一种β榄香烯的制备方法与流程
本发明属于生物医药技术领域,具体涉及一种β榄香烯的制备方法。
技术背景
榄香烯是我国首先从姜科植物温莪术中提取的一种具有抗癌活性的倍半萜烯类化合物,其化学结构中有3个手性碳原子,因此理论上存在8种异构体,目前已鉴定且被文献报道的有α、β、γ、δ四种。其中,β-榄香烯(β-elemene)的反式异构体是发挥抗癌活性的主要物质,其化学名称为(1s,2s,4r)-1-甲基-1-乙烯基-2,4-二异丙烯基环己烷,分子结构如下:
研究表明,β榄香烯可用于腺癌、人神经胶质瘤、肝癌、肺癌、结肠癌、胃癌、卵巢癌等多种疾病的治疗,具有抗癌谱广、疗效确切,毒副作用轻微,无骨髓抑制等优点。目前国内上市的β榄香烯为二类新药,主要用于恶性浆膜腔积液、肺癌、消化道肿瘤、脑瘤及其他浅表性肿瘤的治疗。
目前β榄香烯原料药主要通过天然植物提取分离或化学合成这两种途径制备得到。化学合成β榄香烯存在一些问题,比如工艺步骤复杂,产率低,且在立体异构体选择控制上存在难度,因此目前仅停留在文献研究的层面,无法满足工业大生产要求。国内已上市的榄香烯注射液主要采用水蒸气蒸馏法,从莪术、香茅等植物挥发油中获得β榄香烯原料。然而,天然植物中榄香烯含量一般较低,通常需要耗费大量的植物药材资源,同时经过复杂的分离、纯化步骤才能得到符合纯度要求的榄香烯提取物。例如:莪术挥发油中榄香烯的含量在5%左右,李勇等利用水蒸气蒸馏法从蓬莪术、广西莪术、温莪术中提取挥发油得到榄香烯相对含量分别为6.47%、9.81%、16.91%。可见相对于目前大量的临床需求,现有的药源及较低的提取效率远远不能满足现状[榄香烯的中国菊科植物资源报告,中国中西医结合杂志,2015,35(6)]。
生物合成β-榄香烯是利用重组微生物的自身代谢途径,使其在适当的发酵液中进行发酵培养,合成吉马烯a(germacrenea),再利用吉玛烯a在加热条件下会产生重排反应的特点[(+)-germacreneabiosynthesis,plantphysiol.(1998)117:1381-1392],得到β榄香烯,如附图3所示。
生物发酵制备β榄香烯的方法无需耗费大量的植物资源,同时避免了复杂的化学合成步骤,具有成本低、产率高等优点,为β榄香烯原料药的工业生产提供了新的方向。通过生物发酵得到的吉马烯a存在于含有大量菌丝以及多种营养成分的培养基发酵液中,吉马烯a的分离是生产β-榄香烯的关键步骤之一。然而,现有技术暂未提供一种可供产业化应用的分离方法,极大的制约了β榄香烯生物合成方法的产业化实施。为了能解决现有技术存在的问题,有必要开发一种有效的分离纯化方法,用于制备吉玛烯a发酵产物,并进一步制备得到高纯度的β榄香烯。
技术实现要素:
为了解决现有技术存在的上述问题,本发明的目的之一是提供一种β榄香烯的制备方法。具体的,本发明提供如下技术方案。
一种β榄香烯的制备方法,包括如下步骤:将含有吉玛烯a的发酵液分离,得到的固体用含醇溶剂进行第一次提取,分离,提取后的固体再次用含醇溶剂或烷烃进行第二次提取,合并两次提取液,洗涤、干燥、浓缩后,加热转化为β榄香烯。
