超支化聚合物改性无机纳米成核剂及其制备方法和应用与流程
本发明涉及高分子改性加工领域,尤其是一种超支化聚合物改性无机纳米成核剂及其制备方法和应用。
背景技术:
调控聚烯烃树脂的结晶是提高其性能和更多功能化的重要手段。成核剂是一种促进聚合物结晶并改善其晶粒结构的改性剂。在聚烯烃树脂中加入成核剂可加速聚烯烃树脂的结晶速度、增加聚烯烃树脂的各项力学性能和加工性能、提高聚烯烃树脂的热力学性能。纳米成核剂是近年来发展快速的一种新型成核剂。现有技术对于无机纳米粒子成核剂有一定的研究基础,例如申请号为201110245797.8,申请号为201110245797.8及申请号为201810653093.6的中国专利申请中都使用了无机纳米成核剂。纳米粒子的小尺寸效应和强界面可以对聚烯烃树脂起到增强、增韧的效果。但固体无机颗粒纳米化之后,比表面积大、其表面的原子自由能急剧增强、表面活性增大、处于热力学非稳态,易于和自身及其他原子结合,形成团聚体,从而降低其自身成核效果。因此,无机纳米颗粒的表面包覆聚合物提高其在聚丙烯中的分散度是一种提高成核效果的有效手段。
现有的无机纳米颗粒表面聚合物包覆技术都是活性单体基于表面能在无机纳米粒子可以表面聚合或聚集得到。由于聚合过程比较复杂,在反应体系中不仅仅会产生核-壳结构的复合纳米粒子,还会产生聚合物包覆不完整的无机纳米粒子、独立的聚合物纳米粒子、聚合物包裹的多个无机纳米粒子聚集体等无效颗粒。因此,该方法制备的成核剂的成核能力提升有限。
如何提升成核剂的成核效果,还需要进一步改进。因此,限制聚合单体只在无机纳米粒子表面发生聚合反应是控制聚合物改性纳米粒子分散性高、不易聚集的难点。
技术实现要素:
本发明的目的至少在于解决上述所提到的技术问题。发明人在研究过程中创造性的想到限制聚合单体只在无机纳米粒子表面发生聚合反应,获得核-壳结构的复合纳米粒子,降低聚合物包覆不完整的无机纳米粒子、独立的聚合物纳米粒子、聚合物包裹的多个无机纳米粒子聚集体等无效颗粒的产生;为此,本发明提供了一种超支化聚合物改性无机纳米成核剂及其制备方法和应用。首先制备休眠聚合单体,其在未被氨基激活情况下,不会发生聚合;然后通过与氨基修饰的无机纳米粒子反应,激活休眠聚合单体基团,并在光引发剂和紫外光照的作用下通过巯基-不饱和烷基之间的迈克尔加成聚合,在无机纳米颗粒表面形成具有超支化结构的聚合物,即获得超支化改性无机纳米成核剂。所述的超支化聚合物改性无机纳米成核剂通过共混加工即可有效提高聚烯烃树脂的综合性能。
具体而言,本发明提供了如下技术方案:
在本发明的第一方面,本发明提供了一种超支化聚合物改性无机纳米成核剂的制备方法,包括:(1)将预定比例的高半胱氨酸硫内酯盐酸盐、α-不饱和烷基酰氯和有机碱混合,进行第一反应,以便获得聚合单体;(2)将氨基偶联剂和无机纳米粒子混合,进行第二反应,以便获得氨基修饰的无机纳米粒子;(3)将所述氨基修饰的无机纳米粒子、所述聚合单体和光引发剂混合,在紫外光照射下进行第三反应,以便获得超支化聚合物改性无机纳米成核剂。
