苯甲腈衍生物、发光材料和使用该发光材料的发光元件
本发明涉及一种发光特性优异的2,3,4,5,6-五取代苯甲腈化合物、发光材料和使用该发光材料的发光元件。
本申请基于2018年9月5日在日本申请的日本特愿2018-165955号和2019年2月1日在日本申请的日本特愿2019-017156要求优先权,并将其内容援引于此。
背景技术:
已知某种咔唑-9-基取代苯甲腈化合物能够作为发光材料使用。
例如,专利文献1公开了3,5-二(3,6-二苯基-9h-咔唑-9-基)-2,4,6-三(4-氰基苯基)-苯甲腈等。专利文献2公开了2,3,5,6-四(3,6-二苯基-9h-咔唑-9-基)-4-(4-氰基苯基)-苯甲腈等。专利文献3公开了2,3,5,6-四(9h-咔唑-9-基)-4-苯基-苯甲腈等。
现有技术文献
专利文献
专利文献1:日本特表2016-539182号公报
专利文献2:wo2016/138077a
专利文献3:wo2014/183080a
技术实现要素:
本发明的课题在于提供一种发光特性优异的2,3,4,5,6-五取代苯甲腈化合物(以下,有时称为“本发明化合物”)、发光材料和使用该发光材料的发光元件。
为了解决上述课题,从而完成了包含以下方式的本发明。
〔1〕式(i)表示的化合物。
式(i)中,
l各自独立地为取代或非取代的芳基、或者取代或非取代的杂芳基,n表示l的数量,为1或2,
q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基、取代或非取代的3,6-二苯基-9h-咔唑-9-基、或者取代或非取代的3-苯基-6-叔丁基-9h-咔唑-9-基,且
m表示q的数量,为5-n。
〔2〕根据上述〔1〕所述的化合物,由式(iia)表示。
式(iia)中,l为取代或非取代的芳基、或者取代或非取代的杂芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基。
〔3〕根据上述〔1〕所述的化合物,由式(iib)表示。
式(iib)中,l为取代或非取代的芳基、或者取代或非取代的杂芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基。
〔4〕根据上述〔1〕所述的化合物,由式(iic)表示。
式(iic)中,l为取代或非取代的芳基、或者取代或非取代的杂芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基。
〔5〕根据上述〔1〕所述的化合物,由式(iiia)表示。
式(iiia)中,l各自独立地为取代或非取代的芳基、或者取代或非取代的杂芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
〔6〕根据上述〔1〕所述的化合物,由式(iiib)表示。
式(iiib)中,l各自独立地为取代或非取代的芳基、或者取代或非取代的杂芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
〔7〕根据上述〔1〕所述的化合物,由式(iiic)表示。
式(iiic)中,l各自独立地为取代或非取代的芳基、或者取代或非取代的杂芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
〔8〕根据上述〔1〕所述的化合物,由式(iva)表示。
式(iva)中,l各自独立地为取代或非取代的芳基、或者取代或非取代的杂芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
〔9〕根据上述〔1〕~〔8〕中任一项所述的化合物,其中,l为取代或非取代的含氮或含氧的5元环或6元环杂芳基。
〔10〕根据上述〔1〕~〔8〕中任一项所述的化合物,其中,l为取代或非取代的苯基、取代或非取代的联苯基、取代或非取代的萘基、取代或非取代的蒽基、取代或非取代的菲基、取代或非取代的吡啶基、取代或非取代的嘧啶基、取代或非取代的呋喃基、取代或非取代的噻吩基、取代或非取代的
〔11〕根据上述〔1〕~〔8〕中任一项所述的化合物,其中,l为取代或非取代的苯基、取代或非取代的吡啶基、或者取代或非取代的嘧啶基。
〔12〕一种发光材料,含有上述〔1〕~〔11〕中任一项所述的化合物。
〔13〕一种发光元件,含有上述〔12〕所述的发光材料。
本发明化合物作为发光材料是有用的。本发明的发光材料中有发射延迟荧光的材料。含有本发明的发光材料的发光元件能够实现优异的发光效率。
附图说明
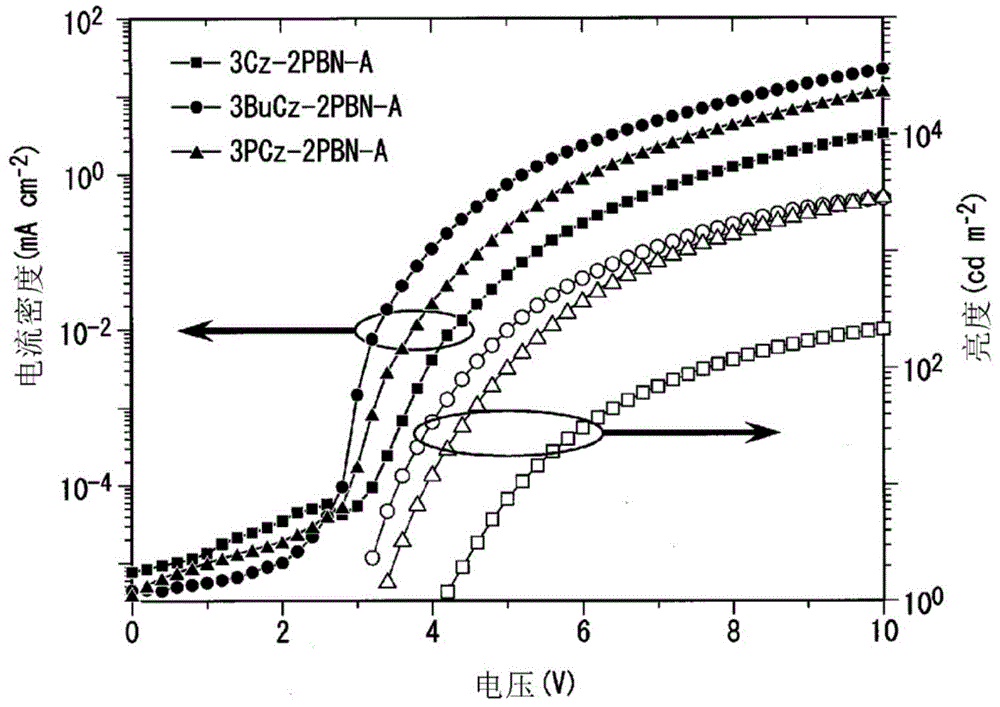
图1是表示3cz-2pbn-a、3bucz-2pbn-a和3pcz-2pbn-a的电压-电流密度-亮度的特性的图。
图2是表示3cz-2pbn-a、3bucz-2pbn-a和3pcz-2pbn-a的亮度-外量子效率的图。
图3是表示3cz-2pbn-b、3bucz-2pbn-b和3pcz-2pbn-b的电压-电流密度-亮度的特性的图。
图4是表示3cz-2pbn-b、3bucz-2pbn-b和3pcz-2pbn-b的亮度-外量子效率的图。
图5是表示4cz-1pbn-a和4bucz-1pbn-a的电压-电流密度-亮度的特性的图。
图6是表示4cz-1pbn-a和4bucz-1pbn-a的亮度-外量子效率的图。
图7是表示3bucz-2pbn-c、3pcz-2pbn-c、3bucz-2pbn-d和3pcz-2pbn-d的电压-电流密度-亮度的特性的图。
图8是表示3bucz-2pbn-c、3pcz-2pbn-c、3bucz-2pbn-d和3pcz-2pbn-d的亮度-外量子效率的图。
图9是表示4bucz-1pbn-a、4bucz-1pbn-b和4bucz-1pbn-c的电压-电流密度-亮度的特性的图。
图10是表示4bucz-1pbn-a、4bucz-1pbn-b和4bucz-1pbn-c的亮度-外量子效率的图。
图11是表示4x-bcz-pbn-bu、4x-bcz-pbn-ome、4x-bcz-pbn-sme和4x-bcz-pbn-cn的电压-电流密度-亮度的特性的图。
图12是表示4x-bcz-pbn-bu、4x-bcz-pbn-ome、4x-bcz-pbn-sme和4x-bcz-pbn-cn的亮度-外量子效率的图。
图13是表示4x-bcz-pbn-co2me、4x-bcz-pbn-mesbn和4x-bcz-pbn-ipn的电压-电流密度-亮度的特性的图。
图14是表示4x-bcz-pbn-co2me、4x-bcz-pbn-mesbn和4x-bcz-pbn-ipn的亮度-外量子效率的图。
图15是表示4x-bcz-pbn-2py、4x-bcz-pbn-3py、4x-bcz-pbn-4py和4x-bcz-pbn-5pm的电压-电流密度-亮度的特性的图。
图16是表示4x-bcz-pbn-2py、4x-bcz-pbn-3py、4x-bcz-pbn-4py和4x-bcz-pbn-5pm的亮度-外量子效率的图。
图17是表示3y-bcz-pbn-tbu、3y-bcz-pbn-ome和3y-bcz-pbn-sme的电压-电流密度-亮度的特性的图。
图18是表示3y-bcz-pbn-tbu、3y-bcz-pbn-ome和3y-bcz-pbn-sme的亮度-外量子效率的图。
图19是表示3f-bcz-pbn-tbu、3f-bcz-pbn-ome和3f-bcz-pbn-sme的电压-电流密度-亮度的特性的图。
图20是表示3f-bcz-pbn-tbu、3f-bcz-pbn-ome和3f-bcz-pbn-sme的亮度-外量子效率的图。
具体实施方式
本发明的2,3,4,5,6-五取代苯甲腈化合物为式(i)表示的化合物。
式(i)中,
l各自独立地为取代或非取代的芳基,
n表示l的数量,为1或2,
q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基、取代或非取代的3,6-二苯基-9h-咔唑-9-基、或者取代或非取代的3-苯基-6-叔丁基-9h-咔唑-9-基,且
m表示q的数量,为5-n。
本发明的2,3,4,5,6-五取代苯甲腈化合物优选为式(iia)、式(iib)、式(iic)、式(iiia)、式(iiib)、式(iiic)或式(iva)表示的化合物,更优选为式(iia)表示的化合物。本发明的2,3,4,5,6-五取代苯甲腈化合物也可以为式(iiid)或式(iiie)表示的化合物。
式(iia)中,l为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基。
式(iib)中,l为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基。
式(iic)中,l为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基。
式(iiia)中,l各自独立地为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
式(iiib)中,l各自独立地为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
式(iiic)中,l各自独立地为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
式(iiid)中,l各自独立地为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
式(iiie)中,l各自独立地为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
式(iva)中,l各自独立地为取代或非取代的芳基,且q各自独立地为取代或非取代的3,6-二叔丁基-9h-咔唑-9-基或者取代或非取代的3,6-二苯基-9h-咔唑-9-基。
取代或非取代的3,6-二叔丁基-9h-咔唑-9-基优选为式(a)表示的基团。
取代或非取代的3,6-二苯基-9h-咔唑-9-基优选为式(b)表示的基团。
取代或非取代的3-苯基-6-叔丁基-9h-咔唑-9-基优选为式(c)表示的基团。
式(a)、(b)和(c)中,r1、r2、r3、r4、r5和r6各自独立地为氢原子或取代基,*为键合部分。
本发明中,术语“非取代(unsubstituted)”是指仅作为母核的基团。仅用作为母核的基团的名称记载时,只要没有特别说明,就为“非取代”的含义。
