美罗培南侧链中间体及其制备方法与流程
本发明涉及一种美罗培南侧链中间体及其制备方法,属于药物化学
技术领域:
。
背景技术:
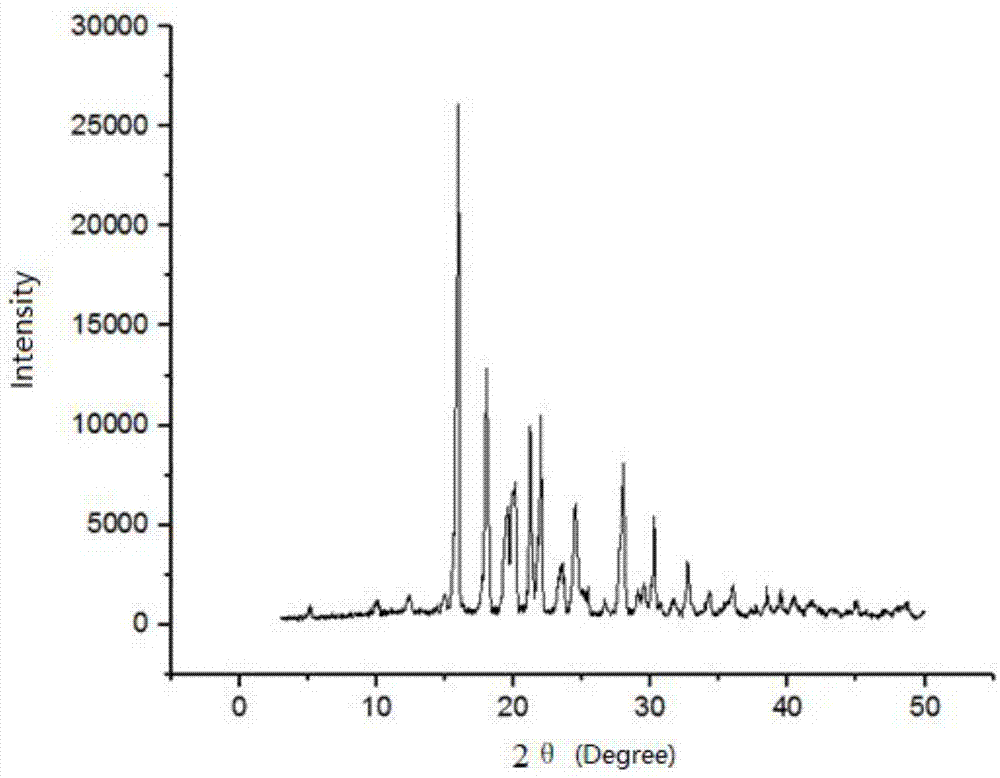
:美罗培南是由dainipponsumitomopharma研发,于1995年首次在意大利上市,它是第一个能单独用药的碳青霉烯类抗生素,同时也是首个1β-甲基碳青霉烯类抗生素。目前该药物主要是通过化学全合成来制备,主要工艺过程包括关键侧链合成和骨架母核的合成。目前文献报道的美罗培南关键侧链(式ⅱ化合物)的合成均以反式-4-羟基脯氨酸(式ⅲ化合物)为起始原料,并对其氨基位进行适当保护[保护剂包括对氯甲酸对硝基苄酯(pnz.c1)、烯丙氧羰酰氯和boc酸酐等],经多步反应得到4位构型翻转的中间体或硫醇内酯,再经适当反应即得目标侧链。自从专利jp5186476a(1993年7月27日)及其同族专利us5322952(1994年7月21日)、ep0551993(1993年7月27日)等首次报道了以(2s,4r)-1((对硝基苄氧基)羰基)-4-羟基吡咯烷-2-羧酸为原料,通过硫内酯环合的方法得到目标侧链以来,绝大部分文献均采用对硝基苄氧羰基作为氨基的保护基团。对于化合物(2s,4r)-1((对硝基苄氧基)羰基)-4-羟基吡咯烷-2-羧酸(化合物ⅰ)虽然其中有很多合成方法,但所有文献均没有提到化合物ⅰ的水合物情况以及晶型情况。而以化合物ⅰ为原料进行后续反应时,对水份的要求比较苛刻,一般要求水份≤0.1%,而合成工艺中一旦生成化合物ⅰ的水合物,水份则极难达标。技术实现要素:针对现有技术的不足,本发明的目的是提供一种美罗培南侧链中间体,能完全满足后续反应的需求,有利于目标美罗培南侧链工业化大规模生产的实现。同时,本发明还提供其制备方法,科学合理,简单易行。本发明所述的美罗培南侧链中间体,结构式如下:化合物ⅰx-ray粉末衍射2θ为15.96,18.04,20.1,21.22,21.96,24.48,28.0,30.26处有特征吸收峰;化合物ⅰ差式扫描量热仪dsc特征吸热峰,最大吸热熔融峰温度为136.71℃。所述的化合物ⅰ水合物x-ray粉末衍射2θ为11.02,12.8,16.0,20.16,22.18,21.96,22.76,25.62处有特征吸收峰;差示扫描量热仪dsc特征吸热峰,最大吸热熔融峰温度为80.08℃。差式扫描量热仪测试条件为30-130℃,升温速率5℃/min,开盖,无氮气环境。