本发明提供了一种超支化聚合物改性无机纳米成核剂的制备方法。首先,其通过将高半胱氨酸硫内酯盐酸盐、α-不饱和烷基酰氯和有机碱反应,制备聚合单体,所形成的该聚合单体为硫代内酯结构,其在未被氨基激活情况下,自身不含巯基末端,无法在光敏剂和紫外光照下聚合,因此避免了反应体系中产生不含无机纳米粒子的超支化聚合物粒子,因此所形成的该聚合单体在本文中也可以称为“休眠聚合单体”。另外,通过氨基偶联剂对无机纳米粒子进行改性,获得氨基修饰的无机纳米粒子。氨基修饰的无机纳米粒子表面的氨基可以激活休眠聚合单体开环产生巯基和不饱和烷基末端,从而可以在光引发剂和紫外光照的作用下通过巯基-不饱和烷基之间的迈克尔加成聚合,在无机纳米颗粒表面形成具有超支化结构的聚合物,即文中所提到的超支化聚合物改性无机纳米成核剂。所制备的超支化聚合物改性无机纳米成核剂具有和聚烯烃树脂相容性更好、分散性更高的优势。而且由于纳米粒子表面的强界面效应,巯基和不饱和烷基之间的反应更容易发生在单个纳米粒子表面,而不是纳米粒子之间。因此,也避免了反应体系中产生超支化聚合物包裹多个无机纳米粒子的聚集体,从而提高超支化聚合物改性纳米粒子的成核效果。
进一步地,以上所提供的超支化聚合物改性无机纳米成核剂的制备方法还可以进一步包括如下技术特征:
进一步地,步骤(1)中所述的高半胱氨酸硫内酯盐酸盐、α-不饱和烷基酰氯和有机碱的摩尔质量比为1:1:1~1.2。有机碱作为缚酸剂,能够及时中和反应过程中的酸。根据本发明的优选实施例,合适含量的有机碱不影响反应的进程。
进一步地,所述的有机碱包括选自三乙胺、吡啶、二甲基吡啶中的任意一种。所述的有机碱优选为三乙胺。
进一步地,所述的α-不饱和烷基酰氯为烯基酰氯和炔基酰氯中的至少一种。根据本发明的实施例,所述的α-不饱和烷基酰氯包括但不限于乙烯基酰氯、甲基丙烯酰氯、丁烯酰氯、4-戊烯酰氯、己烯酰氯和乙炔基酰氯等。根据本发明的优选实施例,所述的α-不饱和烷基酰氯为4-戊烯酰氯。
进一步地,步骤(1)中所述高半胱氨酸硫内酯盐酸盐、所述α-不饱和烷基酰氯和有机碱在中极性溶剂中进行所述第一反应。在进行第一反应时,需要在合适的溶剂中进行,溶剂的选取原则为能够溶解休眠单体,但是不能溶解有机碱盐酸盐,从而易于提纯;而且溶剂的选择对反应程度并无特定影响。根据本发明的具体实施例,所述中极性溶剂包括选自乙醚、四氢呋喃和二氯甲烷中的至少一种。
进一步地,步骤(1)中所述第一反应的反应温度为-20~10℃,反应时间为2~6h。发明人在研究过程中发现,在进行第一反应时,采用低反应温度可以减少副反应的发生,提高产率,而且对反应程度无影响。所以在-20~10℃条件下进行第一反应,可以保证反应的顺利进行,而且能够减少副反应的发生,提高产率。根据本发明的具体实施例,步骤(1)中所述第一反应的反应温度为-10~10摄氏度。根据本发明的具体实施例,步骤(1)中所述第一反应的反应温度为-5~10摄氏度。根据本发明的具体实施例,步骤(1)中所述第一反应的反应温度为-5~5摄氏度。根据本发明的具体实施例,所述第一反应的反应时间为2~4小时。
进一步地,步骤(2)中所述的无机纳米粒子和所述氨基偶联剂的质量比为100~1000:1。氨基偶联剂的使用量会影响到最终所获得的超支化聚合物改性无机纳米成核剂的粒径及其成核效果。发明人在研究过程中发现,当无机纳米粒子高于或低于上述比例时,获得的超支化聚合物改性无机纳米成核剂粒径都将偏小,成核效果会急剧下降。