本发明β榄香烯制备方法中,所述含醇溶剂为低级醇或醇溶液,优选为c1-c3醇或醇溶液,进一步优选自甲醇、无水乙醇、95%乙醇、丙醇、异丙醇中的一种或多种;所述提取溶剂a的用量为发酵液体积的0.5倍及以上,例如0.75倍、1倍、1.25倍或更高,优选为0.5-1.25倍;
本发明β榄香烯制备方法中,所述第二次提取的溶剂优选为烷烃,所述烷烃选自正己烷、环己烷、石油醚、正十二烷中的一种或多种;所述第二次提取的溶剂用量为发酵液体积的0.25倍及以上,例如0.375倍、0.5倍、0.625倍、0.75倍、1倍或更高,优选为0.375-0.75倍。
优选地,本发明β榄香烯制备方法中,将所述含醇溶液提取得到的提取液进行浓缩,并用烷烃反萃后再合并提取液;所述反萃步骤中,烷烃优选正己烷、石油醚、环己烷中的一种或多种。
优选的,本发明β榄香烯的制备方法中,所述加热转化温度为100-180℃,优选120-150℃,加热时间优选为1h;
进一步,前述加热转化得到的β榄香烯可采用适当的纯化方法,例如减压精馏等,进行进一步提纯;本发明中优选的减压精馏工艺参数如下:以不锈钢网θ环为塔柱填料,塔板数10~15,塔釜温度100~130℃,回流比为0.5~3.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃。
本发明的优点在于:
(1)能够有效分离生物发酵液中的吉玛烯a产物,分离提取率高;
(2)β榄香烯转化率和回收率高,为β榄香烯原料药的工业化生产提供新的选择;
(3)工艺简便,成本低、效率高,可操作性强,适于产业应用。
附图说明
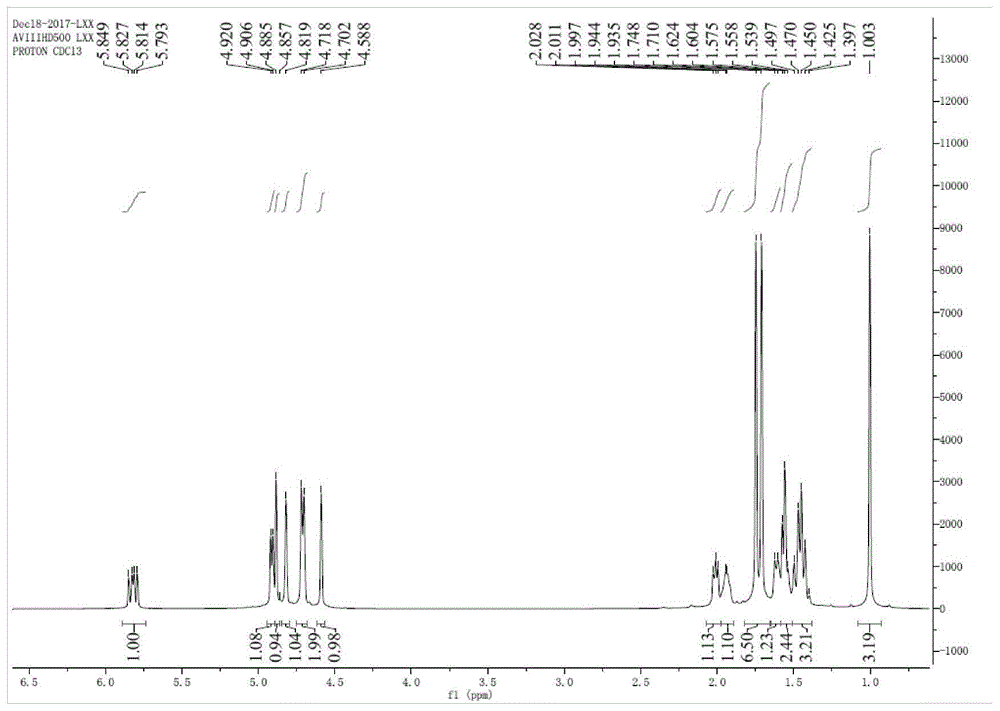
图1为本发明制备方法制备得到的β榄香烯产物的1hnmr谱图
图2为本发明制备方法制备得到的β榄香烯产物的13cnmr谱图
图3为吉玛稀a加热转化得到β榄香烯的化学反应式
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。
本发明所述β榄香烯制备方法可用于分离生物发酵液中的吉玛烯a,并进一步热转化得到β榄香烯;本发明人尝试过不同菌种和/或不同发酵工艺得到的生物发酵液,均适用本发明的制备方法。具体的,本申请采用cn108060092a中公开的生物发酵工艺制备的吉玛烯a发酵液作为举例说明。
一、产物中β榄香烯含量检测方法
供试品溶液制备:将本发明制备的β榄香烯产物用正己烷稀释100倍过有机尼龙膜(0.45μm)作为供试品溶液。
对照品溶液制备:精密称取50mgβ-榄香烯标准品(购于中国食品药品检定研究院,货号100268-201402)至50ml容量瓶中,无水乙醇稀释定容作为对照品溶液。
检测条件:安捷伦气相色谱agilent7890b,测定条件:进样口温度250℃,进样体积1μl,不分流;色谱柱:安捷伦hp-5ms(30m*0.25mm);色谱条件:45℃,1min,10℃/min到300℃,300℃保温5min,fid检测器。
二、吉玛稀a含量检测方法
供试品溶液制备:
(1)发酵液:取发酵液20ml置于50ml离心管中,加入5ml正十二烷,放置摇床中震荡萃取8小时以上,然后静置5min,取上层(含乳化层)2ml置于离心管中,12000rpm离心1min使有机相和水相分层,取上清液用正己烷稀释100倍过有机尼龙膜(0.45μm)作为供试品溶液。