另一方面,术语“取代(substituted)”是指成为母核的基团中的任一氢原子被与母核相同或不同结构的基团取代。因此,“取代基”是指键合于作为母核的基团的另一基团。取代基可以为1个,也可以为2个以上。2个以上的取代基可以相同,也可以不同。
“取代基”只要是化学上允许且具有本发明的效果,就没有特别限制。
作为可成为“取代基”的基团的具体例,可以举出以下的基团。
氟基、氯基、溴基、碘基等卤素基;
甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、正己基等c1~20烷基(优选为c1~6烷基);
乙烯基、1-丙烯基、2-丙烯基、1-丁烯基、2-丁烯基、3-丁烯基、1-甲基-2-丙烯基、2-甲基-2-丙烯基、1-戊烯基、2-戊烯基、3-戊烯基、4-戊烯基、1-甲基-2-丁烯基、2-甲基-2-丁烯基、1-己烯基、2-己烯基、3-己烯基、4-己烯基、5-己烯基等c2~10烯基(优选c2~6烯基);
乙炔基、1-丙炔基、2-丙炔基、1-丁炔基、2-丁炔基、3-丁炔基、1-甲基-2-丙炔基、2-甲基-3-丁炔基、1-戊炔基、2-戊炔基、3-戊炔基、4-戊炔基、1-甲基-2-丁炔基、2-甲基-3-戊炔基、1-己炔基、1,1-二甲基-2-丁炔基等c2~10炔基(c2~6炔基);
环丙基、环丁基、环戊基、环己基、环庚基、立方烷基等c3~8环烷基;
2-环丙烯基、2-环戊烯基、3-环己烯基、4-环辛烯基等c3~8环烯基;
苯基、萘基等c6~40芳基(优选c6~10芳基);
吡咯基、呋喃基、噻吩基、咪唑基、吡唑基、
吡啶基、吡嗪基、嘧啶基、哒嗪基、三嗪基等6元环的杂芳基;
吲哚基、苯并呋喃基、苯并噻吩基、苯并咪唑基、苯并
环氧乙烷基、四氢呋喃基、二氧戊环基、二氧六环基(ジオキラニル基)等环状醚基;
氮丙啶基、吡咯烷基、哌啶基、哌嗪基、吗啉基等环状氨基;
羟基;氧代基;
甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、仲丁氧基、异丁氧基、叔丁氧基等c1~20烷氧基(优选c1~6烷氧基);
乙烯氧基、烯丙氧基、丙烯氧基、丁烯氧基等c2~6烯氧基;
乙炔氧基、炔丙氧基等c2~6炔氧基;
苯氧基、萘氧基等c6~10芳氧基;
噻唑氧基、吡啶氧基等5~6元环的杂芳氧基;
羧基;
甲酰基;乙酰基、丙酰基等c1~6烷基羰基;
甲酰氧基;乙酰氧基、丙酰氧基等c1~6烷基羰基氧基;
甲氧羰基、乙氧羰基、正丙氧羰基、异丙氧羰基、正丁氧羰基、叔丁氧羰基等c1~6烷氧羰基;
氯甲基、氯乙基、三氟甲基、1,2-二氯正丙基、1-氟正丁基、全氟正戊基等c1~6卤代烷基;
2-氯-1-丙烯基、2-氟-1-丁烯基等c2~6卤代烯基;
4,4-二氯-1-丁炔基、4-氟-1-戊炔基、5-溴-2-戊炔基等c2~6卤代炔基;
3,3-二氟环丁基等c3~6卤代环烷基;
2-氯正丙氧基、2,3-二氯丁氧基、三氟甲氧基、2,2,2-三氟乙氧基等c1~6卤代烷氧基;
2-氯丙烯氧基、3-溴丁烯氧基等c2~6卤代烯氧基;
氯乙酰基、三氟乙酰基、三氯乙酰基等c1~6卤代烷基羰基;
氰基;硝基;氨基;
甲基氨基、二甲基氨基、二乙基氨基等c1~20烷基氨基(优选c1~6烷基氨基);
苯胺基、萘基氨基等c6~40芳基氨基(优选c6~10芳基氨基);
甲酰氨基;乙酰氨基、丙酰氨基、丁酰氨基、异丙基羰基氨基等c1~6烷基羰基氨基;
甲氧羰基氨基、乙氧羰基氨基、正丙氧羰基氨基、异丙氧羰基氨基等c1~6烷氧羰基氨基;
s,s-二甲基亚磺酰亚胺基(s,s-dimethylsulfoximinogroup)等c1~6烷基亚磺酰亚胺基;
氨基羰基;
甲基氨基羰基、二甲基氨基羰基、乙基氨基羰基、异丙基氨基羰基等c1~6烷基氨基羰基;
亚氨基甲基、(1-亚氨基)乙基、(1-亚氨基)-正丙基等亚氨基c1~6烷基;
羟基亚氨基甲基、(1-羟基亚氨基)乙基、(1-羟基亚氨基)丙基等羟基亚氨基c1~6烷基;
甲氧基亚氨基甲基、(1-甲氧基亚氨基)乙基等c1~6烷氧基亚氨基c1~6烷基;
巯基;
甲硫基、乙硫基、正丙硫基、异丙硫基、正丁硫基、异丁硫基、仲丁硫基、叔丁硫基等c1~20烷硫基(优选c1~6烷硫基);
三氟甲硫基、2,2,2-三氟乙硫基等c1~6卤代烷硫基;
乙烯基硫基、烯丙基硫基等c2~6烯基硫基;
乙炔基硫基、丙炔基硫基等c2~6炔基硫基;
甲基亚磺酰基、乙基亚磺酰基、叔丁基亚磺酰基等c1~6烷基亚磺酰基;
三氟甲基亚磺酰基、2,2,2-三氟乙基亚磺酰基等c1~6卤代烷基亚磺酰基;
烯丙基亚磺酰基等c2~6烯基亚磺酰基;
丙炔基亚磺酰基等c2~6炔基亚磺酰基;
甲基磺酰基、乙基磺酰基、叔丁基磺酰基等c1~6烷基磺酰基;
三氟甲基磺酰基、2,2,2-三氟乙基磺酰基等c1~6卤代烷基磺酰基;
烯丙基磺酰基等c2~6烯基磺酰基;
丙炔基磺酰基等c2~6炔基磺酰基;
乙酰胺基、n-甲基酰胺基、n-乙基酰胺基、n-(正丙基)酰胺基、n-(正丁基)酰胺基、n-异丁基酰胺基、n-(仲丁基)酰胺基、n-(叔丁基)酰胺基、n,n-二甲基酰胺基、n,n-二乙基酰胺基、n,n-二(正丙基)酰胺基、n,n-二(正丁基)酰胺基、n,n-二异丁基酰胺基、n-甲基乙酰胺基、n-乙基乙酰胺基、n-(正丙基)乙酰胺基、n-(正丁基)乙酰胺基、n-异丁基乙酰胺基、n-(仲丁基)乙酰胺基、n-(叔丁基)乙酰胺基、n,n-二甲基乙酰胺基、n,n-二乙基乙酰胺基、n,n-二(正丙基)乙酰胺基、n,n-二(正丁基)乙酰胺基、n,n-二异丁基乙酰胺基等c2~20烷基酰胺基;
苯基酰胺基、萘基酰胺基、苯基乙酰胺基、萘基乙酰胺基等c6~20芳基酰胺基;
三甲基甲硅烷基、三乙基甲硅烷基、叔丁基二甲基甲硅烷基等三c1~10烷基甲硅烷基(优选三c1~6烷基甲硅烷基);
三苯基甲硅烷基等三c6~10芳基甲硅烷基;
另外,对于这些“取代基”,上述取代基中的任一氢原子可以被不同结构的基团取代。
“c1~6”等术语表示作为母核的基团的碳原子数为1~6个等。该碳原子数不包含取代基中存在的碳原子的个数。例如,乙氧基丁基的作为母核的基团为丁基,由于取代基为乙氧基,因此分类为c2烷氧基c4烷基。
作为优选的取代基,可举出羟基、卤素基团、氰基、碳原子数1~20的烷基、碳原子数1~20的烷氧基、碳原子数1~20的烷硫基、碳原子数1~20的烷基取代氨基、碳原子数2~20的酰基、碳原子数6~40的芳基、碳原子数3~40的杂芳基、碳原子数12~40的二芳基氨基、碳原子数12~40的取代或非取代的咔唑基、碳原子数2~10的烯基、碳原子数2~10的炔基、碳原子数2~10的烷氧羰基、碳原子数1~10的烷基磺酰基、碳原子数1~10的卤代烷基、酰胺基、碳原子数2~10的烷基酰胺基、碳原子数3~20的三烷基甲硅烷基、碳原子数4~20的三烷基甲硅烷基烷基、碳原子数5~20的三烷基甲硅烷基烯基、碳原子数5~20的三烷基甲硅烷基炔基和硝基。
作为更优选的取代基,可以举出卤素基团、氰基、碳原子数1~20的烷基、碳原子数1~20的烷氧基、碳原子数6~40的芳基、碳原子数3~40的杂芳基、碳原子数12~40的二芳基氨基、碳原子数12~40的咔唑基。
作为进一步优选的取代基,可以举出氟基、氯基、氰基、碳原子数1~10的烷基、碳原子数1~10的烷氧基、碳原子数1~10的二烷基氨基、碳原子数6~15的芳基、碳原子数3~12的杂芳基。这些取代基中的可进一步取代的取代基可以被上述取代基所取代。
作为l中的取代或非取代的芳基,可以优选举出取代或非取代的苯基、取代或非取代的联苯基、取代或非取代的萘基、取代或非取代的蒽基、或者取代或非取代的菲基。其中,更优选取代或非取代的苯基。
本发明化合物不特别受其制造方法限定,例如,可以将相当于对应的取代基的化合物作为起始原料,利用专利文献1、专利文献2中记载的方法,或者利用实施例中记载的方法而得到。
所合成的本发明化合物的纯化可以通过基于柱色谱的纯化、基于硅胶、活性炭、活性白土等的吸附纯化、基于溶剂的重结晶、晶析法等而进行。化合物的鉴定可以通过nmr分析等而进行。
本发明化合物可以作为发光材料使用。本发明的发光材料可以提供有机光致发光元件、有机电致发光元件等发光元件。本发明化合物由于具有辅助其他发光材料(主体材料)的发光的功能,因此能够掺杂于其他发光材料中使用。
作为本发明的发光元件之一的有机光致发光元件是在基板上设置含有本发明的发光材料的发光层而成的。发光层可以利用旋涂法等涂布法、喷墨印刷法等印刷法、蒸镀法等而得到。
本发明的有机电致发光元件是在阳极与阴极之间设置有机层而成的。本发明中的“有机层”是指位于阳极与阴极之间的实质上由有机物构成的层,这些层可以在不损害本发明的发光元件的性能的范围含有无机物。
作为本发明的有机电致发光元件的一个实施方式的结构,可以举出在基板上依次由阳极、空穴注入层、空穴传输层、电子阻挡层、发光层、空穴阻挡层、电子传输层、阴极构成的结构,另外在电子传输层与阴极之间进一步具有电子注入层的结构。在这些多层结构中可以省略几层有机层,例如也可以为在基板上依次为阳极、空穴传输层、发光层、电子传输层、电子注入层、阴极的结构,在基板上依次为阳极、空穴传输层、发光层、电子传输层、阴极的结构。
本发明的发光材料不仅可以掺杂于发光层,也可以掺杂于空穴注入层、空穴传输层、电子阻挡层、空穴阻挡层、电子传输层或电子注入层。
基板为发光元件的支承体,可使用硅板、石英板、玻璃板、金属板、金属箔、树脂膜、树脂片等。特别优选玻璃板、聚酯、聚甲基丙烯酸酯、聚碳酸酯、聚砜等透明合成树脂的板。使用合成树脂基板时,需要注意气体阻隔性。如果基板的气体阻隔性过低,则有时发光元件因通过基板的外部空气而劣化。因此,优选在合成树脂基板的某一侧或两侧设置致密的硅氧化膜等而确保气体阻隔性。
在基板上设置阳极。阳极一般使用功函数大的材料。作为阳极用材料,例如,可以举出铝、金、银、镍、钯、铂等金属;铟氧化物、锡氧化物、ito、氧化锌、in2o3-zno、igzo等金属氧化物、碘化铜等卤化金属、炭黑、或聚(3-甲基噻吩)、聚吡咯、聚苯胺等导电性高分子等。阳极的形成通常大多通过溅射法、真空蒸镀法等而进行。另外,在银等金属微粒、碘化铜等的微粒、炭黑、导电性的金属氧化物微粒、导电性高分子微粉末等的情况下,也可以通过分散于适当的粘结剂树脂溶液,并涂布于基板上而形成阳极。此外,为导电性高分子时,也可以通过电解聚合而直接在基板上形成薄膜,或者在基板上涂布导电性高分子而形成阳极。
阳极也可以通过层叠不同的2种以上的物质而形成。阳极的厚度根据所需要的的透明性而不同。需要透明性时,优选使可见光的透射率通常为60%以上、优选为80%以上,该情况下,厚度通常为10~1000nm,优选为10~200nm。可以为不透明的情况下,阳极可以与基板的厚度为同等程度。阳极的薄层电阻优选为几百ω/□以上。