另外,无机纳米粒子种类在一定程度上也会影响到最终在聚烯烃树脂中的成核效果,如:二氧化硅促进聚丙烯α晶的形成,提高聚丙烯树脂的拉伸模量;轻质碳酸钙促进聚丙烯β晶的形成,提高聚丙烯树脂弯曲模量。因此,选择合适的无机纳米粒子,可以获得理想的成核剂。根据本发明的实施例,所述的无机纳米粒子是无机氧化物、无机盐、金属氧化物、金属盐、过渡金属氧化物、过渡金属盐等无机颗粒。根据本发明的具体实施例,所述的无机纳米粒子包括但不限于:二氧化硅、碳酸钙、水滑石、氧化锌、锆氧化物、铝氧化物、钛氧化物、硅酸盐、硅铝酸盐无机颗粒。
根据本发明的实施例,所述的无机纳米粒子粒径为1~2000纳米。根据本发明的实施例,所述的无机纳米粒子的粒径为1~1500纳米,10~1200纳米,50~1000纳米,100~1000纳米,100~800纳米,100~700纳米,100~600纳米或者100~500纳米。
进一步地,所述的氨基偶联剂是含氨基的硅烷偶联剂。
进一步地,所述的光引发剂为水溶性光引发剂,光引发剂的选用影响能够在一定程度上影响反应程度,对超支化聚合物改性纳米粒子最终粒径有较大影响。根据本发明的实施例,所述的光引发剂包括但不限于商品名为:quantacurebtc、quantacurebpq、quantacureabp、quantacureqtx、quantacurebms、darocur2959。根据本发明的优选实施例,所述的光引发剂为darocur2959。
进一步地,所述的紫外光的为波长为220~420nm,特别是250-370nm的电磁辐射,更优选的,波长为365nm的电磁辐射。本发明中紫外光可通过例如包含氯化氙(xecl,308nm)、氟化氙(xef,351nm)、氟化氪(krf,249nm)、氯化氪(krcl,222nm)、氟化氩(arf,193nm)或xe2(172nm)作为激光激活介质的本领域熟练技术人员已知的所有设备,如led、受激准分子辐射器(excimerradiator),或uv荧光管产生。优选的,采用受激准分子辐射器,因为其可通过脉冲操作调节功率从10%到100%之间变化,从而更好调控聚合过程。
进一步地,所述第三反应的时间为0.2-2小时。由此可以快速方便获得获得超支化聚合物改性无机纳米成核剂。
进一步地,所述氨基修饰的无机纳米粒子和所述聚合单体的重量比为1:10~100。通常来说,聚合单体的用量越大,最终得到的超支化聚合物改性的无机纳米成核剂的粒径越大。在该比例条件下能够获得粒径合适的超支化聚合物改性的无机纳米成核剂。
进一步地,所述氨基修饰的无机纳米粒子和所述光引发剂的重量比为1:0.05~0.5。由此能够有效促进光引发反应。
在本发明的第二方面,本发明提供了一种超支化聚合物改性无机纳米成核剂,所述超支化聚合物改性无机纳米成核剂根据本发明第一方面任一实施例所述的方法制备获得。
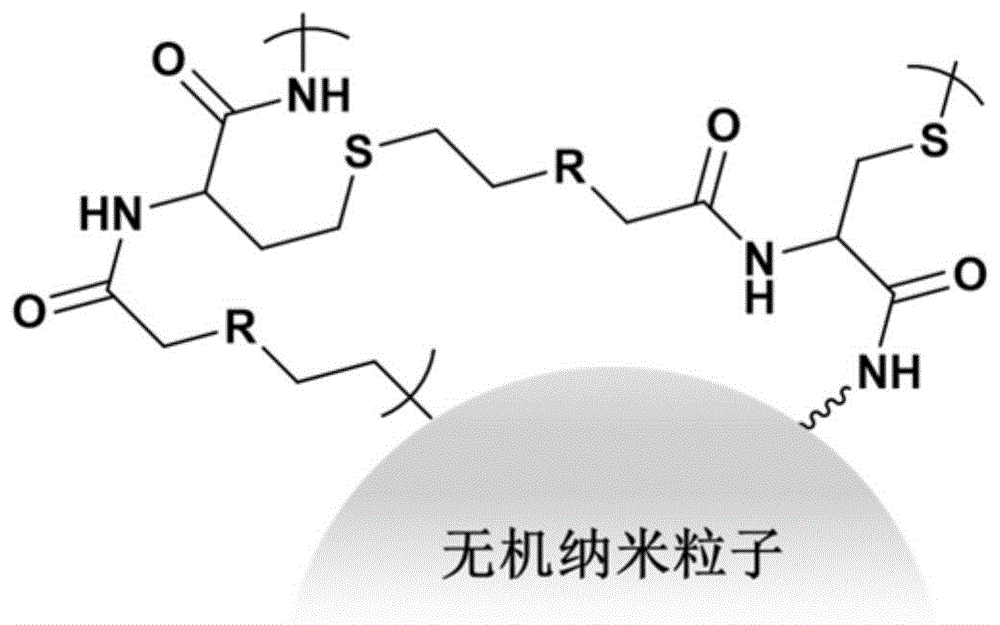
