抗新生血管的化合物、其中间体及其用途的制作方法
本申请是2014年04月09日提交的发明名称为“抗新生血管的化合物、其中间体及其用途”的第201480000555.4号中国专利申请(国际申请号pct/cn2014/074977)的分案申请。本发明涉及一种新生血管抑制剂类和/或蛋白激酶抑制剂类化合物和所述化合物的用途。
背景技术:
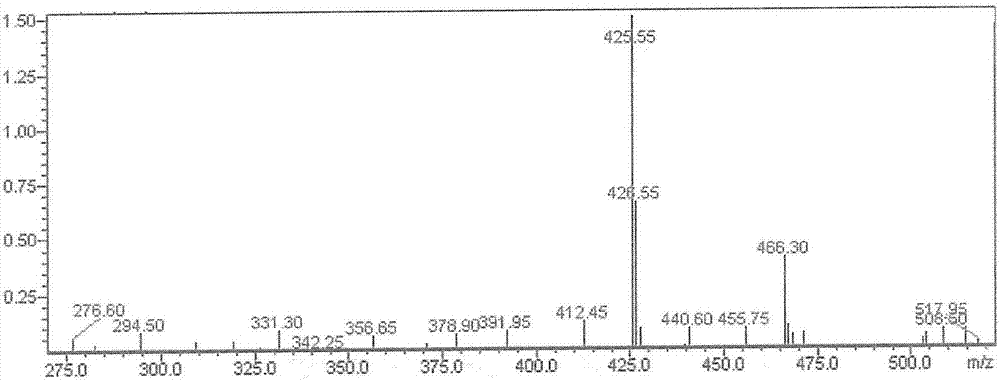
:新生血管(angiogenesis),是从已有血管发芽生成新血管的过程。这一过程与血管内皮细胞迁移和增殖相关。新生血管与多种人类重大疾病有关,如恶性肿瘤。目前发现,眼部血管新生性疾病,包括年龄相关性黄斑变性(amd)、糖尿病视网膜病变、新生血管性青光眼等,这类疾病的共同特点都在于眼部新生血管的异常增生(金晓,等,抗vegf药物在眼科疾病中的应用及机制研究进展,中外医疗,2012年)。其中,黄斑变性主要有干性和湿性两种,湿性黄斑变性(amd)的特点是,脉络膜的新生血管进入视网膜下以及继而发生的出血、渗出及水肿等病理变化。湿性黄斑变性将迅速丧失视力,较干性更为严重。目前,在湿性黄斑变性的治疗方面已有较好的进展。早期的激光烧烁止血被血管内皮因子拮抗剂所代替,但因后者效果不佳,很快被光动力疗法所取代。光动力疗法虽然提高了疗效,但仍不理想。近年又出现了新的血管内皮因子拮抗剂——雷珠单抗(lucentis),是一种人源性vegf亚型单克隆抗体片段的重组体,可减少新生血管生成。2006年,该药物被美国fda批准用于治疗湿性黄斑变性,疗效良好;同时,目前也发现该类抗vegf药物对糖尿病视网膜病变、新生血管性青光眼有治疗作用。但由于雷珠单抗为抗体药,价格极高,它还不能在全世界普及。因此,研究疗效良好、价格低廉的小分子新生血管抑制剂药物是当今国际制药界激烈竞争的焦点。蛋白激酶(proteinkinases)又称蛋白质磷酸化酶(proteinphosphakinase),是一类催化蛋白质磷酸化反应的酶。它能把腺苷三磷酸(atp)上的γ-磷酸转移到蛋白质分子的氨基酸残基上,例如某些丝氨酸、苏氨酸或酪氨酸残基的羟基上,从而改变蛋白质、酶的构象和活性。蛋白质的磷酸化是多种信号传导途径的重要环节,细胞内大部分重要的生命活过程都离不开蛋白质磷酸化。蛋白激酶分为5类:蛋白丝氨酸/苏氨酸激酶、蛋白酪氨酸激酶、蛋白组氨酸激酶、蛋白色氨酸激酶和蛋白天冬氨酰基/谷氨酰基激酶。蛋白激酶在细胞过程的调节和维持中起到了重要作用,在许多疾病状态中都观察到了激酶活性异常,所述疾病状态包括:恶性肿瘤,免疫性疾病、心血管疾病、糖尿病、感染性疾病、关节炎和其它免疫紊乱、神经系统疾病如老年性痴呆症、阿默海茨症(ad)等,现已发与超过400种人类疾病与蛋白激酶相关。其中,vegfr(血管内皮细胞生长因子受体)家族成员为受体酪氨酸激酶,如vegfr1、vegfr2等,该类受体在恶性肿瘤的生长、转移中以及血管增生性疾病(如黄斑变性、肿瘤)等疾病的发展过程中有重要影响。pdgfr(血小板衍生生长因子受体)家族成员为受体酪氨酸激酶,如pdgfrα和pdgfrβ以及集落刺激因子1受体、干细胞生长因子受体kit等,目前也发现该类激酶与肿瘤的发生、发展有密切联系。如pdgfr的异常表达已在黑色素瘤、脑膜瘤、神经内分泌肿瘤、卵巢癌、前列腺癌、肺癌和胰腺癌中有发现,kit的异常活化则是许多肿瘤发生、发展的直接诱因。fgfr家族成员(成纤维细胞生长因子受体),包括fgfr1、fgfr2等,它们与癌症有着密切关系,如fgfr2的异常活化已经在子宫内膜癌、宫颈癌、乳腺癌、肺癌以及胃癌中发现。src激酶家族是具有酪氨酸蛋白激酶活性的蛋白质,它作为一个癌基因蛋白起初发现与rous肉瘤逆转录病毒,目前已发现对src抑制,可对癌症或其他疾病有一定的治疗或改善作用。p38丝裂原活化蛋白激酶(mapk)通路是细胞内应激反应信号通路,与炎症反应密切相关。因此,仍存在开发新型的蛋白激酶抑制剂的需求。技术实现要素:本发明的目的在于提供了一种作为新生血管抑制剂和/或蛋白激酶抑制剂的新型化合物,用于制备该化合物的中间体化合物,以及该化合物的用途。本发明提供了如式i所示的化合物及其药学上可接受的盐或前体药物,其结构式如下:其中,r1选自h、氨基、羟基或巯基;r2选自h、氨基、羟基、巯基或-(ch2)nnhr8,其中,n=1-5,r8为h或c1-3的烷基;r3选自h或c1-6烷基;r4、r5、r6分别独立选自h、卤素、c1-6的烷基或卤素取代烷基;r7选自h、c1-6的烷基或卤素;或者,r2、r3与其相连的碳原子共同构成取代或非取代的含1-2个杂原子的五元或六元环,所述杂原子为n、o或s,其取代基为c1-6的烷基。进一步地,r2选自h、氨基或-(ch2)nnhr8,其中,n=1-3,r8为h或c1-2的烷基;r3选自h或c1-2烷基;r4、r5、r6分别独立选自卤素、c1-2的烷基或卤素取代烷基;r7选自h或卤素;或者,r2、r3与其相连的碳原子共同构成取代或非取代的含1个n的五元或六元环,取代基为c1-3的烷基。更进一步地,r2选自氨基或-(ch2)nnhr8;或者,r2、r3与其相连的碳原子共同构成取代或非取代的含1个n的五元或六元环,取代基为c1-3的烷基。更进一步地,所述卤素为f或cl。根据本发明的优选实施方式,r1、r2和r3中至少一个为氨基,其余为h;r4、r5和r6相同且选自f或cl;r7为h。优选地,所述化合物为:本发明化合物的制备方法可以是任何合适的方法。本发明的化合物可优选通过式ii所示的中间体化合物进行制备。因此,本发明还提供一种用于制备式i化合物的如下式ii所示的中间体化合物:其中,r4、r5、r6分别独立选自h、卤素、c1-6的烷基或卤素取代烷基;r7选自h、c1-6的烷基或卤素。