作为根据需要而设置的空穴注入层,除了以酞菁铜为代表的卟啉化合物以外,还可以使用萘二胺衍生物、星爆型三苯基胺衍生物、具有在分子中具有3个以上的三苯基胺结构且由单键或不含有杂原子的2价基团连接而成的结构的芳基胺化合物等三苯基胺三聚体和四聚体、六氰基氮杂苯并菲(hexacyanoazatriphenylene)这样的受体性杂环化合物、涂布型高分子材料。这些材料除了蒸镀法以外,还可以利用旋涂法、喷墨法等公知的方法来进行薄膜形成。
作为根据需要而设置的空穴传输层中使用的空穴输送材料,优选自阳极的空穴注入效率高且能够高效地传输被注入的空穴。因此,优选离子化势能小,相对于可见光的光线的透明性高,而且空穴迁移率大,进而稳定性优异,在制造时、使用时不易产生成为陷阱的杂质。除上述的一般的要求以外,考虑到车载显示用的应用时,元件进一步优选耐热性高。因此,优选tg具有70℃以上的值的材料。
作为根据需要而设置的空穴传输层,可以举出三唑衍生物、
更具体而言,可以举出含有间咔唑基苯基的化合物、n,n’-二苯基-n,n’-二(间甲苯基)-联苯胺(以下,简称为tpd)、n,n’-二苯基-n,n’-二(α-萘基)-联苯胺(以下,简称为npd)、n,n,n’,n’-四联苯基联苯胺等联苯胺衍生物、1,1-双[(二-4-甲苯基氨基)苯基]环己烷(以下,简称为tapc)、各种三苯基胺三聚体和四聚体、咔唑衍生物等。它们可以单独使用1种或者组合2种以上使用。空穴传输层可以为单层结构的膜,也可以为层叠结构的膜。另外,作为空穴的注入·传输层,可以使用聚(3,4-乙撑二氧噻吩)(以下,简称为pedot)/聚(苯乙烯磺酸盐)(以下,简称为pss)等涂布型的高分子材料。这些材料除了蒸镀法以外,还可以利用旋涂法、喷墨法等公知的方法来进行薄膜形成。
另外,在空穴注入层或空穴传输层中,可以使用对上述层中通常使用的材料进一步p掺杂有三溴苯基六氯锑酸铵的物质、在其部分结构中具有pd结构的高分子化合物等。作为空穴注入·传输性的主体材料,可以使用ppf、ppt、cbp、tcta、mcp等咔唑衍生物等。
以下举出可用作空穴注入材料的优选的化合物(hi1)~(hi7)。
以下举出可用作空穴传输材料的优选的化合物(ht1)~(ht38)。
作为根据需要而设置的电子阻挡层,可以使用4,4’,4”-三(n-咔唑基)三苯基胺(以下,简称为tcta)、9,9-双[4-(咔唑-9-基)苯基]芴、1,3-双(咔唑-9-基)苯(以下,简称为mcp)、2,2-双(4-咔唑-9-基苯基)金刚烷(以下,简称为ad-cz)等咔唑衍生物、以9-[4-(咔唑-9-基)苯基]-9-[4-(三苯基甲硅烷基)苯基]-9h-芴为代表的具有三苯基甲硅烷基和三芳基胺结构的化合物等具有电子阻挡作用的化合物。它们可以单独使用1种或者组合2种以上使用。电子阻挡层可以为单层结构的膜,也可以为层叠结构的膜。这些材料除了蒸镀法以外,还可以利用旋涂法、喷墨法等公知的方法而进行薄膜形成。
以下举出可用作电子阻止材料的优选的化合物(es1)~(es5)。
发光层是具有通过分别从阳极和阴极注入的空穴和电子复合而产生激子、进行发光的功能的层。发光层可以由本发明的发光材料单独形成,也可以在主体材料中掺杂本发明的发光材料而形成。作为主体材料的例子,可以举出ppf、ppt、三(8-羟基喹啉)铝(以下,简称为alq3)等喹啉醇衍生物的金属配合物、蒽衍生物、双苯乙烯基苯衍生物、芘衍生物、
使用主体材料时,可以使发光层含有的本发明的发光材料的量的下限优选为0.1质量%,更优选为1质量%,上限优选为50质量%,更优选为20质量%,进一步优选为10质量%。
以下举出可用作发光层的主体材料的优选的化合物(el1)~(el42)。
作为根据需要而设置的空穴阻挡层,可以举出具有联吡啶基和邻三苯基结构的化合物、浴铜灵(以下,简称为bcp)等菲咯啉衍生物、双(2-甲基-8-喹啉根合)-4-苯基苯酚铝(iii)(以下,简称为balq)等喹啉醇衍生物的金属配合物、各种的稀土配合物、
以下举出可用作空穴阻挡材料的优选的化合物(hs1)~(hs11)。
作为根据需要而设置的电子传输层,除了以alq3、balq为代表的喹啉醇衍生物的金属配合物以外,还可以使用各种金属配合物、三唑衍生物、三嗪衍生物、
作为根据需要而设置的电子注入层,可以使用氟化锂、氟化铯等碱金属盐、氟化镁等碱土金属盐、氧化铝等金属氧化物(可简称为“金属氧化物”)等,在电子传输层和阴极的优选的选择中,可以省略它。
电子注入层或电子传输层中,可以使用对上述层中通常使用的材料进一步n掺杂有铯等金属的物质。
以下举出可用作电子传输材料的优选的化合物(et1)~(et30)。
以下举出可用作电子注入材料的优选的化合物(ei1)~(ei4)。
以下举出可用作稳定化材料的优选的化合物(st1)~(st5)。
阴极一般使用功函数小的材料。作为阴极用材料,例如,可使用钠、钠-钾合金、锂、锡、镁、镁/铜混合物、镁/铝混合物、镁/铟混合物、铝/氧化铝混合物、铟、钙、铝、银、锂/铝混合物、镁银合金、镁铟合金、铝镁合金等。通过使用透明导电性材料能够得到透明或半透明的阴极。阴极的厚度通常为10~5000nm,优选为50~200nm。阴极的薄层电阻优选为几百ω/□以上。
为了保护由低功函数金属形成的阴极,在其上进一步层叠铝、银、镍、铬、金、铂等功函数高且对大气稳定的金属层时,增加元件的稳定性,因而优选。另外,为了提高阴极与邻接的有机层(例如电子传输层、电子注入层)的接触,可以在两者之间设置阴极界面层。作为阴极界面层所使用的材料,可以举出芳香族二胺化合物、喹吖啶酮化合物、萘并萘衍生物、有机硅化合物、有机磷化合物、具有n-苯基咔唑骨架的化合物、n-乙烯基咔唑聚合物等。
本发明的发光元件也可以用于单一的元件、由以阵列状配置的结构构成的元件、阳极和阴极以x-y矩阵状配置的结构中的任一者。
实施例
以下,示出本发明化合物的合成的一个例子,并示出本发明化合物所起到的效果的例子。
(实施例1)
〔2,4,6-三(9h-咔唑-9-基)-3,5-二苯基-苯甲腈(3cz-2pbn-a)的合成〕
将碳酸钾(15.36g,111.1mmol)和9h-咔唑(13.5g,80.8mmol)加入到氮置换后的100ml的三口烧瓶中,进一步加入脱水n-甲基-2-吡咯烷酮100ml,以室温搅拌1小时。在氮气流下向其中加入2,4,6-三氟-3,5-二苯基苯甲腈(6.25g,20.2mmol),以80℃搅拌23小时。接着,恢复到室温,加入甲醇,通过过滤而除去固体。其后,向滤液中加入水,将析出的晶体用丙酮和己烷清洗,减压下干燥,以收量1.40g、收率9.2%得到目标物(3cz-2pbn-a)的黄色固体。
1h-nmr(400mhz,dmso-d6,δ):8.07(d,j=8.0hz,4h),7.81-7.78(m,8h),7.49(td,j=8.0hz,0.8hz,4h),7.37(td,j=7.2hz,0.8hz,2h),7.23(t,j=7.6hz,4h),7.06(t,j=8.0hz,2h),6.81(dd,j=7.2hz,1.2hz,4h),6.55(tt,j=7.6hz,1.6hz,2h),6.44(t,j=7.6hz,4h)
〔发光评价〕
利用真空蒸镀法(5.0×10-4pa以下)在形成了膜厚50nm的由铟·锡氧化物(ito)构成的阳极的玻璃基板上依次层叠10nm厚的hat-cn层、40nm厚的tapc层、10nm厚的ccp层、10nm厚的mcp层和20nm厚的发光层。
使用ppf作为发光层的主体材料,使用2,4,6-三(9h-咔唑-9-基)-3,5-二苯基-苯甲腈(3cz-2pbn-a)作为掺杂材料。将掺杂材料浓度设定为12.0重量%。
接下来,利用真空蒸镀法依次层叠10nm厚的ppf层、40nm厚的b3pypb层、1nm厚的8-羟基喹啉锂膜和100nm厚的铝膜而形成阴极,得到有机发光二极管(oled)。将结果示于图1和2。该有机发光二极管的外量子效率的最大值(eqemax)为13.1%。
(实施例2)
〔2,4,6-三(3,6-二苯基-9h-咔唑-9-基)-3,5-二苯基-苯甲腈(3pcz-2pbn-a)的合成〕
将碳酸钾(2.50g,17.8mmol)和3,6-二苯基-9h-咔唑(4.10g,12.9mmol)加入到氮置换后的100ml的三口烧瓶中,进一步加入脱水n-甲基-2-吡咯烷酮16.2ml,以室温搅拌1小时。在氮气流下向其中加入2,4,6-三氟ー3,5-二苯基苯甲腈(1.00g,3.23mmol),以80℃搅拌16小时。接下来,恢复到室温,加入甲醇,通过过滤而除去固体。其后,向滤液中加入水,将析出的晶体用丙酮和己烷清洗,将固体在减压下干燥,以收量1.20g、收率30.8%得到目标物(3pcz-2pbn-a)的绿色固体。
1h-nmr(400mhz,dmso-d6,δ):8.59(d,j=1.6hz,4h),8.37(d,j=2.0hz,2h),8.02-7.99(m,6h),7.87-7.75(m,18h),7.52-7.45(m,12h),7.37-7.30(m,6h),7.03(dd,j=7.0hz,1.2hz,4h),6.66-6.58(m,6h)
〔发光评价〕
将掺杂材料换成2,4,6-三(3,6-二苯基-9h-咔唑-9-基)-3,5-二苯基-苯甲腈(3pcz-2pbn-a),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图1和2。eqemax为28.1%。
(实施例3)
〔2,4,6-三(3,6-二叔丁基-9h-咔唑-9-基)-3,5-二苯基-苯甲腈(3bucz-2pbn-a)的合成〕
将碳酸钾(2.83g,20.5mmol)和3,6-二叔丁基-咔唑(4.16g,14.9mmol)加入到氮置换后的100ml的三口烧瓶中,进一步加入脱水n-甲基-2-吡咯烷酮20ml,以室温搅拌1小时。在氮气流下向其中加入2,4,6-三氟-3,5-二苯基苯甲腈(1.15g,3.72mmol),以80℃搅拌32小时。接下来,恢复到室温,加入甲醇,通过过滤而除去固体。其后,向滤液中加入水,使析出的晶体溶解于氯仿,进行水洗。其后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯=19/1)对浓缩物进行纯化,以收量1.20g、收率29.7%得到目标物(3bucz-2pbn-a)的淡黄色固体。