在本发明的第三方面,本发明提供了一种超支化聚合物改性无机纳米成核剂,其具有无机纳米粒子的核和超支化聚合物的外表面,其结构如下所示:
其中,r为烷基,a1为与无机纳米粒子连接处。根据本发明的实施例,r可以为正烷基或者为异构烷基。
在本发明的第四方面,本发明提供了一种聚烯烃树脂,所述聚烯烃树脂包括本发明第二方面或者本发明第三方面所述的超支化聚合物改性无机纳米成核剂。
进一步地,所提供的聚烯烃树脂还可以进一步包括如下技术特征:
根据本发明的实施例,所述聚烯烃树脂中所述超支化聚合物改性无机纳米成核剂的含量为0.01%~1%。
根据本发明的实施例,所述聚烯烃树脂中所述超支化聚合物改性无机纳米成核剂的含量为0.05%~0.1%。
超支化聚合物改性无机纳米成核剂可直接在聚烯烃树脂加工过程中共混,即可赋予聚烯烃树脂更优异的力学性能和加工性能;聚烯烃树脂制件形式包括但不限于:薄膜、发泡材料、片材等;其加工方式包括但不限于:注塑、滚塑、压延、吹膜、模压等。根据本发明的实施例,所述聚烯烃树脂用于薄膜、发泡材料或者片材。
本发明所取得的有益效果为:
(1)本发明合成的休眠聚合单体为硫代内酯结构,在未被氨基激活情况下,自身不含巯基末端,无法在光敏剂和紫外光照下聚合,因此避免了反应体系中产生不含无机纳米粒子的超支化聚合物粒子。
(2)本发明中改性无机纳米粒子表面的氨基可以激活休眠聚合单体开环产生巯基和不饱和烷基末端,从而在光引发剂和紫外光照的作用下通过巯基-不饱和烷基之间的迈克尔加成聚合,从而在无机纳米颗粒表面形成具有超支化结构的聚合物。这种超支化聚合物修饰的纳米粒子成核剂和聚烯烃树脂的相容性更好、分散性更高。
(3)由于纳米粒子表面的强界面效应,巯基和不饱和烷基之间的反应更容易发生在单个纳米粒子表面,而不是纳米粒子之间。因此,也避免了反应体系中产生超支化聚合物包裹多个无机纳米粒子的聚集体,从而提高超支化聚合物改性纳米粒子的成核效果。
附图说明
图1是根据本发明的实施例提供的一种超支化聚合物改性无机纳米成核剂的结构示意图。
图2是根据本发明的实施例提供的一种超支化聚合物改性无机纳米成核剂的结构示意图。
具体实施方式
下面详细描述本发明的实施例,需要说明的是,所描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
本发明提供了一种超支化聚合物改性无机纳米成核剂的制备方法,包括:
(1)将预定比例的高半胱氨酸硫内酯盐酸盐、α-不饱和烷基酰氯和有机碱混合,在中极性溶剂中进行第一反应,以便获得聚合单体;
(2)将氨基偶联剂和无机纳米粒子混合,进行第二反应,以便获得氨基修饰的无机纳米粒子;
(3)将所述氨基修饰的无机纳米粒子、所述聚合单体和光引发剂混合,在紫外光照射下进行第三反应,以便获得超支化聚合物改性无机纳米成核剂。
高半胱氨酸硫内酯盐酸盐和α-不饱和烷基酰氯反应制备休眠聚合单体;无机纳米粒子激活休眠单体,并在光引发剂和紫外光的作用下引发巯基-不饱和烷基之间的迈克尔加成反应,形成超支化聚合物表面改性。该方法使用的休眠聚合单体为硫代内酯结构,自身无法聚合,只能和无机纳米粒子表面氨基反应后得到巯基末端,再在紫外光照和光引发剂条件下才能聚合,避免了一般聚合改性方法不可控引起的无机纳米粒子易团聚、分散性差的缺陷。