进一步地,r4、r5、r6分别独立选自卤素、c1-2的烷基或卤素取代烷基;r7选自h或卤素。更进一步地,r4、r5和r6相同、且选自f或cl;r7为h。所述式ii所示中间体化合物的制备方法可包括如下步骤:其中,r4、r5、r6和r7的定义同上。本发明式i化合物的优选制备方法包括如下反应步骤:其中,r1、r2和r3的定义同上。进一步地,本发明优选实施方式的化合物的制备方法包括如下反应步骤:(1)式7所示中间体化合物的合成:其中,r4、r5、r6的定义同上;(2)目标化合物的合成:其中,r1、r2和r3的定义同上。本发明还提供了上述化合物、其药学上可接受的盐或前体药物在制备用于抑制异常增生的新生血管的药物中的用途。其中,所述用于抑制异常增生的新生血管的药物为血管内皮生长受体2(vegfr2)抑制剂。进一步地,所述用于抑制异常增生的新生血管的药物是抗眼部新生血管的药物。更进一步地,所述用于抑制异常增生的新生血管的药物是脉络膜新生血管抑制剂。其中,所述用于抑制异常增生的新生血管的药物是用于治疗或预防湿性黄斑变性、糖尿病视网膜病变或新生血管性青光眼的药物。其中,优选所述药物为眼用制剂。进一步地,所述眼用制剂为滴眼剂、眼膏剂或眼用注射液。更进一步地,所述眼用注射液为玻璃体内注射液。本发明还提供了上述化合物、其药学上可接受的盐或前体药物在制备治疗与异常增生的新生血管相关的疾病的药物中的用途。其中,所述与异常增生新生血管相关的疾病为血管内皮生长受体2(vegfr2)异常导致的疾病。进一步地,所述与异常增生的新生血管相关的疾病是与眼部新生血管相关的疾病。进一步地,所述与异常增生的新生血管相关的疾病是与脉络膜新生血管相关的疾病。更进一步地,所述与异常增生的新生血管相关的疾病是湿性黄斑变性、糖尿病视网膜病变或新生血管性青光眼。本发明还提供一种抑制异常增生的新生血管或者与新生血管相关疾病的治疗方法,包括对需要治疗的患者给药有效量的本发明的化合物、其药学上可接受的盐或前体药物,或下文所述的药物组合物。进一步地,所述抑制异常增生的新生血管是指抑制眼部异常增生的新生血管;所述与异常增生的新生血管相关的疾病是与眼部新生血管相关的疾病。更进一步地,所述抑制异常增生的新生血管是指抑制脉络膜新生血管;所述与异常增生的新生血管相关的疾病是与脉络膜新生血管相关的疾病。其中,所述抑制异常增生的新生血管或者与异常增生的新生血管相关疾病的治疗方法具体是指治疗或预防湿性黄斑变性、糖尿病视网膜病变或新生血管性青光眼的方法。所述给药是指直接向眼部外用给药或者进行眼玻璃体内或结膜下注射。本发明还提供了上述化合物、其药学上可接受的盐或前体药物在制备蛋白激酶抑制剂类药物中的用途。本发明还提供了上述化合物、其药学上可接受的盐或前体药物在制备治疗蛋白激酶异常导致的疾病的药物中的用途。本发明还提供一种蛋白激酶异常导致的疾病的治疗方法,包括对需要治疗的患者给药有效量的本发明的化合物、其药学上可接受的盐或前体药物,或下文所述的药物组合物。所述蛋白激酶为vegfr2、pdgfr-β、kit、aurora-b、fgfr2、src、jak2或p38-α,优选为vegfr2、kit或pdgfr-β。所述蛋白激酶异常导致的疾病是指炎症或恶性肿瘤。本发明还提供了一种药物组合物,含有有效量的上述化合物及其药学上可接受的盐或前体药物。进一步地,所述组合物为眼用制剂。除了本发明提供的上述化合物以外,所述眼用制剂中还可以包含其他已知具有相似治疗用途的药物。进一步地,所述眼用制剂为滴眼剂、眼膏剂或眼用注射液。更进一步地,所述眼用注射液为玻璃体内或结膜下注射液。在本领域中已知的方法可制备本发明化合物的盐。用酸处理,或与合适的阴离子交换剂,与上述化合物可成盐。本发明的化合物的药学上可接受的盐,可以从上述化合物具有碱性氮原子的有机或无机酸加成盐。优选地,合适的无机酸包括但不限于,氢卤酸(如盐酸),硫酸,或者磷酸。优选地,合适的有机酸包括,但不限于,羧酸,磷酸,磺酸或氨基羧酸,例如乙酸,丙酸,辛酸,癸酸,十二烷酸,羟基乙酸,乳酸,富马酸,琥珀酸,己二酸,庚二酸,辛二酸,壬二酸,苹果酸,酒石酸,柠檬酸,氨基酸,如谷氨酸或天冬氨酸,马来酸,羟基酸,甲基马来酸,环己烷羧酸,金刚烷羧酸,苯甲酸酸,水杨酸,4氨基水杨酸,邻苯二甲酸,苯乙酸,扁桃酸,肉桂酸,甲烷或乙烷磺酸磺酸,2-羟基乙磺酸,乙烷-1,2-二磺酸,苯磺酸,2-萘磺酸,1,5-萘二磺酸,2-甲基苯磺酸,对甲基苯磺酸,乙基硫酸,十二烷基硫酸的酸,n环己基氨基乙酸,n-甲基-n-乙基-n-丙基-氨基磺酸,或其它有机酸,如抗坏血酸。另外,它也可以药学上不可接受的盐用于分离或纯化中,例如苦味酸盐或高氯酸盐。但是,用于治疗用途的,只能是药学上可接受的盐或游离化合物,以适用的药物制剂的形式。本发明所述药学上可接受的前体药物,是指所述化合物经过化学结构修饰后得到的在体内经酶或非酶的转化释放出活性成分而发挥药效的化合物。本发明的一种实施方式中,还包括了同位素标记的上述化合物或其药学上可接受的盐,所述同位素标记化合物是指与本文中所列化合物相同,但是其中的一个或多个原子被另一个原子取代,该原子的原子质量或质量数不同于自然界中常见的原子质量或质量数。可以引入化合物中的同位素包括氢、碳、氮、氧、硫,即2h,3h、13c、14c、15n、17o、18o、35s。含有上述同位素和/或其它原子同位素的化合物及其立体异构体,以及该化合物、立体异构体的可药用的盐均应包含在本发明范围之内。本发明中的关键中间体和化合物进行分离和纯化,所使用的方式是有机化学中常用的分离和纯化方法且所述方法的实例包括过滤、萃取、干燥、旋干和各种类型的色谱。可选择地,可以使中间体不经纯化即进行下一步反应。