1h-nmr(400mhz,cdcl3,δ):7.94(d,j=2.0hz,4h),7.73(d,j=1.6hz,2h),7.42(dd,j=8.8hz,1.6hz,4h),7.27(dd,j=8.6hz,2.0hz,4h),7.13(d,j=8hz,4h),6.98(d,j=8.8hz,2h),6.63(d,j=7.8hz,4h),6.50(t,j=6.4hz,2h),6.38(t,j=7.6hz,4h),1.39(s,36h),1.31(s,18h)
〔发光评价〕
将掺杂材料换成2,4,6-三(3,6-二叔丁基-9h-咔唑-9-基)-3,5-二苯基-苯甲腈(3bucz-2pbn-a),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图1和2。eqemax为22.6%。
将为相同的2,3-苯基取代的骨架的实施例1、实施例2和实施例3进行对比时,可知本发明化合物(实施例2和实施例3)的eqemax相对较高,作为发光材料是有用的。
(实施例4)
〔2,3,5-三(9h-咔唑-9-基)-4,6-二苯基-苯甲腈(3cz-2pbn-b)的合成〕
将碳酸钾(2.76g,20.0mmol)和9h-咔唑(2.42g,14.5mmol)加入到氮置换后的100ml的三口烧瓶中,进一步加入脱水n-甲基-2-吡咯烷酮18ml,以室温搅拌1小时。在氮气流下使2,3,5-三氟-4,6-二苯基苯甲腈(1.12g,3.6mmol)溶解于脱水n-甲基-2-吡咯烷酮18ml并加入到其中,以100℃搅拌20小时。接下来,恢复到室温,加入水和乙酸乙酯,将有机层分液。进一步将水层用乙酸乙酯萃取2次,将混合的有机层用水清洗3次,接着用饱和食盐水清洗2次。将有机层用硫酸镁脱水,进行过滤,浓缩滤液,由此得到粗产物。利用硅胶柱色谱法(洗脱液:正己烷/乙酸乙酯)对粗产物进行纯化而得到粗纯化物。进一步,在粗纯化物中加入丙酮/正己烷,进行超声波照射。其后,进行过滤,用正己烷清洗,由此以收率90.5%得到2.46g的目标物(3cz-2pbn-b)的淡黄色固体。
1h-nmr(400mhz,dmso-d6,δ):7.88(d,j=8.0hz,2h),7.85(d,j=8.0hz,2h),7.82(d,j=8.0hz,2h),7.80(d,j=8.0hz,2h),7.64(d,j=8.4hz,2h),7.60(d,j=7.6hz,2h),7.41-7.37(m,2h),7.35-7.32(m,2h),7.25-7.21(m,2h),7.12-7.03(m,9h),6.89(t,j=7.6hz,2h),6.68(d,j=7.2hz,2h),6.42(t,j=7.6hz,1h),6.27(t,j=7.6hz,2h)
〔发光评价〕
将掺杂材料换成2,3,5-三(9h-咔唑-9-基)-4,6-二苯基-苯甲腈(3cz-2pbn-b),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图3和4。eqemax为16.7%。
(实施例5)
〔2,3,5-三(3,6-二苯基-9h-咔唑-9-基)-4,6-二苯基-苯甲腈(3pcz-2pbn-b)的合成〕
将碳酸钾(0.74g,5.4mmol)和3,6-二苯基-9h-咔唑(1.12g,3.5mmol)加入到氮置换后的100ml的三口烧瓶中,进一步加入脱水n-甲基-2-吡咯烷酮8.0ml,以室温搅拌1小时。在氮气流下向其中加入2,3,5-三氟-4,6-二苯基苯甲腈(0.24g,0.8mmol),以100℃搅拌4天。接下来,恢复到室温,加入水和乙酸乙酯,将有机层分液。进一步将水层用乙酸乙酯萃取2次,将混合的有机层用水清洗3次,接着用饱和食盐水清洗2次。将有机层用硫酸镁脱水,进行过滤,浓缩滤液,由此得到粗产物。利用硅胶柱色谱法(洗脱液:正己烷/二氯甲烷)对粗产物进行纯化,由此得到粗纯化物。进一步,在粗纯化物中加入二氯甲烷/乙醚/正己烷,进行超声波照射。其后,进行过滤,用正己烷清洗,由此以收率96.1%得到0.90g的作为目标物(3pcz-2pbn-b)的黄色固体。
1h-nmr(400mhz,dmso-d6,δ):8.44(d,j=2.0hz,2h),8.27(d,j=1.6hz,2h),8.05(d,j=1.6hz,2h),8.00(d,j=8.8hz,2h),7.89(d,j=8.8hz,2h),7.82-7.79(m,6h),7.73(d,j=8.8hz,2h),7.63(d,j=7.6hz,4h,),7.56(d,j=7.2hz,4h),7.53-7.47(m,8h),7.43-7.26(m,16h),7.19-7.15(m,3h),6.97(d,j=7.2hz,2h),6.58-6.48(m,3h)
〔发光评价〕
将掺杂材料换成2,3,5-三(3,6-二苯基-9h-咔唑-9-基)-4,6-二苯基-苯甲腈(3pcz-2pbn-b),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图3和4。eqemax为35.1%。
(实施例6)
〔2,3,5-三(3,6-二叔丁基-9h-咔唑-9-基)-4,6-二苯基-苯甲腈(3bucz-2pbn-b)的合成〕
将碳酸钾(4.04g,29.2mmol)和3,6-二苯基-9h-咔唑(5.47g,19.6mmol)加入到氮置换后的200ml的三口烧瓶中,进一步加入脱水n-甲基-2-吡咯烷酮32.0ml,以室温搅拌1小时。在氮气流下向其中加入2,3,5-三氟-4,6-二苯基苯甲腈(1.00g,3.2mmol),以100℃搅拌3天。接下来,恢复到室温,加入水和乙酸乙酯,将有机层分液。进一步将水层用乙酸乙酯萃取2次,将混合的有机层用水清洗3次,接着用饱和食盐水清洗2次。将有机层用硫酸镁脱水,进行过滤,浓缩滤液,由此得到粗产物。利用硅胶柱色谱法(洗脱液:正己烷/二氯甲烷)对粗产物进行纯化,由此得到粗纯化物。进一步,在粗纯化物中加入正己烷,进行超声波照射。其后,进行过滤,用正己烷清洗,以收率79.3%得到2.79g的作为目标物(3bucz-2pbn-b)的淡黄白色固体。
1h-nmr(400mhz,dmso-d6,δ):7.90(d,j=2.0hz,2h),7.74(d,j=2.0hz,2h),7.58(d,j=8.8hz,2h),7.50(d,j=1.6hz,2h),7.39(td,j=8.8hz,2.0hz,4h),7.27(d,j=8.4hz,2h),7.18(d,j=8.8hz,2h),7.11-7.05(m,5h),6.97(dd,j=8.8hz,2.0hz,2h),6.78(d,7.2hz,2h),6.47(t,j=8.0hz,1h),6.36(t,j=8.0hz,2h),1.35(s,18h),1.31(s,18h),1.22(s,18h)
〔发光评价〕
将掺杂材料换成2,3,5-三(3,6-二叔丁基-9h-咔唑-9-基)-4,6-二苯基-苯甲腈(3bucz-2pbn-b),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图3和4。eqemax为24.4%。
将为相同的1,4苯基取代的骨架的实施例4、实施例5和实施例6进行对比时,可知本发明化合物(实施例5和实施例6)的eqemax相对较高,作为发光材料是有用的。
(实施例7)
〔2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-苯基-苯甲腈(4bucz-1pbn-a)的合成〕
向氮置换后的烧瓶中加入乙酸钯(ii)(0.64g,2.85mmol)、碳酸银(15.8g,57.3mmol)和二苯基碘
1hnmr(400mhz,cdcl3):δ7.56-7.52(m,3h),7.48-7.45(m,2h).
向氮置换后的烧瓶中加入3,6-二叔丁基咔唑(1.37g,4.90mmol)和n,n-二甲基甲酰胺(10ml)。其后,以室温加入添加了叔丁醇钾(0.55g,4.90mmol)的n,n-二甲基甲酰胺(5ml),以室温搅拌30分钟。其后,用10分钟滴加加入了化合物1(0.3g,1.19mmol)的n,n-二甲基甲酰胺(10ml)。其后以80℃搅拌10小时。恢复到室温,加入水(20ml),经过30分钟后加入氯仿进行萃取。向有机层中加入硫酸钠使其干燥,利用柱色谱法(乙酸乙酯:己烷=1:9)进行纯化,得到黄色固体的化合物2(4bucz-1pbn-a)(1.32g,86%)。
1hnmr(400mhz,dmso-d6):δ7.76(d,j=1.2hz,4h),7.56(d,j=1.2hz,4h),7.46(d,j=8.4hz,4h),7.42(d,j=8.8hz,4h),7.09(dd,j=8.8,1.2hz,6h),7.02(dd,j=8.4,1.6hz,4h),6.60-6.57(m,3h),1.32(s,36h),1.26(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-苯基-苯甲腈(4bucz-1pbn-a),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图5和6。eqemax为40.1%。
(实施例8)
〔2,3,5,6-四(9h-咔唑-9-基)-4-苯基-苯甲腈(4cz-1pbn-a)的合成〕
向氮置换后的烧瓶中加入乙酸钯(ii)(0.64g,2.85mmol)、碳酸银(15.8g,57.3mmol)和二苯基碘
向氮置换后的烧瓶中加入咔唑(1.09g,6.52mmol)和n,n-二甲基甲酰胺(10ml)。其后,以室温加入添加了叔丁醇钾(0.73g,6.52mmol)的n,n-二甲基甲酰胺(5ml),以室温搅拌30分钟。其后,用10分钟滴加加入了化合物1(0.4g,1.59mmol)的n,n-二甲基甲酰胺(10ml)。其后,以80℃搅拌10小时。接下来,恢复到室温,加入水(20ml),经过30分钟后加入氯仿进行萃取。向有机层中加入硫酸钠使其干燥,利用柱色谱法(乙酸乙酯:己烷=1:9)进行纯化,得到黄色固体的化合物2(4cz-1pbn-a)(1.18g,88%)。
1hnmr(400mhz,dmso-d6):δ7.92(d,j=8.4hz,4h),7.89-7.87(m,8h),7.65(d,j=7.2hz,4h),7.24(td,j=7.2,1.2hz,4h),7.15-7.08(m,8h),6.94(td,j=7.4,0.8hz,4h),6.72(dd,j=8.4,1.2hz,2h),6.44(tt,j=8.0,1.2hz,1h),6.31(t,j=7.8hz,2h).