其中步骤(1)和步骤(2)的顺序不做特殊要求,可以先进行步骤(1),也可以先进行步骤(2),或者步骤(1)和步骤(2)同时进行或者交叉进行。
步骤(1)中,反应温度-20~10℃,反应温度为2~6h,搅拌速度50~200rpm。在进行完第一反应后,过滤去除反应有机碱盐酸盐固体,减压蒸去溶剂得到聚合单体。反应过程中,低反应温度可以减少副反应的发生,提高产率,但对反应程度无影响;保持一定的搅拌速度和反应时间可提高反应程度,但继续增加搅拌速度和延长反应时间对反应程度无影响。因此反应温度-20~10℃,反应温度为2~6h,搅拌速度50~200rpm进行,可以获得理想的反应效果。
在本发明的至少一些实施方式中,步骤(2)中可以在高混机上高速搅拌10-30min。
在本发明的至少一些实施方式中,所述的有机碱为三乙胺、吡啶、二甲基吡啶中的任一种。优选的,所述的有机碱为三乙胺。
所述的α-不饱和烷基酰氯为烯基酰氯或炔基酰氯,包括但不限于:甲基丙烯酰氯、丁烯酰氯、4-戊烯酰氯、己烯酰氯和乙炔酰氯等。
溶剂的选取原则为溶解休眠单体,不能溶解三乙胺盐酸盐,从而易于提纯;溶剂的选择对反应程度并无特定影响;所述的中极性溶剂是:乙醚、四氢呋喃和二氯甲烷中的任一种。
所述的光引发剂为水溶性光引发剂,包括但不限于商品名为:quantacurebtc、quantacurebpq、quantacureabp、quantacureqtx、quantacurebms、darocur2959中的任一种;优选的,所述的光引发剂为darocur2959;光引发剂的选用一定程度上会影响反应程度,对超支化聚合物改性纳米粒子最终粒径有较大影响。
在本发明的至少一些实施方式中,所述的高半胱氨酸硫内酯盐酸盐、乙烯基酰氯和有机碱的摩尔质量比为1:1:1~1.2。
在本发明的至少一些实施方式中,所述的无机纳米粒子和所述氨基偶联剂的质量比为100~1000:1。氨基偶联剂的使用量影响最终所获得的超支化聚合物改性无机纳米颗粒成核剂的粒径。当无机纳米粒子高于或低于上述比例时,获得的超支化聚合物改性无机纳米成核剂粒径都将偏小,成核效果会急剧下降。在本发明的至少一些实施方式中,所述的无机纳米粒子和所述氨基偶联剂的质量比为100~800:1。在本发明的至少一些实施方式中,所述的无机纳米粒子和所述氨基偶联剂的质量比为100~700:1。在本发明的至少一些实施方式中,所述的无机纳米粒子和所述氨基偶联剂的质量比为100~600:1。在本发明的另一些实施方式中,所述的无机纳米粒子和所述氨基偶联剂的质量比为100~500:1。在本发明的又一些实施方式中,所述的无机纳米粒子和所述氨基偶联剂的质量比为100~400:1。
无机纳米粒子种类影响最终在聚烯烃树脂中的成核效果,如:二氧化硅促进聚丙烯α晶的形成,提高聚丙烯树脂的拉伸模量;轻质碳酸钙促进聚丙烯β晶的形成,提高聚丙烯树脂弯曲模量。在本发明的至少一些实施方式中,所述的无机纳米粒子是无机氧化物、无机盐、金属氧化物、金属盐、过渡金属氧化物、过渡金属盐等无机颗粒。包括但不限于:二氧化硅、碳酸钙、水滑石、氧化锌、锆氧化物、铝氧化物、钛氧化物、硅酸盐、硅铝酸盐无机颗粒。所述的无机纳米粒子粒径可以为1~2000纳米。