本发明化合物具有良好的抗异常增生新生血管作用,该类化合物是通过抑制vegfr2(又名kdr)产生活性。该类化合物可用于对异常增生新生血管、kdr、fgfr2等蛋白激酶异常所致疾病的治疗,如湿性黄斑变性、炎症、恶性肿瘤等。附图说明图1是化合物2-1的质谱图。图2是化合物2-2的质谱图。图3是化合物2-2的核磁图。图4是化合物2-3的质谱图。图5是化合物2-4的质谱图。图6是化合物2-5的质谱图。图7显示的是斑马鱼脊椎血管发育的荧光显微照片,其中7a是斑马鱼脊椎血管发育正常(阴性对照)的荧光显微照片;7b是用1um化合物2-2处理后,斑马鱼脊椎血管发育被抑制(100%)的荧光显微照片。图8显示的是本发明化合物2-1、2-2和2-4(即图中kdr2)在体外对vegfr2抑制作用的结果图。图9显示的是抑制脉络膜新生血管形成的zeiss荧光显微照片,其中9a是化合物2-2在1um浓度下显著抑制脉络膜新生血管形成的zeiss荧光显微照片;9b是以pbs作为阴性对照的zeiss荧光显微照片。图10是本发明化合物的剂量效应曲线,其中10a为星形孢菌素(staurosporine)的阳性对照;10b为化合物2-1;10c为化合物2-2。图11显示的是化合物2-2对小鼠眼部角膜新生血管影响的照片,其中11a为用化合物2-2处理的小鼠的右眼;11b为用pbs作为对照处理的小鼠的左眼。图12显示的是化合物2-2对兔眼部角膜新生血管影响的照片,其中12a为用化合物2-2处理;12b为用pbs作为对照处理;12c为用化合物2-1处理。具体实施方式本发明提供了包括但不限于以下实施方案:1、如式i所示的化合物及其药学上可接受的盐或前体药物:其中,r1选自h、氨基、羟基或巯基;r2选自h、氨基、羟基、巯基或-(ch2)nnhr8,其中,n=1-5,r8为h或c1-3的烷基;r3选自h或c1-6烷基;r4、r5、r6分别独立选自h、卤素、c1-6的烷基或卤素取代烷基;r7选自h、c1-6的烷基或卤素;或者,r2、r3与其相连的碳原子共同构成取代或非取代的含1-2个杂原子的五元或六元环,所述杂原子为n、o或s,其取代基为c1-6的烷基。2、根据实施方案1所述的化合物及其药学上可接受的盐或前体药物,其特征在于:r2选自h、氨基或-(ch2)nnhr8,其中,n=1-3,r8为h或c1-2的烷基;r3选自h或c1-2烷基;r4、r5、r6分别独立选自卤素、c1-2的烷基或卤素取代烷基;r7选自h或卤素;或者,r2、r3与其相连的碳原子共同构成取代或非取代的含1个n的五元或六元环,取代基为c1-3的烷基,优选地,所述卤素为f或cl。3、根据实施方案2所述的化合物及其药学上可接受的盐或前体药物,其特征在于:r2选自氨基或-(ch2)nnhr8;或者,r2、r3与其相连的碳原子共同构成取代或非取代的含1个n的五元或六元环,取代基为c1-3的烷基。4、根据实施方案2所述的化合物及其药学上可接受的盐或前体药物,其特征在于:r1、r2和r3中至少一个为氨基,其余为h;r4、r5和r6相同且选自f或cl;r7为h。5、根据实施方案1-4任意一项所述的化合物及其药学上可接受的盐或前体药物,其特征在于:所述化合物为如下之一:6、实施方案1-5任意一项所述化合物及其药学上可接受的盐或前体药物在制备用于抑制异常增生的新生血管的药物或制备治疗与异常增生的新生血管相关疾病的药物中的用途。7、根据实施方案6所述的用途,其特征在于:所述用于抑制异常增生的新生血管的药物为vegfr2抑制剂。8、根据实施方案6所述的用途,其特征在于:所述用于抑制异常增生的新生血管的药物是抗眼部新生血管的药物,所述与异常增生的新生血管相关疾病是与眼部新生血管相关的疾病。9、根据实施方案6所述的用途,其特征在于:所述用于抑制异常增生的新生血管的药物是脉络膜新生血管抑制剂,所述与异常增生的新生血管相关疾病是与脉络膜新生血管相关的疾病。10、根据实施方案8或9所述的用途,其特征在于:所述用于抑制异常增生的新生血管的药物是用于治疗或预防湿性黄斑变性、糖尿病视网膜病变或新生血管性青光眼的药物,所述与异常增生的新生血管相关疾病是湿性黄斑变性、糖尿病视网膜病变或新生血管性青光眼。11、实施方案1-5任意一项所述化合物及其药学上可接受的盐或前体药物在制备蛋白激酶抑制剂类药物或制备治疗蛋白激酶异常导致的疾病的药物中的用途。12、根据实施方案11所述的用途,其特征在于:所述蛋白激酶为vegfr2、pdgfr-β、kit、aurora-b、fgfr2、src、jak2或p38-α,优选为vegfr2、kit或pdgfr-β。13、一种药物组合物,其特征在于:含有有效量的实施方案1-5任意一项所述化合物及其药学上可接受的盐或前体药物。14、根据实施方案13所述的药物组合物,其特征在于所述组合物为眼用制剂。15、一种由下式ii表示的中间体化合物:其中r4、r5和r6分别独立选自h、卤素、c1-6的烷基或卤素取代烷基,r7选自h、c1-6的烷基或卤素;优选r4、r5和r6分别独立选自卤素、c1-2的烷基或卤素取代烷基,r7选自h或卤素;更优选r4、r5和r6相同、且选自f或cl;r7为h。实施例1中间体化合物7的制备第1步:氮气下0℃,将乙基乙烯基醚(50克,0.69mol)缓慢与草酰氯(132.3克,1.04摩尔)混合.将混合物在0℃下搅拌2小时,然后加热至室温并维持12小时。蒸馏除去过量的草酰氯,将残余物在120℃加热30分钟,然后通过真空蒸馏纯化,得到纯的化合物1(49克,产率:53%),为浅黄色油状物。步骤2:亚硫酰氯(66ml,0.91mol)在0℃、搅拌下加入到化合物2(50克,0.365mol)的甲醇(1l)溶液中。加完后,将反应混合物搅拌,在40℃过夜。