〔发光评价〕
将掺杂材料换成2,3,5,6-四(9h-咔唑-9-基)-4-苯基-苯甲腈(4cz-1pbn-a),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图5和6。eqemax为26.1%。
将为相同的4-苯基取代的骨架的实施例7和实施例8进行对比时,可知本发明化合物(实施例7)的eqemax相对较高,作为发光材料是有用的。
(合成例9)
〔4’,5’,6’-三氟-[1,1’:3’,1”-三联苯基]-2’-甲腈(2pbn-c)的合成〕
向200ml茄形烧瓶中加入3,4,5-三氟苯甲腈(3.00g,19.1mmol)、溴苯(6.00g,38.2mmol)、2-乙基己酸(280mg,1.91mmol)、碳酸钾(7.91g,57.3mmmol)和二甲苯45ml。进行脱气和氩置换。其中加入三环己基膦(20%甲苯溶液5.10ml,2.87mmol)和乙酸钯(214mg,0.96mmmol),以140℃搅拌18小时。恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。对滤液进行水洗。其后,加入硫酸镁使其干燥,用旋转蒸发仪进行浓缩。将浓缩物用正己烷/乙酸乙酯=9/1进行清洗,得到4.43g的作为目标物(2pbn-c)的白色晶体。(收率75.1%)
1h-nmr(400mhz,cdcl3,δ):7.55~7.47(m,10h)
(实施例9)
〔3,4,5-三(3,6-二叔丁基-9h-咔唑-9-基)-2,6-二苯基-苯甲腈(3bucz-2pbn-c)的合成〕
向100ml的三口烧瓶中加入4’,5’,6’-三氟-[1,1’:3’,1”-三联苯基]-2’-甲腈(0.75g,2.43mmol)、3,6-二(叔丁基)咔唑(2.71g,9.70mmol)、叔丁醇钾(1.33g,10.9mmol)和n-甲基-2-吡咯烷酮15ml,以100℃搅拌71小时。向其中加入冰水100ml,对析出物进行过滤。将滤出物溶解于醚,进行水洗。其后,用硫酸镁使其干燥并进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯=19/1)对浓缩物进行分离纯化,得到1.25g(收率47.3%)的目标物(3bucz-2pbn-c)。
1h-nmr(400mhz,cdcl3,δ):7.45(d,4h),7.30(d,2h),7.28(d,2h),7.13(d,2h),7.04(m,6h),6.88(d,4h),6.87(d,4h),6.67(d,2h),6.49(dd,2h),1.24(s,36h),1.17(s,18h)
〔发光评价〕
将掺杂材料换成3,4,5-三(3,6-二叔丁基-9h-咔唑-9-基)-2,6-二苯基-苯甲腈(3bucz-2pbn-c),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图7和8。eqemax为12.0%。
(实施例10)〔3,4,5-三(3,6-二苯基-9h-咔唑-9-基)-2,6-二苯基-苯甲腈(3pcz-2pbn-c)的合成〕
向50ml的三口烧瓶中加入4’,5’,6’-三氟-[1,1’:3’,1”-三联苯基]-2’-甲腈(0.36g,1.17mmol)、3,6-二苯基咔唑(1.50g,4.70mmol)、叔丁醇钾(0.64g,5.24mmol)和n-甲基-2-吡咯烷酮8ml,以140℃搅拌18.5小时。向其中加入冰水100ml,对析出物进行过滤。将滤出物溶解于氯仿,进行水洗。其后,用硫酸镁使其干燥,进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/3)对浓缩物进行分离纯化,得到0.75g(收率53.2%)的目标物(3pcz-2pbn-c)。
1h-nmr(400mhz,cdcl3,δ):7.80(s,4h),7.47-7.41(m,14h),7.36-7.32(m,8h),7.28-7.21(m,22h),7.16-7.14(m,6h),7.09(d,2h),6.86(dd,2h)
〔发光评价〕
将掺杂材料换成3,4,5-三(3,6-二苯基-9h-咔唑-9-基)-2,6-二苯基-苯甲腈(3pcz-2pbn-c),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图7和8。eqemax为21.6%。
(合成例11)
〔3’,5’,6’-三氟-[1,1’:4’,1”-三联苯基]-2’-甲腈(2pbn-d)的合成〕
向200ml茄形烧瓶中加入2,4,5-三氟苯甲腈(3.00g,19.1mmol)、溴苯(6.00g,38.2mmol)、2-乙基己酸(280mg,1.91mmol)、碳酸钾(7.91g,57.3mmmol)和二甲苯45ml。进行脱气和氩置换。向其中加入三环己基膦(20%甲苯溶液5.10ml,2.87mmol)和乙酸钯(214mg,0.96mmmol),以140℃搅拌15小时。恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。对滤液进行水洗。其后,加入硫酸镁使其干燥,用旋转蒸发仪进行浓缩。在浓缩物中加入氯仿150ml进行加热溶解。向其中加入正己烷300ml进行冷却。将析出的白色固体过滤,得到4.40g(收率74.6%)的目标物(2pbn-d)。
1h-nmr(400mhz,cdcl3,δ):7.54~7.47(m,10h)
(实施例11)
〔3,4,6-三(3,6-二苯基-9h-咔唑-9-基)-2,5-二苯基-苯甲腈(3pcz-2pbn-d)的合成〕
将3,6-二苯基咔唑(1.75g,5.5mmol)加入到氮置换后的100ml的二口烧瓶中,溶解于n-甲基-2-吡咯烷酮10ml。向其中加入叔丁醇钾(0.65g,5.8mmol),以室温搅拌1小时。在氮气流下将3’,5’,6’-三氟-[1,1’:4’,1”-三联苯基]-2’-甲腈(0.42g,1.4mmol)悬浮于n-甲基-2-吡咯烷酮10ml并加入到其中,以120℃搅拌20小时。接下来,恢复到室温,加入水和乙酸乙酯,将有机层分液。进一步将水层用乙酸乙酯萃取2次,将混合的有机层用水清洗3次,接着用饱和食盐水清洗2次。将有机层用硫酸镁脱水,进行过滤和浓缩,由此得到粗产物。利用硅胶柱色谱法(洗脱液:正己烷/二氯甲烷)对粗产物进行纯化而得到粗纯化物。再次利用硅胶柱色谱法(洗脱液:正己烷/甲苯)对粗纯化物进行纯化,由此以收率56.7%得到0.93g的目标物(3pcz-2pbn-d)。
1h-nmr(400mhz,cdcl3,δ):8.32(d,j=2.0hz,2h),7.33-7.70(m,8h),7.66(d,j=2.0hz,2h),7.53-7.26(m,30h),7.26-7.21(m,5h),7.13(dd,j=8.4hz,2.0hz,2h),7.05(d,j=8.4hz,2h),6.99(d,j=8.8hz,2h,),6.91-6.89(m,2h),6.67-6.57(m,3h)
〔发光评价〕
将掺杂材料换成3,4,6-三(3,6-二苯基-9h-咔唑-9-基)-2,5-二苯基-苯甲腈(3pcz-2pbn-d),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图7和8。eqemax为20.7%。
(实施例12)
〔3,4,6-三(3,6-二叔丁基-9h-咔唑-9-基)-2,5-二苯基-苯甲腈(3bucz-2pbn-d)的合成〕
将3,6-二叔丁基咔唑(3.62g,13.0mmol)加入到氮置换后的300ml的四口烧瓶中,溶解于n-甲基-2-吡咯烷酮65ml。向其中加入叔丁醇钾(1.58g,14.1mmol),以室温搅拌1小时。在氮气流下向其中加入3’,5’,6’-三氟-[1,1’:4’,1”-三联苯基]-2’-甲腈(1.00g,3.2mmol),以120℃搅拌6小时。其后,以130℃搅拌14小时。接下来,恢复到室温,加入水和乙酸乙酯,将有机层分液。进一步将水层用乙酸乙酯萃取2次,将混合的有机层用水清洗3次,接着用饱和食盐水清洗2次。将有机层用硫酸镁脱水,进行过滤和浓缩,由此得到粗产物。利用硅胶柱色谱法(洗脱液:正己烷/二氯甲烷,正己烷/甲苯)对粗产物进行纯化,由此以收率49.2%得到1.73g的目标物(3bucz-2pbn-d)。
1h-nmr(400mhz,cdcl3,δ):8.03(d,j=1.2hz,2h),7.44-7.40(m,6h),7.35(d,j=1.6hz,2h),7.17(d,j=8.4hz,2h),7.10-7.06(m,3h),6.83-6.76(m,6h),6.66(d,j=8.4hz,2h),6.62(d,j=8.8hz,2h),6.56-6.46(m,3h),1.43(s,18h),1.29(s,18h),1.25(s,18h)
〔发光评价〕
将掺杂材料换成3,4,6-三(3,6-二叔丁基-9h-咔唑-9-基)-2,5-二苯基-苯甲腈(3bucz-2pbn-d),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图7和8。eqemax为15.6%。
(合成例13)
〔2,4,5,6-四氟-[1,1’-联苯基]-3-甲腈(1pbn-c)的合成〕
向300ml茄形烧瓶中加入1,3-二溴四氟苯(6.50g,21.1mmol)、苯基硼酸(2.73g,22.4mmol)、碳酸钾(8.74g,63.3mmol)、水26ml和四氢呋喃65ml。进行脱气,接着进行氩置换。其后,加入pd(pph3)4(0.56g,0.63mmol),加热回流下搅拌17小时。使得到的液体恢复到室温,加入乙醚100ml进行萃取。其后,用50ml的水进行2次水洗,用硫酸镁使其干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷)对浓缩物进行分离纯化,得到2.93g(收率45.5%)的3-溴-2,4,5,6-四氟-1,1’-联苯。
1h-nmr(400mhz,cdcl3,δ):7.50~7.38(m,5h)
向200ml茄形烧瓶中加入3-溴-2,4,5,6-四氟-1,1’-联苯(2.97g,9.74mmol)、氰化铜(1.74g,19.4mmol)、碘化钠(0.29g,1.93mmol)和n-甲基-2-吡咯烷酮30ml,以150℃搅拌22.5小时。冷却至室温。向其中加入乙醚50ml,用10%氨水进行清洗,接着用水进行清洗。其后,加入硫酸镁使其干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对浓缩物进行分离纯化,得到2.05g(收率83.3%)的目标物2,4,5,6-四氟-[1,1’-联苯基]-3-甲腈。
1h-nmr(400mhz,cdcl3,δ):7.51-7.48(m,3h),7.40-7.37(m,2h)
(实施例13)
〔2,4,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-3-甲腈(4bucz-1pbn-c)的合成〕
向50ml的茄形烧瓶中加入2,4,5,6-四氟-[1,1’-联苯基]-3-甲腈(0.25g,1.00mmol)、3,6-二叔丁基-咔唑(1.25g,4.47mmol)和脱水dmf10ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.20g,5.00mmol)。其后,以室温搅拌2小时。将得到的液体注入到冰水中,对析出物进行过滤。用醚使滤出物溶解,进行水洗。其后,用硫酸镁使其干燥,接着进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对浓缩物进行分离纯化。将得到的纯化物用2-丙醇进行清洗,得到1.14g(收率88.9%)的目标物(4bucz-1pbn-c)。
1h-nmr(400mhz,cdcl3,δ):8.01(d,2h),7.59(s,2h),7.46~7.44(m,4h),7.18(dd,4h),6.98(d,4h),6.91(dd,4h),6.67~6.64(m,4h),6.55~6.52(3h,m),6.43(dd,2h),1.43(s,18h),1.30(s,18h),1.22(s,18h),1.21(s,18h)
〔发光评价〕
将掺杂材料换成2,4,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-3-甲腈(4bucz-1pbn-c),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图9和10。eqemax为26.0%。
(实施例14)
〔3,4,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-2-甲腈(4bucz-1pbn-b)的合成〕
向氮置换后的烧瓶中加入乙酸钯(ii)(0.13g,0.58mmol)、碳酸银(3.15g,11.4mmol)和二苯基碘
1hnmr(400mhz,cdcl3):δ7.58-7.53(m,3h),7.51-7.45(m,3h)
向氮置换后的烧瓶中加入3,6-二叔丁基咔唑(1.37g,4.90mmol)和n,n-二甲基甲酰胺(10ml)。其后,以室温加入叔丁醇钾(0.55g,4.90mmol)和n,n-二甲基甲酰胺(5ml)的混合物,以室温搅拌30分钟。其后,用10分钟滴加化合物1(0.30g,1.19mmol)和n,n-二甲基甲酰胺(10ml)的混合物。其后,以80℃搅拌10小时。其后,恢复到室温,加入水(20ml),30分钟后加入氯仿进行萃取。向有机层中加入硫酸钠使其干燥,利用柱色谱法(乙酸乙酯:己烷=1:9)进行纯化,得到黄色固体的化合物2(4bucz-1pbn-b)(1.24g,81%)。
1hnmr(400mhz,dmso-d6):δ7.73(d,j=1.0hz,2h),7.52(d,j=6.8hz,4h),7.32-7.27(m,4h),7.21-7.11(m,11h),6.99(d,j=8.4hz,2h),6.87(d,j=8.8hz,2h),6.63-6.59(m,4h),1.33(s,18h),1.27(s,18h),1.16(d,j=8.0hz,36h)
〔发光评价〕
将掺杂材料换成3,4,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-2-甲腈(4bucz-1pbn-b),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图9和10。eqemax为22.3%。