在本发明的至少一些实施方式中,所述氨基修饰的无机纳米粒子和所述聚合单体的重量比为1:(10~100)。一般而言,单体的用量越大,最终得到的超支化聚合物修饰纳米成核剂粒径越大。在本发明的至少一些实施方式中,所述氨基修饰的无机纳米粒子和所述聚合单体的重量比为1:(10~90),1:(10~80),或者1:(10~60)等。
在本发明的至少一些实施方式中,所述氨基修饰的无机纳米粒子和光引发剂的重量比为1:0.05~0.5。在本发明的至少一些实施方式中,所述氨基修饰的无机纳米粒子和光引发剂的重量比为1:0.1~0.5。在本发明的至少一些实施方式中,所述氨基修饰的无机纳米粒子和光引发剂的重量比为1:0.2~0.5。在本发明的至少一些实施方式中,所述氨基修饰的无机纳米粒子和光引发剂的重量比为1:0.2~0.4。根据本发明的实施例,所述的紫外光为波长为220~420nm,特别是250-370nm的电磁辐射,更优选的,波长为365nm的电磁辐射。本发明中紫外光可通过例如包含氯化氙(xecl,308nm)、氟化氙(xef,351nm)、氟化氪(krf,249nm)、氯化氪(krcl,222nm)、氟化氩(arf,193nm)或xe2(172nm)作为激光激活介质的本领域熟练技术人员已知的所有设备,如led、受激准分子辐射器(excimerradiator),或uv荧光管产生。优选的,采用受激准分子辐射器,因为其可通过脉冲操作调节功率从10%到100%之间变化,从而更好调控聚合过程。
在本发明的至少一些实施方式中,所提供的超支化聚合物改性无机纳米成核剂如图1所示。在本发明的至少一些实施方式中,所提供的超支化聚合物改性无机纳米成核剂如图2所示。其中图1和图2中所示出的r为正烷基或异构烷基。例如r可以为甲基,乙基,丙基等。
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
实施例1
将高半胱氨酸硫内酯盐酸盐、丁烯酰氯和有机碱按照1:1:1的摩尔质量比加入100l反应容器中,加入60l四氢呋喃溶解,搅拌反应6h。反应温度0℃,搅拌速度200rpm。反应结束后,过滤除去三乙胺盐酸盐沉淀,减压蒸馏除溶剂,得到休眠聚合单体。
配制质量分数为10%的氨基硅烷偶联剂,二氧化硅纳米颗粒和氨基硅烷偶联剂溶液按100:1的质量比在高混机上混合均匀,搅拌时间30min,得到氨基修饰二氧化硅粒子。
氨基修饰二氧化硅粒子、休眠聚合单体、光引发剂darocur2959按照1:10:0.1的质量比加入100l反应容器中,加入60l甲醇后搅拌分散,在紫外光(365nm)光照下反应1h,过滤收集沉淀得到超支化聚合物改性纳米成核剂。
超支化聚合物改性纳米成核剂和聚丙烯按照0.1:100的质量比混合均匀后,通过双螺杆挤出机在210℃,螺杆转速100rpm条件下熔融挤出造粒并注塑拉伸测试标准样条。
实施例2
实验过程和实施例1基本相同,区别之处在于:用甲基丙烯酰氯替换丁烯酰氯,利用轻质碳酸钙替换二氧化硅。休眠聚合单体的反应温度为10℃,反应时间为2小时,搅拌速度为50rpm。另外,轻质碳酸钙和氨基偶联剂的重量比为1000:1。