(2)提取或萃取的上清液:取样品用正己烷稀释10-50倍,过有机尼龙膜(0.45μm)作为供试品溶液。
对照品溶液制备:气相色谱的进样口温度在250℃,在此温度下供试品溶液里的吉玛烯a会发生重排反应,转化成β-榄香烯,因此本实验中采用β-榄香烯作为对照品,精密称取50mgβ-榄香烯标准品(购买于中国食品药品检定研究院,货号100268-201402)至50ml容量瓶中,无水乙醇稀释定容作为对照品溶液。
检测条件:安捷伦气相色谱agilent7890b,测定条件:进样口温度250℃,进样体积1μl,不分流;色谱柱:安捷伦hp-5ms(30m*0.25mm);色谱条件:45℃,1min,10℃/min到300℃,300℃保温5min,fid检测器。
三、产物中β榄香烯结构验证
化合物结构如下:
结构确证方法及结果:
取精馏纯化后的β-榄香烯产物送检nmr、ms。该化合物1hnmr谱(附图1)显示存在24个氢原子,13cnmr谱(附图2)显示存在15个碳原子包括6个双键c原子(δc150.4,150.3,147.7,112.1,109.9,108.3),与ms数据分子离子峰204([m]+)吻合,提示该化合物为倍半萜类化合物,且分子式为c15h24。根据不饱和度计算,该化合物还存在一个环。1hnmr(cdcl3,600mhz):1.00(3h,s,h-15)、1.45(3h,m,h-2,3a)、1.64(1h,m,h-5a)、1.56(2h,m,h-3b,5b)、1.71(3h,s,h-11)、1.76(3h,s,h-14)、1.91(1h,m,h-6)、2.01(1h,m,h-4)、4.59(1h,s,h-10a)、4.82(1h,s,h-10b)、4.70(1h,s,h-13a)、4.72(1h,s,h-13b)、4.89(1h,d,j=12.6hz,h-8a)、4.90(1h,d,j=20.4hz,h-8b)、5.82(1h,dd,j=20.4、12.6hz,h-7)。13cnmr(cdcl3,125mhz):16.6(c-15)、21.1(c-14)、24.8(c-11)、26.8(c-5)、32.9(c-3)、39.8(c-1)、39.9(c-2)、45.7(c-6)、52.8(c-4)、108.3(c-13)、109.9(c-8)、112.1(c-10)、147.7(c-9)、150.3(c-12)、150.4(c-7)。以上数据与文献[榄香烯原料药的化学成分,沈阳药科大学学报,2009,26(8)]中化合物1(-)β-榄香烯的nmr和ms数据一致,故确证该化合物为(-)β-榄香烯。
试验例1
取生物合成制备的吉玛烯a发酵液(吉马烯a含量8.50g/l)三份,分别采用实验室玻璃精馏装置、工业160l提取罐、工业100l单效浓缩器进行蒸馏提取,间歇补水,提取总时长约15小时。将蒸馏出的液体经分液漏斗或油水分离器分出油相,对于严重乳化的部分,采用高速(12000r/min)离心机离心10min,记录挥发油的总体积,取样分析吉马烯a含量,计算提取率。
往挥发油中加入无水硫酸钠干燥,滤除干燥剂,挥发油置于120℃油浴中,减压脱除残余溶剂,再升温至150℃保温反应1小时,精馏收集β-榄香烯,称重取样分析含量,计算回收率。实验结果表1所示。