(实施例15)
〔2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4-甲腈(4bucz-1pbn-a)的合成〕
利用与实施例7同样的方法得到目标物(4bucz-1pbn-a)。
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4-甲腈(4bucz-1pbn-a),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图9和10。
(合成例16)4’-(叔丁基)-2,3,5,6-四氟-[1,1’-联苯基]-4-甲腈的合成
向50ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(0.50g,2.86mmol)、4-叔丁基溴苯(0.64g,3.00mmol)、2-乙基己酸(41.0mg,0.29mmol)、碳酸钾(0.59g,4.29mmmol)、二甲苯10ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.45ml,0.25mmol)、乙酸钯(19.2mg,0.09mmmol),以140℃搅拌16小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对残渣进行分离纯化,得到0.50g的目标物的晶体。(收率56.8%)
1h-nmr(400mhz,cdcl3,δ):7.53(d,2h),7.40(d,2h),1.36(s,9h)
(实施例16)4’-(叔丁基)-2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4-甲腈(4x-bcz-pbn-bu)
向100ml的茄形烧瓶中加入4’-(叔丁基)-2,3,5,6-四氟-[1,1’-联苯基]-4-甲腈(0.50g,1.63mmol)、3,6-二叔丁基-咔唑(2.05g,7.34mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.33g,8.15mmol)后,以室温搅拌5小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物1.66g(收率75.8%)。
1h-nmr(400mhz,cdcl3,δ):7.55(d,4h),7.42(d,4h),6.94(dd,4h),6.88(d,4h),6.78(dd,6h),6.62(d,4h),6.48(d,2h),1.35(s,36h),1.39(s,36h),0.71(s,9h)
〔发光评价〕
将掺杂材料换成4’-(叔丁基)-2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4-甲腈(4x-bcz-pbn-bu),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图11和12。eqemax为26.5%。
(合成例17)4’-甲氧基-2,3,5,6-四氟-[1,1’-联苯基]-4-甲腈的合成
向50ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(0.80g,4.57mmol)、4-甲氧基溴苯(0.90g,4.80mmol)、2-乙基己酸(66.0mg,0.46mmol)、碳酸钾(0.95g,6.86mmmol)、二甲苯10ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.72ml,0.41mmol)、乙酸钯(31.0mg,0.14mmmol),以140℃搅拌17.5小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对残渣进行分离纯化,得到0.63g的目标物的晶体。(收率49.2%)
1h-nmr(400mhz,cdcl3,δ):7.41(d,2h),7.03(d,2h),3.87(s,3h)
(实施例17)2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4’-甲氧基-[1,1’-联苯基]-4-甲腈(4x-bcz-pbn-ome)
向100ml的茄形烧瓶中加入4’-甲氧基-2,3,5,6-四氟-[1,1’-联苯基]-4-甲腈(0.50g,1.78mmol)、3,6-二叔丁基-咔唑(2.24g,8.01mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.36g,8.90mmol)后,以室温搅拌3小时,进而以80℃搅拌2小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物1.93g(收率82.5%)。
1h-nmr(400mhz,cdcl3,δ):7.54(d,4h),7.43(d,4h),6.93(dd,4h),6.86(d,4h),6.81(dd,6h),6.63(d,4h),6.06(d,2h),3.23(s,3h),1.34(s,36h),1.30(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4’-甲氧基-[1,1’-联苯基]-4-甲腈(4x-bcz-pbn-ome),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图11和12。eqemax为28.5%。
(合成例18)4’-甲硫基-2,3,5,6-四氟-[1,1’-联苯基]-4-甲腈的合成
向50ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(0.80g,4.57mmol)、4-甲硫基溴苯(0.97g,4.80mmol)、2-乙基己酸(66.0mg,0.46mmol)、碳酸钾(0.95g,6.86mmmol)、二甲苯10ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.72ml,0.41mmol)、乙酸钯(31.0mg,0.14mmmol),以140℃搅拌17.5小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对残渣进行分离纯化,得到目标物的晶体0.68g。(收率50.0%)
1h-nmr(400mhz,cdcl3,δ):7.37(d,2h),7.34(d,2h),2.53(s,3h)
(实施例18)2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4’-甲硫基-[1,1’-联苯基]-4-甲腈(4x-bcz-pbn-sme)
向100ml的茄形烧瓶中加入4’-甲硫基-2,3,5,6-四氟-[1,1’-联苯基]-4-甲腈(0.50g,1.68mmol)、3,6-二叔丁基-咔唑(2.12g,7.56mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.34g,8.40mmol)后,以室温搅拌4小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯=19/1)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物2.08g(收率92.9%)。
1h-nmr(400mhz,cdcl3,δ):7.56(d,4h),7.44(d,4h),6.95(dd,4h),6.85(d,4h),6.83(dd,6h),6.64(d,4h),6.42(d,2h),1.96(s,3h),1.36(s,36h),1.32(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4’-甲硫基-[1,1’-联苯基]-4-甲腈(4x-bcz-pbn-sme),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图11和12。eqemax为29.0%。
(合成例19)2,3,5,6-四氟-[1,1’-联苯基]-4,4’-二甲腈的合成
向50ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(0.80g,4.57mmol)、4-氰基溴苯(0.87g,4.80mmol)、2-乙基己酸(66.0mg,0.46mmol)、碳酸钾(0.95g,6.86mmmol)、二甲苯15ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.72ml,0.41mmol)、乙酸钯(31.0mg,0.14mmmol),以140℃搅拌18小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/苯=1/1)对残渣进行分离纯化,得到0.79g的目标物的晶体。(收率62.7%)
1h-nmr(400mhz,cdcl3,δ):7.83(d,2h),7.58(d,2h)
(实施例19)2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4,4’-二甲腈(4x-bcz-pbn-cn)
向50ml的茄形烧瓶中加入2,3,5,6-四氟-[1,1’-联苯基]-4,4’-二甲腈(0.25g,0.91mmol)、3,6-二叔丁基-咔唑(1.27g,4.55mmol)、脱水dmf10ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.18g,4.53mmol)后,以室温搅拌2小时,进而以80℃搅拌5小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯=1/1)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物1.12g(收率94.1%)。
1h-nmr(400mhz,cdcl3,δ):7.57(d,4h),7.46(d,4h),7.03(d,2h),6.96(dd,4h),6.87(d,4h),6.84(dd,6h),6.59(d,4h),1.35(s,36h),1.31(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4,4’-二甲腈(4x-bcz-pbn-cn),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图11和12。eqemax为31.6%。
(合成例20)4’-氰基-2’,3’,5’,6’-四氟-[1,1’-联苯基]-4-甲酸甲酯的合成
向100ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(1.00g,5.71mmol)、4-甲氧羰基溴苯(1.29g,6.00mmol)、2-乙基己酸(82.0mg,0.57mmol)、碳酸钾(1.18g,8.56mmmol)、二甲苯20ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.90ml,0.51mmol)、乙酸钯(38.5mg,0.17mmmol),以140℃搅拌18小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/苯=1/1)对残渣进行分离纯化,得到1.29g的目标物的晶体。(收率72.9%)
1h-nmr(400mhz,cdcl3,δ):8.18(d,2h),7.54(d,2h),3.96(s,3h)
(实施例20)4’-氰基-2’,3’,5’,6’-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4-甲酸甲酯(4x-bcz-pbn-co2me)
向100ml的茄形烧瓶中加入4-(4-甲氧羰基苯基)-2,3,5,6-四氟苯甲腈(0.50g,1.78mmol)、3,6-二叔丁基-咔唑(2.24g,8.01mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.36g,8.90mmol)后,以室温搅拌3小时,进而以80℃搅拌2小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物1.93g(收率82.5%)。
1h-nmr(400mhz,cdcl3,δ):7.54(d,4h),7.43(d,4h),6.93(dd,4h),6.86(d,4h),6.81(dd,6h),6.63(d,4h),6.06(d,2h),3.23(s,3h),1.34(s,36h),1.30(s,36h)
〔发光评价〕
将掺杂材料换成4’-氰基-2’,3’,5’,6’-四(3,6-二叔丁基-9h-咔唑-9-基)-[1,1’-联苯基]-4-甲酸甲酯(4x-bcz-pbn-co2me),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图13和14。eqemax为30.4%。
(实施例21)4x-bcz-pbn-mesbn的合成
向200ml的三口烧瓶中加入2,4,6-三甲基苯基硼酸(2.62g,16.0mmol)、4-溴-2,3,5,6-四氟苯甲腈(2.03g,8.0mmol)、磷酸钾(6.80g,32.0mmol)、三(二亚苄基丙酮)二钯(0)(0.37g,0.40mmol)、sphos(0.66g,1.61mmol)、脱水甲苯100ml,进行脱气和氮置换后,以120℃搅拌22小时。将反应液恢复到室温,加入甲苯使用硅藻土滤出不溶物。浓缩滤液,利用硅胶柱色谱法(正己烷/二氯甲烷)对残渣进行纯化,得到1.09g的无色油状物的前体。(收率46.5%)
1h-nmr(400mhz,cdcl3,δ):7.01(s,2h),2.35(s,3h),2.05(s,6h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(2.12g,7.6mmol),溶解于脱水n-甲基-2-吡咯烷酮30ml,加入叔丁醇钾(0.82g,7.3mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下将前体(0.50g,1.70mmol)溶解于脱水n-甲基-2-吡咯烷酮5ml而加入,以100℃搅拌3小时,将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于二氯甲烷,用硫酸镁干燥并进行浓缩。在残渣中加入乙酸乙酯,进行超声波照射,滤取所析出的固体,馏去溶剂,由此得到2.05g的淡黄绿色固体的目标物(收率90.3%)。