实施例3
实验过程和实施例1基本相同,区别之处在于:用丁炔酰氯替换丁烯酰氯,利用水滑石替换二氧化硅。休眠聚合单体的反应温度为-10℃,反应时间为4小时,搅拌速度为100rpm。另外,水滑石和氨基偶联剂的重量比为500:1。
实施例4
实验过程和实施例1基本相同,区别之处在于:用光引发剂quantacureqtx替换darocur2959。紫外光照反应时间调整为0.5h。另外,氨基修饰二氧化硅粒子、休眠聚合单体、quantacureqtx质量比调整为1:10:0.05。
实施例5
实验过程和实施例1基本相同,区别之处在于:用光引发剂quantacurebms替换darocur2959。紫外光照反应时间调整为2h。另外,氨基修饰二氧化硅粒子、休眠聚合单体、quantacurebms质量比调整为1:100:0.5。
对比例1
聚丙烯(熔融指数2~8g/10min,市场可购买的大庆石化t30g、抚顺石化t30s、泉州石化l5d98、泉州石化h3030及类似熔指聚丙烯牌号)通过注塑得到拉伸测试标准样条。
对比例2
本对比例通过在聚合反应过程中加入乙二胺,将高半胱氨酸硫内酯盐酸盐转变为活性单体。该活性单体在紫外光照下既可以和无机纳米粒子反应,也可以自身聚合,还可以交联多个无机纳米粒子。配制质量分数为10%的氨基硅烷偶联剂,二氧化硅纳米颗粒和氨基硅烷偶联剂溶液按100:1的质量比在高混机上混合均匀,搅拌时间30min,得到氨基修饰二氧化硅粒子。
氨基修饰二氧化硅粒子、高半胱氨酸硫内酯盐酸盐、乙二胺、光引发剂darocur2959按照1:10:10:0.1的质量比加入100l反应容器中,加入60l甲醇后搅拌分散,在紫外光(365nm)光照下反应1h,过滤收集沉淀得到超支化聚合物改性纳米成核剂。
对比例3
实验过程和实施例1基本相同,区别之处在于:二氧化硅纳米颗粒和氨基偶联剂的重量比调整为10:1。
对比例4
实验过程和实施例1基本相同,区别之处在于:高半胱氨酸硫内酯盐酸盐、丁烯酰氯和有机碱在四氢呋喃中的反应温度为80℃。
分别将各实施例和对比例所制备得到的超支化聚合物改性纳米成核剂和聚丙烯按照0.1:100的质量比混合均匀后,通过双螺杆挤出机在210℃,螺杆转速100rpm条件下熔融挤出造粒并注塑拉伸测试标准样条,并进行力学性能测试,其结果如表1所示。
表1为实施例和对比例的力学性能测试结果(基体树脂为h3030)
由表1可以看出,本发明超支化聚合物改性无机纳米成核剂通过共混熔融挤出提高聚丙烯的杨氏模量。相对于单纯的聚丙烯对比例1,本发明制备的超支化聚合物改性无机纳米成核剂在聚丙烯基体树脂中有优秀的成核效果,提升聚丙烯塑料制品的力学性能。相对于有活性单体的对比例2,本发明制备的拉伸测试样条杨氏模量要更高。这说明了:本发明中休眠单体聚合形成的超支化聚合物可以有效的控制聚合程度,提高纳米成核剂在聚丙烯中的分散程度,提升聚丙烯塑料制品的力学性能。
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
- 还没有人留言评论。精彩留言会获得点赞!