表1水蒸气蒸馏提取发酵液效果
注:gm---吉马烯a(gemenea),em---β-榄香烯(elemene),下同
gm提取率=(挥发油中gm含量/发酵液中gm含量)*100%
em回收率=(em重量*em纯度/发酵液中gm含量)*100%
实验表明,采用蒸馏法提取发酵液,会出现不同程度的乳化现象,提取过程中容易产生大量泡沫,严重影响了提取进程,同时也造成蒸馏液里面含有较多杂质,且在长时间蒸馏过程中易产生副反应生成新杂质,导致gm提取率及β榄香烯回收率均较低。
试验例2
取生物合成制备的吉马烯a发酵液400ml(吉马烯a含量10.25g/l)六份,分别加入400ml的无水乙醇、甲醇、正丁醇、石油醚、正十二烷和95%乙醇+正己烷(1:1,v/v),振摇搅拌提取30min。对于未乳化的样品,静置沉降10小时,记录上清液体积,取上清液过滤后用气相色谱方法分析含量,并以实验室通常的过滤方法考察过滤操作的难易度。对于严重乳化的样品,采用高速离心机离心10min,记录上层上清液体积,取上清液用气相色谱方法分析含量并计算本步骤提取率。实验结果表2所示。
表2有机溶剂直接提取发酵液效果
注:提取率a=(上清液中gm含量a/发酵液中gm含量)*100%
实验表明,采用正丁醇、石油醚、正十二烷、95%与正己烷的混合溶剂(1:1,v/v)等有机溶剂提取发酵液,出现不同程度的乳化现象,常规过滤方式难以分离乳化层和有机溶剂层,虽然在实验室可采用高速(12000r/min)离心设备将乳化层分离,但是工业上发酵液的体量一般在吨位级别,目前暂时没有能够满足如此大批量分离要求的高速离心设备。
采用无水乙醇和甲醇提取时,虽未出现乳化,但提取液过滤困难,可操作性差。
试验例3
取生物合成制备的吉马烯a发酵液400ml(吉马烯a含量10.25g/l)6份,以高速离心机于3000转离心20min,弃去上层液体,底部固体沉降物(即菌丝)分别加入300ml的95%乙醇、无水乙醇、甲醇、异丙醇、石油醚、正十二烷,振摇搅拌30min,对可沉降样品过滤,对乳化样品离心,取上清液分析吉玛烯a含量,计算本步骤提取率。实验结果如表3所示。
表3不同溶剂提取菌丝效果对比
注:提取率a=(上清液中gm含量a/发酵液中gm含量)*100%
试验例4
取生物合成制备的吉马烯a发酵液40ml(吉马烯a含量10.25g/l)6份,以高速离心机于3000转离心20min,弃去上层溶液,底部固体沉降物分别加入5、10、20、30、40、50ml的95%乙醇、振摇搅拌30min,离心沉降,取上清液分析吉玛烯a含量,计算本步骤提取率a。实验结果如表4所示。
表4不同溶剂用量萃取菌丝效果对比
注:提取率a=(上清液中gm含量a/发酵液中gm含量)*100%;
试验例5
将生物合成制备的吉马烯a发酵液2400ml(吉马烯a含量10.25g/l)以高速离心机离心,弃去上层溶液得到底部固体沉降物;将底部固体沉降物加入无水乙醇2400ml搅拌提取1小时,过滤,得到乙醇提取液和固体菌丝渣。
将菌丝渣平均分成六份,分别加入200ml95%乙醇、无水乙醇、甲醇、异丙醇、石油醚、环己烷振摇搅拌30min,离心,取上清液用气相色谱法检测分析含量并计算本步骤提取率b。实验结果见表5.