美罗培南侧链中间体(2s,4r)-1((对硝基苄氧基)羰基)-4-羟基吡咯烷-2-羧酸(化合物ⅰ以及水合物)具有如下特征:化合物ⅰx-ray粉末衍射特征吸收峰为:2θ(°)强度(计数)强度/%11.02385017.612.8325014.916.0686031.420.16671030.722.182183010021.961055048.322.76865039.623.9248011.425.62492022.528.5813606.230.1216107.434.0613406.135.4417207.9化合物ⅰ水合物x-ray粉末衍射特征吸收峰为:本发明所述的美罗培南侧链中间体的制备方法,包括以下步骤:反应阶段:低温下向3-羟基脯氨酸和氢氧化钠水溶液中缓慢滴加对硝基苄氧碳基氯,混合,tlc检测反应终点;洗涤阶段:将反应阶段混合物静置,分液后,水层用有机溶剂a洗涤2-3次;析晶过滤阶段:1.将洗涤阶段所得水相转入反应瓶中,加入适量有机溶剂b,低温下缓慢滴加无机酸至ph为2,经养晶、过滤和淋洗后,得到化合物ⅰ湿品;2.将洗涤阶段所得水相转入反应瓶中,低温下加入无机酸调ph至2,经养晶、过滤和淋洗后得到化合物ⅰ水合物;干燥阶段:将得到的化合物ⅰ湿品进行干燥,得到化合物ⅰ;将化合物ⅰ水合物进行干燥,干燥后得到化合物ⅰ。所述的有机溶剂a为甲苯或二氯甲烷中的一种或两种;与反应阶段加入水的体积比为1:2-4。优选的,所述的有机溶剂a为甲苯。所述的反应阶段,低温为-5-5℃。所述的氢氧化钠水溶液质量浓度为5-9%。所述的3-羟基脯氨酸与对硝基苄氧碳基氯的质量比为1:5-8。所述的氢氧化钠与3-羟基脯氨酸的质量比为1:1.1-2.2。所述的析晶阶段所用无机酸为盐酸或硫酸,质量浓度为5-30%。所述的析晶阶段,加入无机酸调酸时,温度为-10-10℃。优选的,所述的无机酸调酸时,温度为0-5℃。所述的析晶阶段中,有机溶剂b为甲醇、乙醇或丙酮中的一种或几种。所述的析晶阶段,有机溶剂b质量为反应阶段所加水和有机溶剂b总质量的10-50%。所述的化合物ⅰ湿品在真空干燥箱内,于60℃下干燥8.0小时,取样检测水分<0.1%时出料;化合物ⅰ水合物使用热风烘箱在40~42℃烘干12h,然后缓慢升温,每小时升高1-2℃,升温至60℃停止升温,升温过程中观察物料状态,每隔15min翻动粉碎物料一次,防止物料结块;稳定2h不结块以后,继续升温至80℃烘干8h,取样检测水分<0.1%,出料。与现有技术相比,本发明具有如下有益效果:1.制得的化合物ⅰ能满足后续反应对其水分的要求,有利于美罗培南侧链工业化生产的实现;2.本发明提供了化合物ⅰ及化合物ⅰ水合物晶型特征和区别,并提供了化合物ⅰ水合物向化合物ⅰ转变的方法;3.本发明通过改变析晶方法,在析晶工序即能得到无水化合物ⅰ,大大降低了后续烘料的难度,节省了大量的人力物力,具有极高的生产价值;4.