1h-nmr(400mhz,cdcl3,δ):7.52(d,j=1.6hz,4h),7.38(s,4h),6.89(dd,j=8.4hz,2.0hz,4h),6.81(d,j=8.8hz,4h),6.73-6.68(m,8h),6.26(s,2h),2.17(s,6h),1.70(s,3h),1.35(s,36h),1.30(s,36h)
〔发光评价〕
将掺杂材料换成4x-bcz-pbn-mesbn,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图13和14。eqemax为29.5%。
(实施例22)4x-bcz-pbn-ipn的合成
向200ml的三口烧瓶中加入3,5-二氰基苯基硼酸频那醇酯(0.51g,2.0mmol)、4-溴-2,3,5,6-四氟苯甲腈(0.62g,2.4mmol)、磷酸钾(1.69g,8.0mmol)、三(二亚苄基丙酮)二钯(0)(90.2mg,0.10mmol)、sphos(162.8mg,0.40mmol)、脱水甲苯25ml,进行脱气和氮置换后,以120℃搅拌22小时。将反应液恢复到室温,加入甲苯使用硅藻土滤出不溶物。浓缩滤液,利用硅胶柱色谱法(正己烷/乙酸乙酯)对残渣进行纯化,以微黄白色固体的形式得到0.18g的前体(收率30.4%)。
1h-nmr(400mhz,cdcl3,δ):8.12(t,j=1.2hz,1h),8.00(d,j=1.2hz,2h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(1.84g,6.6mmol),溶解于脱水n-甲基-2-吡咯烷酮27ml,加入叔丁醇钾(0.71g,6.3mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下将前体(0.45g,1.49mmol)溶解于脱水n-甲基-2-吡咯烷酮5ml而加入,以100℃搅拌3小时。将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于二氯甲烷,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到粗纯化物。在粗纯化物中加入正己烷进行超声波照射,滤取所析出的固体,馏去溶剂,由此得到1.24g的黄色固体的目标物(收率62.0%)。
1h-nmr(400mhz,cdcl3,δ):7.64(d,j=1.6hz,4h),7.50(d,j=1.6hz,4h),7.12(d,j=1.2hz,2h),7.07(dd,j=8.8hz,2.0hz,4h),7.02-6.96(m,9h),6.67(d,j=8.4hz,4h),1.36(s,36h),1.32(s,36h)
〔发光评价〕
将掺杂材料换成4x-bcz-pbn-ipn,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图13和14。eqemax为31.0%。
(合成例23)2,3,5,6-四氟-4-(吡啶-2-基)苯甲腈的合成
向50ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(0.50g,2.86mmol)、2-溴吡啶(0.47g,3.0mmol)、2-乙基己酸(41.0mg,0.29mmol)、碳酸钾(0.59g,4.29mmmol)、二甲苯10ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.45ml,0.25mmol)、乙酸钯(19.2mg,0.09mmmol),以140℃搅拌18小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯=7/3)对残渣进行分离纯化,得到0.56g的目标物的晶体。(收率77.8%)
1h-nmr(400mhz,cdcl3,δ):8.80(d,1h),7.88(m,1h),7.51(d,1h),7.45(m,1h)
(实施例23)2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(吡啶-2-基)苯甲腈(4x-bcz-pbn-2-py)
向100ml的茄形烧瓶中加入2,3,5,6-四氟-4-(吡啶-2-基)苯甲腈(0.50g,1.98mmol)、3,6-二叔丁基-咔唑(2.49g,8.91mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.40g,9.90mmol)后,以室温搅拌5小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/1)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到2.26g的目标物(收率89.0%)。
1h-nmr(400mhz,cdcl3,δ):7.84(d,1h),7.56(d,4h),7.42(d,4h),6.98-6.95(m,5h),6.92(d,4h),6.86-6.83(m,5h),6.77(d,4h),6.43(dd,1h),1.36(s,36h),1.30(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(吡啶-2-基)苯甲腈(4x-bcz-pbn-2-py),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图15和16。eqemax为24.4%。
(合成例24)2,3,5,6-四氟-4-(吡啶-3-基)苯甲腈的合成
向100ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(1.15g,6.57mmol)、3-溴吡啶(1.09g,6.90mmol)、2-乙基己酸(95mg,0.66mmol)、碳酸钾(1.36g,9.86mmmol)、二甲苯20ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液1.04ml,0.59mmol)、乙酸钯(44.0mg,0.18mmmol),以140℃搅拌18小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯=7/3)对残渣进行分离纯化,得到1.16g的目标物的晶体(收率69.9%)。
1h-nmr(400mhz,cdcl3,δ):8.76-8.73(m,2h),7.80(d,1h),7.50-7.47(t,1h)
(实施例24)2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(吡啶-3-基)苯甲腈(4x-bcz-pbn-3-py)
向100ml的茄形烧瓶中加入2,3,5,6-四氟-4-(吡啶-3-基)苯甲腈(0.50g,1.98mmol)、3,6-二叔丁基-咔唑(2.49g,8.91mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.40g,9.90mmol)后,以室温搅拌4.5小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/3)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物2.18g(收率85.8%)。
1h-nmr(400mhz,cdcl3,δ):8.22(d,1h),7.81(d,1h),7.57(d,4h),7.45(d,4h),7.23(d,1h),6.96(dd,4h),6.89(d,4h),6.84(dd,4h),6.62(d,4h),6.49(dd,1h),1.36(s,36h),1.31(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(吡啶-3-基)苯甲腈(4x-bcz-pbn-3-py),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图15和16。eqemax为33.6%。
(合成例25)2,3,5,6-四氟-4-(吡啶-4-基)苯甲腈的合成
向50ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(0.80g,4.57mmol)、4-溴吡啶盐酸盐(0.93g,4.80mmol)、2-乙基己酸(66.0mg,0.46mmol)、碳酸钾(1.58g,11.4mmmol)、二甲苯15ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.72ml,0.41mmol)、乙酸钯(31.0mg,0.14mmmol),以140℃搅拌17小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/苯=1/4)对残渣进行分离纯化,得到0.70g的目标物的晶体(收率60.9%)。
1h-nmr(400mhz,cdcl3,δ):8.81(d,2h),7.38(d,2h)
(实施例25)2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(吡啶-4-基)苯甲腈(4x-bcz-pbn-4-py)
向100ml的茄形烧瓶中加入2,3,5,6-四氟-4-(吡啶-4-基)苯甲腈(0.50g,1.98mmol)、3,6-二叔丁基-咔唑(2.49g,8.91mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.40g,9.90mmol)后,以室温搅拌5小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(苯)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物2.24g(收率88.2%)。
1h-nmr(400mhz,cdcl3,δ):7.82(d,2h),7.56(d,4h),7.45(d,4h),6.95(dd,4h),6.87-6.82(m,10h),6.62(d,4h),1.35(s,36h),1.31(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(吡啶-4-基)苯甲腈(4x-bcz-pbn-4-py),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图15和16。eqemax为30.9%。
(合成例26)2,3,5,6-四氟-4-(嘧啶-5-基)苯甲腈的合成
向100ml茄形烧瓶中加入2,3,5,6-四氟苯甲腈(1.00g,5.71mmol)、5-溴嘧啶(0.95g,6.00mmol)、2-乙基己酸(82.0mg,0.57mmol)、碳酸钾(1.18g,8.57mmmol)、二甲苯20ml。进行脱气和氩置换后,加入三环己基膦(20%甲苯溶液0.90ml,0.50mmol)、乙酸钯(38.52mg,0.18mmmol),以140℃搅拌18小时。将反应液恢复到室温,加入乙酸乙酯,滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥,用旋转蒸发仪进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯=7/3)对残渣进行分离纯化,得到0.96g的目标物的晶体(收率66.5%)。
1h-nmr(400mhz,cdcl3,δ):9.35(s,1h),8.91(s,2h)
(实施例26)2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(嘧啶-5-基)苯甲腈(4x-bcz-pbn-5-pm)
向100ml的茄形烧瓶中加入2,3,5,6-四氟-4-(嘧啶-5-基)苯甲腈(0.50g,1.98mmol)、3,6-二叔丁基-咔唑(2.48g,8.91mmol)、脱水dmf20ml,用冰水浴进行冷却。逐次少量加入60%氢化钠(0.40g,9.90mmol)后,以室温搅拌4小时。将反应液注入到冰水中,过滤所析出的晶体。将晶体溶解于醚,水洗后,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯=2/3)对残渣进行分离纯化。将得到的晶体用2-丙醇进行清洗,得到目标物2.23g(收率87.8%)。
1h-nmr(400mhz,cdcl3,δ):8.39(s,1h),8.27(s,2h),7.57(d,4h),7.47(d,4h),6.97(dd,4h),6.90(d,4h),6.86(dd,4h),6.59(d,4h),1.36(s,36h),1.31(s,36h)
〔发光评价〕
将掺杂材料换成2,3,5,6-四(3,6-二叔丁基-9h-咔唑-9-基)-4-(嘧啶-5-基)苯甲腈(4x-bcz-pbn-5-pm),除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图15和16。eqemax为30.3%。
(实施例27)3y-bcz-pbn-tbu的合成
向100ml茄形烧瓶中加入2,4,6-三氟苯甲腈(1.00g,6.4mmol)、1-溴-4-叔丁基苯(2.86g,13.4mmol)、2-乙基己酸(93.8mg,0.65mmol)、碳酸钾(2.64g,19.1mmol)、脱水二甲苯20ml。进行脱气和氮置换后,加入三环己基膦(20%甲苯溶液1.6ml,0.95mmol)、乙酸钯(72.6mg,0.32mmol),以140℃搅拌24小时。将反应液恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到1.44g的淡褐白色固体的前体(收率53.7%)。