表5二次提取实验效果
注:提取率b=(上清液中gm含量b/发酵液中gm含量)*100%
试验例6
将生物合成制备的吉马烯a发酵液2400ml(吉马烯a含量10.25g/l)以高速离心机离心,弃去上层溶液得到底部固体沉降物;将底部固体沉降物加入无水乙醇2400ml搅拌提取1小时,过滤,得到乙醇提取液和固体菌丝渣。
将前述菌丝渣平均分成六份,分别加入50、100、150、200、250、300ml石油醚振摇搅拌30min,离心,取样上清液检测分析含量并计算本步骤提取率b。实验结果见表6。
表6二次提取过程中不同溶剂用量效果对比
注:提取率b=(上清液中gm含量b/发酵液中gm含量)*100%
试验例7
将实验例6的乙醇提取液平均分成3份,编号为1、2、3,分别减压蒸出乙醇至蒸馏瓶内出现油水分离界面,冷却至室温。将1号分去水层,加入无水硫酸钠干燥,滤除干燥剂得到吉马烯a粗品1号。2号3号分别加入80ml的石油醚和环己烷,震摇萃取,分去水相,有机相加入无水硫酸钠干燥,滤除干燥剂,减压蒸出溶剂得到吉马烯a粗品2、3号。
将吉马烯a粗品1、2、3号取样称重,分别置于120℃油浴中,减压脱除残余溶剂,再升温至150℃保温反应1小时,称重取样分析转化前的吉马烯a和转化后β-榄香烯含量,计算转化率和回收率。结果见表7。
表7不同提取工艺热转化实验
注:gm转化率=(em粗品中em含量/gm粗品中gm含量)*100%;
em回收率=(em粗品中em含量/提取液中gm含量)*100%
实施例1
(1)、将生物合成制备的吉马烯a发酵液40l(吉马烯a含量10.25g/l)以管式离心机离心,弃去上清液得到菌丝沉淀物11.7kg;
(2)、将菌丝沉淀物加入提取罐,以95%乙醇30l搅拌提取1小时,过滤,得到乙醇提取液和菌丝渣。
(3)、将步骤(2)中获得的菌丝渣以20l石油醚搅拌提取0.5小时,过滤分离收集石油醚提取液。
(4)、将步骤(2)中得到的乙醇提取液进行减压浓缩回收乙醇,真空度-0.06mpa~-0.08mpa,收集水油混合物。
(5)、步骤(4)中得到的水油混合物以石油醚4l萃取,得到石油醚萃取液,下层水相排出。
(6)、将步骤(3)中的石油醚提取液和步骤(5)中石油醚萃取液合并,依次以水、盐水洗涤,无水硫酸钠干燥,滤除干燥剂。
(7)、将步骤(6)中干燥后的产物进行减压浓缩回收石油醚,真空度-0.04mpa~-0.08mpa,得到吉马烯a粗品。
(8)、步骤(7)中得到吉马烯a粗品在反应釜中加热转化,维持釜内温度120~150℃,保温1小时,釜内吉马烯a转化为β-榄香烯,得到β-榄香烯粗品。
(9)、将步骤(8)中得到的β-榄香烯粗品在精馏塔中进行减压精馏,塔柱填料为不锈钢网θ环,塔釜温度100~140℃,回流比为0.5~3.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃,得到β-榄香烯258.71g。
色谱分析所得β-榄香烯纯度为97.01%,相对于发酵液吉马烯a的产率(成品中β-榄香烯含量/发酵液中吉玛烯a含量,下同)为61.21%。
实施例2
(1)、将生物合成制备的吉马烯a发酵液40l(吉马烯a含量10.25g/l)以管式离心机离心,弃去上清液得到菌丝沉淀物11.7kg;
(2)、将菌丝沉淀物加入提取罐,以无水乙醇30l搅拌提取1小时,过滤,得到乙醇提取液和菌丝渣。
(3)、将步骤(2)中获得的菌丝渣以20l石油醚搅拌提取0.5小时,过滤分离收集石油醚提取液。
(4)、将步骤(2)中得到的乙醇提取液进行减压浓缩回收乙醇,真空度-0.06mpa~-0.08mpa,得到水油混合物。
(5)、步骤(4)中得到的水油混合物以石油醚4l萃取,得到石油醚萃取液,下层水相排出。
(6)、将步骤(3)中的石油醚提取液和步骤(5)中石油醚萃取液合并,依次以水、盐水洗涤,无水硫酸钠干燥,滤除干燥剂。
(7)、将步骤(6)中干燥后的产物进行减压浓缩回收石油醚,真空度-0.04mpa~-0.08mpa,得到吉马烯a粗品。