用tlc(石油醚/乙酸乙酯(pe/ea)=1:1)检测反应完成后,将混合物蒸发,残留物通过添加na2co3调节ph为8,然后用乙酸乙酯(ea)萃取。有机相用饱和食盐水洗涤,用无水硫酸钠干燥并浓缩,得到化合物3(51.7克,收率:93.8%),为棕色固体,不经进一步纯化直接用于下一个步骤。步骤3:氮气下0℃,将化合物3(10克,66mmol)溶于100ml二氯甲烷(dcm)溶液中,加入吡啶(9.06克,119mmol)和溶于dcm(40ml)中的化合物1(15克,112mmol)。然后将混合物温热至室温并搅拌2.5小时。用tlc(pe/ea=1:1)检测反应完成后,将混合物依次用水、盐水洗涤,用无水na2so4干燥并浓缩,得到粗产物。该产物通过色谱层析(用dcm/甲醇=20:1洗脱)纯化,得到纯的化合物4(9.67克,收率:59%;),为白色固体。步骤4:在0℃下,化合物4(9.67克0.039mol)加入到175毫升浓h2so4中。然后将反应混合物在室温下搅拌6小时。用tlc(pe/ea=1:1)检测反应完成后,将混合物倒入冰-水中。滤出沉淀物,并用乙醚(et2o)洗涤。用乙醇重结晶,得到化合物5(2克,产率:25%),为米色固体。步骤5:0℃、搅拌下,向化合物5(2.0克,9.85毫摩尔)的甲醇(meoh,20ml)溶液中,逐滴加入2nnaoh(24.6毫升,49.25毫摩尔)。加完后,将反应混合物在室温下搅拌过夜。用tlc(dcm/甲醇=15:1)检测,反应完成后,将混合物蒸发,通过加入1n盐酸至ph为2,将残余物酸化,所形成的沉淀物通过过滤收集,然后干燥,得到纯的化合物6(0.8克,43%),为白色固体。步骤6:化合物6(0.8克,4.22毫摩尔),3-(三氟甲基)苯胺(0.75克,4.64毫摩尔),hatu(c10h15f6n6op,1.92克,5.06毫摩尔)和n,n-二异丙基乙胺(dipea,1.64克,12.66毫摩尔)混合于dmf(10ml)中,混合物在氮气气氛、室温下搅拌过夜。用tlc(dcm/甲醇=10:1)检测反应完成后,将反应混合物用水稀释,用ea萃取。用盐水洗涤有机相,用无水na2so4干燥并浓缩,得到粗产物。将粗产物通过硅胶色谱法纯化,得到纯的化合物7(0.85g,收率:60.6%),为黄色固体。实施例2化合物2-1(本文中亦称series2-1)的制备化合物8的制备:将化合物8a(20克,0.15摩尔)和二甲基氨基吡啶(dmap,1.9克,15.4毫摩尔)加入四氢呋喃(thf,750毫升)中搅拌,向溶液中滴加二叔丁基二碳酸酯((boc)2o,75克,0.34摩尔)。滴加结束后,将反应混合物在室温下搅拌过夜。用tlc(pe/ea=3:1)检测反应完成后,将反应混合物浓缩,然后重新悬浮于pe/ea(10:1,200毫升)混合溶剂中,过滤,得到纯的化合物8(50克,100%),为白色固体。化合物9的制备:在氮气下,化合物7(30毫克,0.09毫摩尔)和碳酸铯(58.6毫克,0.18毫摩尔)混合于二甲亚砜(dmso,1ml)溶液中,混合物在室温下搅拌一个半小时,然后加入化合物8(32.8毫克,0.009毫摩尔)。将所得的反应混合物搅拌18小时,用tlc(dcm/甲醇=15:1)检测,反应未完成。将反应混合物在80℃再搅拌5小时。然后将反应混合物用水稀释,用ea萃取。用盐水洗涤有机相,用无水na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物9(10毫克,17.8%),为黄色固体。化合物2-1的制备:化合物9(10毫克,0.019毫摩尔)和三氟乙酸(tfa,0.2ml)的混合物,在氮气、室温下搅拌1小时。用tlc(dcm/甲醇=20:1)检测反应完成。将反应混合物用碳酸钠碱化,用dcm萃取。用盐水洗涤有机相,用无水na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物2-1(4.3毫克,53.75%),为黄色固体,其质谱图参见图1。实施例3化合物2-2(本文亦称series2-2)的制备化合物10的制备:化合物10b的制备:65℃下,向搅拌着的化合物10a(5.0克,45毫摩尔)的吡啶(200毫升)溶液中,逐滴加入(boc)2o(14.7克,67.5毫摩尔)。加完后,将反应物在85℃下搅拌4小时。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物冷却至0℃,加入浓盐酸(100毫升),再加水(50ml)。用ea萃取后,将有机相依次用nahco3溶液和盐水洗涤,用na2so4干燥并浓缩,得到黄色油状物,将其悬浮在et2o中,然后通过过滤收集固体,得到化合物10b(4.3克,收率:45.2%),为白色固体。化合物10c的制备:0℃、氮气保护下,向化合物10b(2.4克,11.4毫摩尔),n,n-二甲基苯胺(6.6毫升)的dcm(84毫升)溶液中滴加三氯氧磷(3.2毫升,34.2毫摩尔)。滴加结束后,将反应混合物在室温下搅拌2小时。用tlc(pe/ea=2:1)检测反应完成。将反应混合物倾入冰水中,然后分离水相和有机相。将有机相依次用nahco3水溶液和盐水洗涤,用na2so4干燥并浓缩,得到粗产物。将粗产物通过硅胶色谱法纯化,得到纯的化合物10c(1.8克,70%),为白色固体。化合物10的制备:化合物10c(100毫克,0.47毫摩尔)和dmap(12毫克,0.09毫摩尔)溶于thf(1毫升)中,在室温下向混合物中滴加(boc)2o(124毫克,0.57毫摩尔)。滴加结束后,将反应混合物在室温下搅拌过夜。用tlc(pe/ea=1:1)检测反应完成。用水稀释该反应混合物,用ea萃取。