1h-nmr(400mhz,cdcl3,δ):7.51(d,j=8.0hz,4h),7.37(d,j=8.4hz,4h),1.37(s,18h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(1.16g,4.2mmol),溶解于脱水n-甲基-2-吡咯烷酮24ml,加入叔丁醇钾(0.44g,3.9mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下加入前体(0.50g,1.19mmol),以100℃搅拌3小时。将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于二氯甲烷,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯)对残渣进行纯化,得到0.81g的目标物的粗晶体。向由同样的方法而得到的粗晶体3.28g中加入乙酸乙酯,进行超声波照射,滤取所析出的晶体,馏去溶剂而得到2.25g的淡黄绿色固体的目标物(收率57.7%)。
1h-nmr(400mhz,cdcl3,δ):7.94(d,j=1.2hz,4h),7.71(d,j=2.0hz,2h),7.38(dd,j=8.8hz,2.0hz,4h),7.21(dd,j=8.8hz,2.0hz,2h),7.10(d,j=8.8hz,4h),6.93(d,j=8.4hz,2h),6.43(d,j=8.0hz,4h),6.33(d,j=8.8hz,4h),1.39(s,36h),1.31(s,18h),0.77(s,18h)
〔发光评价〕
将掺杂材料换成3y-bcz-pbn-tbu,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图17和18。eqemax为24.6%。
(实施例28)3y-bcz-pbn-ome的合成
向100ml茄形烧瓶中加入2,4,6-三氟苯甲腈(1.00g,6.4mmol)、4-溴茴香醚(2.51g,13.4mmol)、2-乙基己酸(92.9mg,0.64mmol)、碳酸钾(2.64g,19.1mmol)、脱水二甲苯20ml。进行脱气和氮置换后,加入三环己基膦(20%甲苯溶液1.6ml,0.95mmol)、乙酸钯(72.4mg,0.32mmol),以140℃搅拌24小时。将反应液恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到0.52g的白色固体的前体(收率22.1%)。
1h-nmr(400mhz,cdcl3,δ):7.37(d,j=8.8hz,4h),7.03-7.00(m,4h),3.87(s,6h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(1.33g,4.8mmol),溶解于脱水n-甲基-2-吡咯烷酮20ml,加入叔丁醇钾(0.44g,3.9mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下将前体(0.51g,1.35mmol)溶解于脱水n-甲基-2-吡咯烷酮7ml而加入,以100℃搅拌3小时。将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于二氯甲烷,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到目标物的粗纯化物1.63g。向由同样的方法而得到的粗纯化物3.33g中加入正己烷/乙酸乙酯,进行超声波照射,滤取所析出的晶体,馏去溶剂,得到2.37g的作为淡黄白色固体的目标物(收率74.1%)。
1h-nmr(400mhz,cdcl3,δ):7.96(d,j=2.0hz,4h),7.76(d,j=2.0hz,2h),7.41(dd,j=8.8hz,2.0hz,4h),7.27(dd,2h),7.11(d,j=8.0hz,4h),6.96(d,j=8.8hz,2h),6.52(d,j=8.8hz,4h),5.92(d,j=9.2hz,4h),3.29(s,6h),1.40(s,36h),1.33(s,18h)
〔发光评价〕
将掺杂材料换成3y-bcz-pbn-ome,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图17和18。eqemax为21.5%。
(实施例29)3y-bcz-pbn-sme的合成
向100ml茄形烧瓶中加入2,4,6-三氟苯甲腈(1.00g,6.4mmol)、4-溴硫代苯甲醚(2.71g,13.3mmol)、2-乙基己酸(94.3mg,0.65mmol)、碳酸钾(2.66g,19.2mmol)、脱水二甲苯20ml。进行脱气和氮置换后,加入三环己基膦(20%甲苯溶液1.6ml,0.95mmol)、乙酸钯(75.6mg,0.34mmol),以140℃搅拌21小时。将反应液恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥并进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯)对残渣进行纯化后,用正己烷/乙酸乙酯进行清洗,得到1.43g的白色固体的前体(收率56.0%)。
1h-nmr(400mhz,cdcl3,δ):7.35(s,8h),2.53(s,6h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(1.95g,7.0mmol),溶解于脱水n-甲基-2-吡咯烷酮32ml,加入叔丁醇钾(0.76g,6.8mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下加入前体(0.80g,2.00mmol),以100℃搅拌3小时。将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于二氯甲烷,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到2.25g的目标物的粗纯化物。在粗纯化物中加入正己烷,进行超声波照射,滤取所析出的晶体,馏去溶剂,得到1.52g的微黄白色固体的目标物(收率64.7%)。
1h-nmr(400mhz,cdcl3,δ):7.96(d,j=2.0hz,4h),7.77(d,j=2.0hz,2h),7.41(dd,j=8.4hz,2.0hz,4h),7.26(dd,j=8.4hz,2.0hz,2h),6.93(d,j=8.0hz,2h),6.50(d,j=8.4hz,4h),6.28(d,j=8.4hz,4h),1.99(s,6h),1.40(s,36h),1.34(s,18h)
〔发光评价〕
将掺杂材料换成3y-bcz-pbn-sme,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图17和18。eqemax为20.9%。
(实施例30)3f-bcz-pbn-tbu的合成
向100ml茄形烧瓶中加入2,3,5-三氟苯甲腈(1.00g,6.4mmol)、1-溴-4-叔丁基苯(2.85g,13.4mmol)、2-乙基己酸(94.7mg,0.66mmol)、碳酸钾(2.64g,19.1mmol)、脱水二甲苯20ml。进行脱气和氮置换后,加入三环己基膦(20%甲苯溶液1.6ml,0.95mmol)、乙酸钯(72.4mg,0.32mmol),以140℃搅拌22小时。将反应液恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到2.54g的白色固体的前体(收率94.7%)。
1h-nmr(400mhz,cdcl3,δ):7.53(d,j=10.4hz,4h),7.45-7.43(m,4h),1.37(s,18h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(1.16g,4.2mmol),溶解于脱水n-甲基-2-吡咯烷酮24ml,加入叔丁醇钾(0.44g,3.9mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下加入前体(0.50g,1.19mmol),以100℃搅拌3小时。将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于二氯甲烷,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/乙酸乙酯)对残渣进行纯化,得到0.76g的粗纯化物。将由同样的方法而得到的粗晶体1.44g溶解于乙酸乙酯并混合,馏去溶剂,得到1.41g的淡黄绿白色固体的目标物(收率49.5%)。
1h-nmr(400mhz,cdcl3,δ):7.80(s,2h),7.54(s,2h),7.38(s,2h),7.26(d,j=8.8hz,2h),7.15(d,j=8.4hz,2h),6.99(t,j=8.0hz,4h),6.91(d,j=8.8hz,4h),6.85(d,j=8.4hz,2h),6.76(d,j=8.0hz,2h),6.59(dd,j=10.4hz,8.8hz,4h),6.38(d,j=8.0hz,2h),1.37(s,18h),1.34(s,18h),1.27(s,18h),1.09(s,9h),0.73(s,9h)
〔发光评价〕
将掺杂材料换成3f-bcz-pbn-tbu,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图19和20。eqemax为26.5%。
(实施例31)3f-bcz-pbn-ome的合成
向100ml茄形烧瓶中加入2,3,5-三氟苯甲腈(1.00g,6.4mmol)、4-溴茴香醚(2.50g,13.4mmol)、2-乙基己酸(96.5mg,0.67mmol)、碳酸钾(2.64g,19.1mmol)、脱水二甲苯20ml。进行脱气和氮置换后,加入三环己基膦(20%甲苯溶液1.6ml,0.95mmol)、乙酸钯(72.3mg,0.32mmol),以140℃搅拌22小时。将反应液恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到1.51g的白色固体的前体(收率64.2%)。
1h-nmr(400mhz,cdcl3,δ):7.46-7.42(m,4h),7.05-7.02(m,4h),3.87(s,6h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(1.51g,5.4mmol),溶解于脱水n-甲基-2-吡咯烷酮25ml,加入叔丁醇钾(0.58g,5.2mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下将前体(0.57g,1.54mmol)溶解于脱水n-甲基-2-吡咯烷酮6ml而加入,以100℃搅拌3小时。将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于乙酸乙酯,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化,得到0.80g的粗纯化物。在由同样的方法而得到的粗纯化物2.07g中加入正己烷/乙酸乙酯,进行超声波照射,滤取所析出的晶体,馏去溶剂,得到1.82g的作为微黄白色固体的目标物(收率64.7%)。
1h-nmr(400mhz,cdcl3,δ):7.85(d,j=1.6hz,2h),7.53(d,j=2.0hz,2h),7.38(d,j=2.0hz,2h),7.31(dd,j=8.8hz,2.0hz,2h),7.19(d,j=8.4hz,2h),7.01(d,j=8.8hz,2h),6.90(dd,j=8.0hz,1.6hz,2h),6.81-6.77(m,4h),6.64-6.55(m,6h),5.94(d,j=8.8hz,2h),3.62(s,3h),3.25(s,3h),1.39(s,18h),1.34(s,18h),1.28(s,18h)
〔发光评价〕
将掺杂材料换成3f-bcz-pbn-ome,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图19和20。eqemax为25.2%。
(实施例32)3f-bcz-pbn-sme的合成
向100ml茄形烧瓶中加入2,3,5-三氟苯甲腈(1.00g,6.4mmol)、4-溴硫代苯甲醚(2.72g,13.4mmol)、2-乙基己酸(95.0mg,0.66mmol)、碳酸钾(2.65g,19.2mmol)、脱水二甲苯20ml。进行脱气和氮置换后,加入三环己基膦(20%甲苯溶液1.6ml,0.95mmol),乙酸钯(73.0mg,0.33mmol),以140℃搅拌21小时。将反应液恢复到室温,加入乙酸乙酯使用硅藻土滤出不溶物。将滤液水洗后,加入硫酸镁进行干燥并进行浓缩。利用硅胶柱色谱法(正己烷/苯)对残渣进行纯化后,用正己烷/乙酸乙酯进行清洗,得到2.05g的作为白色固体的前体(收率80.2%)。
1h-nmr(400mhz,cdcl3,δ):7.42-7.34(m,8h),2.54(s,6h)
向氮置换后的200ml的三口烧瓶中加入3,6-二叔丁基咔唑(1.95g,7.0mmol),溶解于脱水n-甲基-2-吡咯烷酮32ml,加入叔丁醇钾(0.76g,6.8mmol),以室温搅拌1小时。将该混合物进行冰水冷却,在氮气流下加入前体(0.80g,2.00mmol),以100℃搅拌3小时。将反应液进行冰水冷却,加入冷水,滤取所析出的固体。将固体溶解于二氯甲烷,用硫酸镁干燥并进行浓缩。利用硅胶柱色谱法(正己烷/二氯甲烷)对残渣进行纯化,得到粗纯化物。在粗纯化物中加入正己烷/乙醚,进行超声波照射,滤取所析出的晶体,馏去溶剂,得到1.85g的作为淡黄色固体的目标物(收率78.7%)。
1h-nmr(400mhz,cdcl3,δ):7.85(d,j=1.6hz,2h),7.53(d,j=2.0hz,2h),7.38(d,j=1.2hz,2h),7.31(dd,j=8.8hz,2.0hz,2h),7.18(d,j=8.4hz,2h),6.99(d,j=8.8hz,2h),6.92-6.89(m,4h),6.80-6.77(m,4h),6.60(d,j=8.4hz,2h),6.57(d,j=8.4hz,2h),6.29(d,j=8.4hz,2h),2.30(s,3h),1.95(s,3h),1.39(s,18h),1.34(s,18h),1.28(s,18h)
〔发光评价〕
将掺杂材料换成3f-bcz-pbn-sme,除此以外,利用与实施例1相同的方法进行发光评价。将结果示于图19和20。eqemax为22.0%。
产业上的可利用性
能够提供发光特性优异的2,3,4,5,6-五取代苯甲腈化合物、发光材料和使用该发光材料的发光元件。
- 还没有人留言评论。精彩留言会获得点赞!