所述的制备方法,科学合理,简单易行。附图说明图1是实施例1中化合物ⅰ水合物x-ray粉末衍射图谱;图2是实施例1中化合物ⅰ水合物差示扫描量热仪dsc图谱;图3是实施例2中化合物ⅰx-ray粉末衍射图谱;图4是实施例2中化合物ⅰ差示扫描量热仪dsc图谱;图5是实施例3中化合物ⅰx-ray粉末衍射图谱;图6是实施例3中化合物ⅰ差示扫描量热仪dsc图谱。具体实施方式下面结合实施例对本发明做进一步的说明。实施例1在500ml三口瓶中加入纯化水192g,加入氢氧化钠15.3g,搅拌溶清,降温至0℃;加入3-羟基脯氨酸30g,搅拌溶解;降温至-5℃,滴加对硝基氯甲酸苄酯甲苯液约180g,约1h滴毕,滴加完毕ph=9;继续搅拌15min,然后静置20min,分液,得下层水相;向下层水相中加入甲苯63ml×3洗涤三次,静置分液;控制內温10℃,水相滴加6n盐酸,滴加至水相变浑浊停止滴加,ph=4,加入少量晶种,搅拌至有较多晶体析出,继续滴加盐酸至ph=2(共需要6n稀盐酸约52g),降温至0℃,保温1h抽滤,用纯水洗至ph=3,得化合物ⅰ水合物产品。其粉末衍射图谱见图1,其dsc图谱见图2。实施例2(2s,4r)-1((对硝基苄氧基)羰基)-4-羟基吡咯烷-2-羧酸水合物转变为无水化合物。将实施例1中湿品使用热风烘箱在40℃烘干12h,然后缓慢升温(每小时升高1℃),升温至60℃停止升温,升温过程中观察物料状态,每隔15min翻动粉碎物料一次,防止物料结块。稳定2h不结块以后,继续升温,2h升温至80℃继续烘干8h,取样检测水分<0.1%,出料,得化合物ⅰ,即无水(2s,4r)-1((对硝基苄氧基)羰基)-4-羟基吡咯烷-2-羧酸。其粉末衍射图谱见图3,其dsc图谱见图4。实施例3在500ml三口瓶中加入纯化水300g,加入氢氧化钠24g,搅拌溶清,降温至0℃;加入3-羟基脯氨酸32g,搅拌溶解;降温至-5℃,滴加对硝基氯甲酸苄酯甲苯液约144g,控制內温0℃,约1h滴毕,滴加完毕ph=10;继续搅拌15min,然后静置20min,分液,得下层水相;向下层水相中加入甲苯75ml×3洗涤三次,静置分液;向水相中加入甲醇50g,控制內温5℃,滴加6n盐酸,滴加至水相变浑浊停止滴加,ph=4,加入少量晶种,搅拌至有较多晶体析出,继续滴加盐酸至ph=2(共需要6n稀盐酸52g),降温至0℃,保温1h抽滤,用水:甲醇=4:1的质量比混合液洗至ph=3,得化合物ⅰ产品。其粉末衍射图谱见图5,其dsc图谱见图6。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1