用盐水洗涤有机相,用na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物10(70毫克,44.8%),为白色固体。化合物11的制备:将化合物7(50毫克,0.15毫摩尔)和k2co3(62.2毫克,0.45毫摩尔)溶于dmso(3毫升)中,在氮气、室温下搅拌半小时,然后加入化合物10(148.4毫克,0.45毫摩尔)。将所得的反应混合物搅拌5小时后,用tlc(dcm/甲醇=20:1)检测反应完成。将反应混合物用水稀释,用ea萃取。有机相用盐水洗涤,用na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物11(31毫克,33%),为白色固体。化合物2-2的制备:将化合物11(25毫克,0.04毫摩尔)加入tfa(0.2ml)中,在氮气、室温下搅拌半小时。用tlc(dcm/甲醇=20:1)检测反应完成。将反应混合物用碳酸钠碱化,用dcm萃取。有机相用盐水洗涤,用na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物2-2(17毫克,85.3%),为白色固体,其h1nmr谱图和质谱图参见图2和图3。实施例4化合物2-3(本文亦称series2-3)的制备化合物12b的制备:0℃、氮气保护下,在dcm(600毫升)中搅拌溶解化合物12a(50克,0.47摩尔),加入tea(94克,0.93摩尔),然后将溴代乙酸乙酯(94克,0.56摩尔)逐滴加入。将所得的反应混合物在室温下搅拌过夜。用tlc(pe/ea=1:1)检测反应完成。将反应混合物过滤,浓缩滤液,并通过硅胶色谱法(与pe/ea洗脱纯化=20:1~10:1~5:1),得到纯的化合物12b(46克,51%),为黄色油状物。化合物12c的制备:化合物12b(38克,196.65毫摩尔)和tea(29.9克,295毫摩尔)在甲苯(800毫升)中在95℃加热搅拌,然后将乙基4-溴丁酸乙酯(72.9克,373.65毫摩尔)逐滴加入。加完后,将反应混合物加热至回流过夜。用tlc(pe/ea=5:1)检测反应完成。将反应混合物浓缩,用硅胶色谱法(pe/ea=20:1至5:1洗脱)纯化,得到纯的化合物12c(32克,产率:53%),为黄色油状物。化合物12d的制备:化合物12c(32克,104.3毫摩尔)的甲苯(300毫升)溶液中,在0℃加入叔丁醇钾(51.2克,456.3毫摩尔)。滴加结束后,将反应混合物在室温下搅拌1小时。用tlc(pe/ea=5:1)检测反应完成。将反应混合物通过加入2nhcl调节ph=6,然后用ea萃取。有机相,用盐水洗涤,用na2so4干燥并浓缩,得到粗化合物12d为暗黄色油状物(17克,产率:62.5%),不需进一步纯化,直接用于下一步骤。化合物12e的制备:将甲醇钠(meona,11克,161.65毫摩尔)溶解在甲醇(280毫升)中,冷却至5℃,加入甲脒乙酸盐(3.0克,29.15毫摩尔)。将反应混合物搅拌半小时,然后加入化合物12d(17克,65.1毫摩尔)。将反应混合物在40℃下搅拌过夜。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物冷却至室温,蒸发以除去大多数溶剂。将残余物,用ea萃取处理。用盐水洗涤有机相,用na2so4干燥并浓缩,得到粗产物。将粗产物通过硅胶色谱法纯化,得到纯的化合物12e(2.5克,产率:16%),为浅黄色固体。化合物12f的制备:化合物12e(2.5克,10.4毫摩尔),10%pd/c(0.5g)和(boc)2o(2.7g,12.4mmol)在meoh(40ml)中混合,氢气气氛(压力为0.5兆帕)下搅拌下过夜。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物过滤,将滤液浓缩,得到纯的化合物12f(2.6克,100%),为黄色固体。化合物12的制备:化合物12f(1.6克,6.3毫摩尔),三苯基膦(3.33克,12.6毫摩尔)和四氯化碳(2.93克,18.9毫摩尔)在1,2-二氯乙烷(1,2-dce,64毫升)中混合,并加热至70℃,维持1小时。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物浓缩,通过硅胶色谱法纯化,得到纯的化合物12(1.38克,81%),为浅黄色固体。化合物13的制备:在氮气气氛下,化合物7(50毫克,0.15毫摩尔)和k2co3(62.2毫克,0.45毫摩尔)溶于dmso(2ml)中,在室温下搅拌1.5小时,然后加入化合物12(121.4毫克,0.45毫摩尔)。将所得的反应混合物搅拌1.5小时,用tlc(dcm/甲醇=20:1)检测反应完成。将反应混合物用水稀释,用ea萃取。有机相用盐水洗涤,用na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物13(10毫克,12.2%),为白色固体。化合物2-3的制备:化合物13(10毫克,0.018毫摩尔)和tfa(0.1毫升)混合,在氮气气氛、室温下搅拌0.5小时。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物用碳酸钠碱化,用dcm萃取。有机相用盐水洗涤,用na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物2-3(3毫克,35.7%),为白色固体,其质谱图参见图4。实施例5化合物2-4(本文亦称kdr2)的制备第1步:在0℃、氮气气氛下,将乙基乙烯基醚(50克,0.69mol)缓慢加入到草酰氯(132.3克,1.04摩尔)中。