(8)、步骤(7)中得到吉马烯a粗品在反应釜中加热转化,维持釜内温度120~150℃,保温1小时,釜内吉马烯a转化为β-榄香烯,得到β-榄香烯粗品。
(9)、将步骤(8)中得到的β-榄香烯粗品在精馏塔中进行减压精馏,塔柱填料为不锈钢网θ环,塔釜温度100~140℃,回流比为0.5~2.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃,得到β-榄香烯263.22g。色谱检测β-榄香烯含量为97.05%,相对于发酵液中吉马烯a的产率为62.3%。
实施例3
(1)、将生物合成制备的吉马烯a发酵液40l(吉马烯a含量10.25g/l)以管式离心机离心,弃去上清液得到菌丝沉淀物11.7kg;
(2)、将菌丝沉淀物加入提取罐,以甲醇30l搅拌提取1小时,过滤,得到甲醇提取液和菌丝渣。
(3)、将步骤(2)中获得的菌丝渣以20l石油醚搅拌提取0.5小时,过滤分离收集石油醚提取液。
(4)、将步骤(2)中得到的甲醇提取液进行减压浓缩回收甲醇,真空度-0.06mpa~-0.08mpa,得到水油混合物。
(5)、步骤(4)中得到的水油混合物以石油醚4l萃取,得到石油醚萃取液,下层水相排出。
(6)、将步骤(3)中的石油醚提取液和步骤(5)中石油醚萃取液合并,依次以水、盐水洗涤,无水硫酸钠干燥,滤除干燥剂。
(7)、将步骤(6)中干燥后的产物进行减压浓缩回收石油醚,真空度-0.04mpa~-0.08mpa,得到吉马烯a粗品。
(8)、步骤(7)中得到吉马烯a粗品在反应釜中加热转化,维持釜内温度120~150℃,保温1小时,釜内吉马烯a转化为β-榄香烯,得到β-榄香烯粗品。
(9)、将步骤(8)中得到的β-榄香烯粗品在精馏塔中进行减压精馏,塔柱填料为不锈钢网θ环,塔釜温度100~140℃,回流比为0.5~2.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃,得到β-榄香烯261.58g。色谱检测β-榄香烯含量为96.55%,相对于发酵液中吉马烯a的产率为61.60%。
实施例4
(1)、将生物合成制备的吉马烯a发酵液40l(吉马烯a含量10.25g/l)以管式离心机离心,弃去上清液得到菌丝沉淀物11.7kg;
(2)、将菌丝沉淀物加入提取罐,以异丙醇30l搅拌提取1小时,过滤,得到异丙醇提取液和菌丝渣。
(3)、将步骤(2)中获得的菌丝渣以20l石油醚搅拌提取0.5小时,过滤分离收集石油醚提取液。
(4)、将步骤(2)中得到的异丙醇提取液进行减压浓缩回收异丙醇,真空度-0.06mpa~-0.08mpa,得到水油混合物。
(5)、步骤(4)中得到的水油混合物以石油醚4l萃取,得到石油醚萃取液,下层水相排出。
(6)、将步骤(3)中的石油醚提取液和步骤(5)中石油醚萃取液合并,依次以水、盐水洗涤,无水硫酸钠干燥,滤除干燥剂。
(7)、将步骤(6)中干燥后的产物进行减压浓缩回收石油醚,真空度-0.04mpa~-0.08mpa,得到吉马烯a粗品。
(8)、步骤(7)中得到吉马烯a粗品在反应釜中加热转化,维持釜内温度120~150℃,保温1小时,釜内吉马烯a转化为β-榄香烯,得到β-榄香烯粗品。
(9)、将步骤(8)中得到的β-榄香烯粗品在精馏塔中进行减压精馏,塔柱填料为不锈钢网θ环,塔釜温度100~140℃,回流比为0.5~3.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃,得到β-榄香烯243.37g。色谱分析所得β-榄香烯纯度为96.87%,相对于发酵液吉马烯a的产率为57.50%。
实施例5
(1)、将生物合成制备的吉马烯a发酵液40l(吉马烯a含量10.25g/l)以管式离心机离心,弃去上清液得到菌丝沉淀物11.7kg;
(2)、将菌丝沉淀物加入提取罐,以95%乙醇30l搅拌提取1小时,过滤,得到乙醇提取液和菌丝渣。
(3)、将步骤(2)中获得的菌丝渣以20l正己烷搅拌提取0.5小时,过滤分离收集正己烷提取液。
(4)、将步骤(2)中得到的乙醇提取液进行减压浓缩回收乙醇,真空度-0.06mpa~-0.08mpa,得到水油混合物。
