctc20
<210>36
<211>20
<212>dna
<213>人工序列
<400>36
ggtcctcgctctgatcttgg20
<210>37
<211>20
<212>dna
<213>热人工序列
<400>37
attgcttgaagatggctgct20
<210>38
<211>20
<212>dna
<213>人工序列
<400>38
catgcataccttcctccccc20
- 还没有人留言评论。精彩留言会获得点赞!