然后将混合物在0℃下搅拌2小时,温热至室温,放置12小时。蒸馏除去过量的草酰氯,将残余物在120℃加热30分钟,然后通过真空蒸馏纯化,得到纯的化合物1(49克,产率:53%),为浅黄色油状物。步骤2:在0℃下将亚硫酰氯(66ml,0.91mol)加入到搅拌的化合物2(50克,0.365mol)的甲醇(1l)溶液中。加完后,将反应混合物搅拌,在40℃过夜。用tlc(pe/ea=1:1)检测反应完成。将混合物蒸发,残留物通过添加na2co3调节至ph为8,然后用ea萃取碱化。有机相用饱和食盐水洗涤,用无水硫酸钠干燥并浓缩,得到化合物3(51.7克,收率:93.8%),为棕色固体,不经进一步纯化直接用于下一个步骤。步骤3:化合物3(10克,66mmol)溶于100ml的dcm中,在0℃、氮气气氛下加入吡啶(9.06克,119mmol)和化合物1(15克,112mmol)的dcm(40ml)。然后将混合物温热至室温并搅拌2.5hr。用tlc(pe/ea=1:1)检测反应完成。将混合物用水、盐水洗涤,用na2so4干燥并浓缩,得到粗产物。将粗产物通过色谱层析(用dcm/甲醇=20:1洗脱)纯化,得到纯的化合物4(9.67克,收率:59%;),为白色固体。步骤4:在0℃将化合物4(9.67克0.039mol)加入到175毫升浓h2so4中。然后将反应混合物在室温下搅拌6小时。用tlc(pe/ea=1:1)检测反应完成。将混合物倒入冰-水中。滤出沉淀物,并用et2o洗涤。固体用乙醇重结晶,得到化合物5(2克,产率:25%),为米色固体。步骤5:将碳酸钾(0.686克4.97mmol)中加入化合物5(0.504克2.48mmol)的3ml的dmso溶液中,半小时后,加入化合物6(0.768克2.98mmol)。在氮气气氛下将反应混合物加热到100℃过夜。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物用水稀释并用etoac萃取。有机相用饱和食盐水洗涤,用无水硫酸钠干燥并浓缩,得到粗产物。将粗产物通过层析(聚乙烯/ea=5:1至2:1洗脱纯化),得到纯的化合物7(0.683克,产率:65%),为浅黄色固体。步骤6:在0℃下,向化合物7(0.683克,1.6mol)的thf(10ml)溶液中逐滴加入2n氢氧化锂(1.6毫升,3.2mmol)。然后将混合物温热至室温并搅拌过夜。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物蒸发,以除去thf,残余物用水稀释,并用ea萃取,以除去杂质。将水相酸化,通过加入2n盐酸调节至ph为3。白色沉淀物通过过滤收集并干燥,得到纯的化合物8(400mg,收率:61%),为白色固体。第7步:在室温下,向化合物8(100毫克,0.24mmol)和化合物9(47mg0.29mmol)的dmf(3ml)溶液中加入hatu(139mg0.37mmol)和dipea(95毫克0.73mmol)。然后在氮气气氛下,将反应混合物加热至40℃并搅拌过夜。用tlc(dcm/甲醇=10:1)检测反应完成。用ea稀释该混合物,用盐水洗涤,用na2so4干燥,并浓缩,得到粗产物。将车窗外通过预-tlc纯化,得到60mg的化合物10(60mg,收率:45%),为浅黄色固体。第8步:将化合物10(10毫克,0.018mmol)和tfa(0.1毫升)的混合物在室温下搅拌3小时。用tlc(dcm/甲醇=10:1)检测反应完成。蒸发tfa,并将残余物通过na2co3水溶液碱化,用dcm萃取。用盐水洗涤有机相,用na2so4干燥并浓缩,得到粗产物。将其通过预-tlc纯化,得到纯的化合物2-4(5毫克,产率:62%),为黄色固体,其结构鉴定数据参见图5。实施例5化合物2-5(本文亦称series2-5)的制备化合物14c的制备:配备有机械搅拌器和氯化钙管的3l的烧瓶中装入化合物14a(100克,0.716摩尔)和乙醇(1.0l)。将该混合物搅拌20分钟,然后滴加三乙胺(tea,72.5克,0.716摩尔)。将所得混合物搅拌10分钟,然后将丙烯酸乙酯(61.6克,0.716摩尔)加入到上述混合物。将反应混合物在室温下搅拌17小时。然后在室温下逐滴加入(boc)2o(234.5g,1.08mol)。滴加结束后,将反应混合物在室温下搅拌过夜。将反应混合物浓缩以除去大部分乙醇。将残余物溶解在水(3升)中,并用et2o(1升×3)萃取。依次用水、氯化铵(500毫升×3)溶液和盐水(500毫升×3)洗涤,无水na2so4干燥并浓缩,得到粗化合物14c(300克,92%)为黄色油状物,直接用于下一步骤而无需额外的纯化。化合物14d的制备:将钠(27.6克,0.765mol)逐步加入无水乙醇(1.5l)中。当固体钠完全消失,将化合物14c(300克,1.04mol)中加入到溶液中。将反应混合物回流过夜。用tcl(pe/ea=4:1)检测,起始原料完全被消耗。将反应混合物蒸发以除去大部分溶剂,将残余物溶解在水(1l)中,用柠檬酸酸化至ph6。然后将混合物用ea(1升×3)萃取。将萃取液合并,用盐水(1升×3)洗涤,用无水na2so4干燥并蒸发,得到化合物14d(169克,63.4%),为棕色油状物。该粗产物无需进一步纯化,直接用于下一步骤。化合物14e的制备:将meona(2.6克,48.58毫摩尔)溶解在meoh(50ml)中,将反应混合物冷却至5℃,加入甲脒乙酸盐(3.0克,29.15毫摩尔)。将反应混合物搅拌半小时,然后加入化合物14d(5.0克,19.43毫摩尔)。将反应混合物搅拌回流过夜。