(5)、步骤(4)中得到的水油混合物以正己烷4l萃取,得到正己烷萃取液,下层水相排出。
(6)、将步骤(3)中的正己烷提取液和步骤(5)中正己烷萃取液合并,依次以水、盐水洗涤,无水硫酸钠干燥,滤除干燥剂。
(7)、将步骤(6)中干燥后的产物进行减压浓缩回收正己烷,真空度-0.04mpa~-0.08mpa,得到吉马烯a粗品。
(8)、步骤(7)中得到吉马烯a粗品在反应釜中加热转化,维持釜内温度120~150℃,保温1小时,釜内吉马烯a转化为β-榄香烯,得到β-榄香烯粗品。
(9)、将步骤(8)中得到的β-榄香烯粗品在精馏塔中进行减压精馏,塔柱填料为不锈钢网θ环,塔釜温度100~140℃,回流比为0.5~3.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃,得到β-榄香烯236.00g。色谱分析所得β-榄香烯纯度为96.94%,相对于发酵液吉马烯a的产率为55.8%。
实施例6
(1)、将生物合成制备的吉马烯a发酵液40l(吉马烯a含量10.25g/l)以管式离心机离心,弃去上清液得到菌丝沉淀物11.7kg;
(2)、将菌丝沉淀物加入提取罐,以95%乙醇30l搅拌提取1小时,过滤,得到乙醇提取液和菌丝渣。
(3)、将步骤(2)中获得的菌丝渣以20l环己烷搅拌提取0.5小时,过滤分离收集环己烷提取液。
(4)、将步骤(2)中得到的乙醇提取液进行减压浓缩回收乙醇,真空度-0.06mpa~-0.08mpa,得到水油混合物。
(5)、步骤(4)中得到的水油混合物以环己烷4l萃取,得到环己烷萃取液,下层水相排出。
(6)、将步骤(3)中的环己烷提取液和步骤(5)中环己烷萃取液合并,依次以水、盐水洗涤,无水硫酸钠干燥,滤除干燥剂。
(7)、将步骤(6)中干燥后的产物进行减压浓缩回收环己烷,真空度-0.04mpa~-0.08mpa,得到吉马烯a粗品。
(8)、步骤(7)中得到吉马烯a粗品在反应釜中加热转化,维持釜内温度120~150℃,保温1小时,釜内吉马烯a转化为β-榄香烯,得到β-榄香烯粗品。
(9)、将步骤(8)中得到的β-榄香烯粗品在精馏塔中进行减压精馏,塔柱填料为不锈钢网θ环,塔釜温度100~140℃,回流比为0.5~3.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃,得到β-榄香烯273.47g。色谱分析所得β-榄香烯纯度为96.85%,相对于发酵液吉马烯a的产率为64.6%。
实施例7
(1)、将生物合成制备的吉马烯a发酵液180l(吉马烯a含量9.78g/l)以管式离心机离心,弃去上清液得到菌丝沉淀物44.3kg;
(2)、将菌丝沉淀物加入提取罐,以95%乙醇135l搅拌提取1小时,三足离心机过滤,得到乙醇提取液和菌丝渣。
(3)、将步骤(2)中获得的菌丝渣加入提取罐,以90l石油醚搅拌提取0.5小时,以板框过滤机过滤,得到石油醚提取液。
(4)、将步骤(2)中得到的乙醇提取液进行减压浓缩回收乙醇,真空度-0.06mpa,浓缩至残液中乙醇浓度小于5%,残液为吉马烯a的水油混合物。
(5)、步骤(4)中得到的水油混合物转入萃取罐,以石油醚18l萃取,得到石油醚萃取液,下层水相排出。
(6)、将步骤(3)中的石油醚提取液和步骤(5)中石油醚萃取液合并于洗涤罐,以水、盐水洗涤,无水硫酸钠干燥,滤除干燥剂。
(7)、将步骤(6)中干燥后的产物进行减压浓缩回收石油醚,真空度-0.04mpa~-0.08mpa,得到吉马烯a粗品。
(8)、步骤(7)中得到吉马烯a粗品在反应釜中加热转化,维持釜内温度120~150℃,保温1小时,釜内吉马烯a转化为β-榄香烯,得到β-榄香烯粗品。
(9)、将步骤(8)中得到的β-榄香烯粗品在精馏塔中进行减压精馏,塔柱填料为不锈钢网θ环,塔釜温度100~140℃,回流比为0.5~3.0,截取沸点为75-76℃/85pa的馏分,沸程不超过1℃,得到β-榄香烯1.137kg。色谱分析所得β-榄香烯纯度为97.49%,相对于发酵液吉马烯a的产率为62.97%。
- 还没有人留言评论。精彩留言会获得点赞!