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物冷却至室温,蒸发以除去大多数溶剂。将残余物,用ea萃取处理。有机相用盐水洗涤,用无水na2so4干燥,并浓缩,得到粗产物。将粗产物通过硅胶色谱法纯化,得到纯的化合物14e(680mg,收率:14.7%),为浅黄色固体。化合物14的制备:在氮气气氛、0℃、搅拌条件下,在化合物14e(100毫克,0.42毫摩尔)和n,n-二甲基苯胺(0.28毫升)的dcm(4毫升)溶液中加入三氯氧磷(174mg,1.26毫摩尔,)。加完后,将反应混合物倾入冰水中,加入固体碳酸钠,然后用dcm萃取。有机相用盐水洗涤,用无水na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物14(60毫克,55.6%),为白色固体。化合物15的制备:在氮气气氛下,化合物7(50毫克,0.15毫摩尔)和k2co3(62.2毫克,0.45毫摩尔)溶于dmso(2ml),并在室温下搅拌一个半小时,然后加入化合物14(115.4毫克,0.45毫摩尔),再搅拌一个半小时,tlc(dcm/甲醇=20:1)表明反应完成。将反应混合物用水稀释,用ea萃取。用盐水洗涤有机相,用无水na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物15(14毫克,16.7%),为白色固体。化合物2-5的制备:化合物15(14毫克,0.025毫摩尔)和tfa(0.2ml)的混合物在氮气气氛、室温下搅拌0.5小时。用tlc(dcm/甲醇=10:1)检测反应完成。将反应混合物用碳酸钠碱化,用dcm萃取。有机相用盐水洗涤,用无水na2so4干燥并浓缩,得到粗产物。将粗产物通过tlc纯化,得到纯的化合物2-5(2.7mg,收率:24.5%),为白色固体,其质谱图参见图6。以下通过试验例具体说明本发明的有益效果。试验例1斑马鱼血管发育抑制试验斑马鱼(daniorerio,也称zebrafish)为鲤科(cyprinidae)短担尼鱼属(danio)的一种硬骨鱼。其基因与人类基因的相似度高达85%,雌鱼一次可产卵200~300枚,其受精和胚胎发育过程均在体外进行,24小时内就可发育成形,且胚胎通体透明,便于观察体内器官组织的变化。诸多特点使其成为了是国际标准化组织认可的5种鱼类实验动物之一。目前,斑马鱼在人类疾病研究中得到广泛应用,用于心血管系统方面的研究尤为多。斑马鱼可用于筛查小分子化合物对血管形成作用影响。实验方法:该实验使用通常作为筛查化合物对血管形成作用影响的flk1-gfp转基因斑马鱼为动物模型,在荧光显微镜下可以活体观察血管(suk-wonjin,2005年,development)。将出生后的flk1-gfp转基因斑马鱼胚胎经挑选后,放于培养皿中并置于28摄氏度培养箱中抚育3-5天。将本发明化合物、帕唑帕尼(pazopanib,130b,阳性对照)以下表1中的浓度直接加入抚育3-5天的斑马鱼的培养液中;以40umdmso作为阴性对照。24小时后检查脊椎血管的发育情况并用荧光显微镜照相。各化合物对斑马鱼脊椎血管发育的抑制率见表1,其中阴性对照组的血管发育状况设定为抑制率为0%,血管完全不发育的状况设定为抑制率为100%。图7a和图7b分别显示的是阴性对照组用40umdmso处理后斑马鱼的荧光显微照片和用1um化合物2-2处理后斑马鱼的荧光显微照片,其中可见阴性对照组的斑马鱼脊椎血管发育正常,而经化合物2-2处理后,脊椎血管的发育被100%地抑制。表1本发明化合物对flk1-gfp转基因斑马鱼血管发育的抑制作用(n=5)100nm1μm5μm10μm20μm40μm100μm2-110%50%100%100%100%100%100%2-250%100%100%100%100%100%100%2-30%0%0%0%0%5%20%2-40%0%0%0%5%25%70%2-50%0%10%40%80%100%100%130b0%0%85%90%95%95%100%由表1和图7可知,本发明化合物2-1、2-2、2-3、2-4和2-5均对斑马鱼的血管发育有抑制作用。其中,化合物2-1和2-2的活性明显更优。试验例2本发明化合物对vegfr2体外抑制试验检测本发明化合物抑制vegfr2激酶的实验方法如下:1)将原代培养的人脐静脉内皮细胞(huvec)p3-p5转移至6孔板,每孔约2×105细胞;2)待细胞生长至70-80%,加入本发明的化合物2-1、2-2和2-4(各化合物浓度为10nm,100nm和1μm三组),vegf为对照,孵育30min;3)加入vegf(美国cellsignaling公司)50ng/ml进行刺激,持续10min;4)加入非变性裂解液终止反应、收集细胞裂解液、进行蛋白定量;5)sds-page电泳,转硝酸纤维素膜,将膜切下后用5%脱脂奶配制的磷酸盐吐温缓冲液ttbs缓冲液(tris-bufferedsaline0.01%tween20,美国cellsignaling公司ph8.0),封闭2小时;6)洗膜,用抗磷酸化vegfr2抗体(1:1000稀释,美国cellsignaling公司)4℃封闭过夜;7)第2天洗膜后用辣根过氧化物酶标记的山羊抗兔二抗(美国cellsignaling公司)与膜室温共孵育1小时;8)洗膜后用化学发光试剂盒(millipore公司)显色,照相。试验结果参见图8。由图8可知,本发明化合物2-1、2-2和2-4均能抑制vegfr2,其中,化合物2-2效果较好。试验例3本发明化合物对脉络膜新生血管的抑制作用小鼠为c57/bl(美国jaxlab),采用激光诱导的脉络膜新生血管(choroidalneovascularization,cnv)动物模型进行实验。此模型目前也是应用最广的一种被用来研究药物对湿性黄斑变性的cnv形成影响的动物模型。小鼠全部为2-3月大,用avertin麻醉后,1%tropicamide(alcon)行瞳孔放大,采用iridexoculightgl532nm激光光凝(iridex)与裂隙灯输送系统对每只眼作4个光凝烧伤点,参数为功率120mw,光斑尺寸75μm,和持续时间0.1秒。c57/bl小鼠(每个视网膜4个激光点)进行激光光凝诱导cnv形成,只有观察到气泡、表明bruch膜的破裂的激光点纳入研究。激光实施后立即进行注射:右眼注射浓度1um的2-2,左眼注射均注射与药物相同体积的pbs作为对照。注射后5天将动物处死并获取眼球。去除眼前节、玻璃体和视网膜后,制作脉络膜铺片并行异凝集素(isolectin)染色。采用zeiss荧光显微镜系统进行拍照和cnv面积的测量。结果见图9a和图9b。根据图9可知,化合物2-2(1um)能显著抑制脉络膜新生血管形成,可有效治疗或缓解湿性黄斑变性。结论:综合上述试验例可知,本发明化合物具有良好的抗异常增生新生血管作用,该类化合物是通过抑制vegfr2产生活性。该类化合物可用于对新生血管异常所致疾病的治疗,如湿性黄斑变性等。试验例4本发明化合物对激酶的影响1)抑制剂量效应研究化合物2-1、2-2分别溶解于100%dmso溶剂中,稀释为3组系列浓度,dmso在最后测试中均保持浓度为1%。化合物最高浓度为50um。用非选择性蛋白激酶的活性抑制剂星形孢菌素(staurosporine,美国sigma公司)作为参照,其最高浓度为1um。具体测试条件见表2,测试结果见表3a,计算ic50值的结果见表3b和图10。图10a为阳性对照组结果,10b为化合物2-1组的结果,10c为化合物2-2组的结果。表2表3a不同浓度化合物对kdr和pdgfr-β的抑制活性表3b对kdr和pdgfr-β的抑制活性(ic50)由结果可见,本发明化合物2-1和2-2对kdr和pdgfr-β有良好的抑制活性,其中化合物2-2的效果还优于阳性参照星形孢菌素。2)抑制特异性研究本实验中,对化合物2-1和2-2测试了22种激酶活性抑制试验,测试浓度为5000nm,重复两次。待测化合物首先溶于100%dmso中,溶解浓度为最终测试浓度的100倍,所有最终测试时待测液中的dmso浓度都为1%。对于p38-α用sb-202191作为对照,对于pi3k-α用渥曼青霉素(wortmannin)作为对照,对于其他蛋白激酶采用非选择性蛋白激酶的活性抑制剂星形孢菌素(staurosporine)作为对照,测试时浓度10um。具体试验方法和试剂列于下表4:表4表5化合物2-1对激酶两次试验平均抑制率表6化合物2-2对激酶两次试验平均抑制率从以上试验结果可知,化合物2-1和2-2均可选择性高效抑制蛋白激酶kdr,化合物2-1抑制率大于97%,化合物2-2抑制率达100%,另外,这些化合物还对其他几种与炎症、肿瘤等疾病相关的蛋白激酶也有一定抑制作用,其中2-1对kit抑制率约为72%,对pdgfr-β抑制率约为71%,2-1还一定程度上抑制aurora-b(抑制率约31%)、fgfr2(约36%)、src(约21%)和jak2(约21%),其它激酶抑制率则绝大部分小于5%。化合物2-2对fgfr2抑制率约为61%、对pdgfr-β抑制率约为97%、对kit抑制率约为92%、对p38-α抑制率约为80%、对src抑制率约为65%,但对其余蛋白激酶抑制活性绝大部分小于5%。由此可见,和现有研发的小分子激酶抑制剂相比,本发明化合物对kdr具有较高特异性和高效率的抑制作用,且对fgfr2、pdgfr-β等与肿瘤、炎症有密切关联的蛋白激酶也有一定的抑制作用,这就表明化合物2-1、2-2对kdr、fgfr2等几种蛋白激酶异常活化相关的各种肿瘤、炎症及黄斑变性等疾病均有潜在治疗活性。试验例5角膜新生血管抑制作用的研究角膜炎症,各种化学烧伤,外伤,手术伤等会产生病理性新生血管,角膜新生血管会导致严重的视力损害。化学角膜损伤动物模型是广泛应用的药效学研究疾病模型。a:小鼠角膜化学烧伤模型本试验共测试了5只2-3月龄小鼠,小鼠为c57/bl(美国,jaxlab),在眼角膜中央,用75%硝酸银溶液和25%硝酸钾棒诱导化学烧伤造模。化学烧伤之后立即在右眼结膜下注射0.02ml化合物2-2(浓度100um),然后每天滴0.2ml化合物2-2两次(浓度100um),左眼用pbs作为对照,和右眼同样处理。7天后对眼角膜拍照比较。结果参见图11,其中11a为化合物2-2处理的结果,11b为以pbs作为阴性对照处理的结果。由图12所示,化合物2-2处理显著减少了新生血管和出血。b:兔角膜化学烧伤模型本试验共测试了5只5-6月龄新西兰大白兔,麻醉后,在眼角膜中央,右眼用6mm直径圆形滤纸浸0.1mnaoh,诱导3min,化学烧伤造模。化学烧伤之后立即在右眼结膜下注射0.1ml100um化合物2-2,然后每天滴0.5ml100um化合物2-2两次。左眼为化学造模后用pbs作为对照,处理方法同右眼。7天后对眼角膜拍照比较。结果参见图12,其中12a为化合物2-2处理的结果,12b为以pbs作为阴性对照处理的结果,12c为化合物2-1处理的结果。由图12所示,化合物2-1和2-2的处理均显著减少了角膜新生血管。综上所述,本发明化合物具有良好的抗异常增生的新生血管作用,该类化合物是通过抑制vegfr2(又名kdr)产生活性。该类化合物可用于对异常增生的新生血管、kdr、fgfr2等蛋白激酶异常所致疾病的治疗,如湿性黄斑变性、炎症、恶性肿瘤等。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1