一种合成1,3,5-三嗪衍生物的方法与流程
本发明属于有机合成技术领域,特别是涉及一种合成1,3,5-三嗪衍生物的方法。
背景技术:
1,3,5-三嗪是许多具有生物活性的化合物的核心骨架,如杀虫剂、农药、除草剂、抗疟药、抗癌抗菌素、抗病毒药等。此外,1,3,5-三嗪还广泛应用于有机染料、液晶、荧光增白剂、电致发光材料、气体发生剂、固体推进剂、烟火等。
虽然1,3,5-三嗪衍生物具有广泛的应用,但是国内外对1,3,5-三嗪衍生物合成方法的报道仍十分有限。一般1,3,5-三嗪衍生物的合成方法有腈类的三聚、酰亚胺的环化、卤化三嗪在过渡金属催化下的偶联以及特定的多组分反应。以上合成方法往往需要苛刻的反应条件,如微波、高温、金属催化等。目前无催化剂、无碱、无配体条件下合成1,3,5-三嗪衍生物的报道较少。因此,开发出一种由简单的前体化合物合成1,3,5-三嗪衍生物的新的合成方法具有极其重要的意义。
技术实现要素:
本发明的目的是提供一种合成1,3,5-三嗪衍生物的方法,用以解决现有的1,3,5-三嗪衍生物的合成方法往往需要苛刻的反应条件,反应底物复杂的技术问题。
一种合成1,3,5-三嗪酮类化合物的方法,该反应通式如下,包括以下步骤:
步骤(1):以酰胺、草酰氯和脒为反应底物,将其分别加入到二甲苯中,搅拌加热进行反应;
步骤(2):反应结束后进行后处理、分离纯化得到1,3,5-三嗪酮类化合物。
一种合成含有三个不同取代基的2,4,6-三取代-1,3,5-三嗪类化合物的方法,该反应通式如下,包括以下步骤:
步骤(1):以酰胺、草酰氯和脒为反应底物,将其分别加入到二甲苯中,搅拌加热进行反应;
步骤(2):反应结束后进行后处理、分离纯化得到1,3,5-三嗪酮类化合物;
步骤(3):将1,3,5-三嗪酮类化合物、三氯氧磷和三乙胺在乙腈中反应生成2-氯-4,6-二取代1,3,5-三嗪类化合物;
步骤(4):将2-氯-4,6-二取代1,3,5-三嗪类化合物与各种亲核试剂偶联,实现对2-氯-4,6-二取代1,3,5-三嗪中氯原子的取代,从而制备得到含有三个不同取代基的2,4,6-三取代-1,3,5-三嗪类化合物。
优选地,所述r1为烷基、芳基和杂环芳基中的任意一种。
优选地,所述r2为烷基、芳基和杂环芳基中的任意一种。
优选地,所述步骤(1)中酰胺与脒的摩尔比为1-5:1。
优选地,所述步骤(1)中的反应温度为100-130℃,反应时间为12-24h。
优选地,步骤(3)中三氯氧磷、三乙胺、1,3,5-三嗪酮类化合物三者的摩尔比为3:3:1。
优选地,步骤(3)中的反应温度为90℃,反应时间为12h。
本发明的有益效果:本发明合成步骤简单,反应条件温和,合成1,3,5-三嗪酮类化合物不需要添加催化剂、碱、配体,反应条件温和,产率较高,产物选择性好,且在1,3,5-三嗪酮的基础上,可通过几步简单的成熟反应进行进一步衍生化,能够高选择性地制备含有三个不同取代基的2,4,6-三取代-1,3,5-三嗪类化合物。本发明适用范围广,反应收率较高,为1,3,5-三嗪衍生物的合成提供了一种新的途径,具有较强的工业应用前景。
附图说明
图1是实施例1制备的4,6-二苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图2是实施例1制备的4,6-二苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图3是实施例2制备的4-苯基-6-(对甲苯基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图4是实施例2制备的4-苯基-6-(对甲苯基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图5是实施例3制备的6-(4-甲氧基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图6是实施例3制备的6-(4-甲氧基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图7是实施例4制备的6-(4-羟基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图8是实施例4制备的6-(4-羟基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图9是实施例5制备的6-(4-氟苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图10是实施例5制备的6-(4-氟苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图11是实施例6制备的6-(4-氯苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图12是实施例6制备的6-(4-氯苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图13是实施例7制备的6-(3-氯苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图14是实施例7制备的6-(3-氯苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图15是实施例8制备的6-(2-氯苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图16是实施例8制备的6-(2-氯苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图17是实施例9制备的4-(3-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图18是实施例9制备的4-(3-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图19是实施例10制备的4-(4-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图20是实施例10制备的4-(4-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图21是实施例11制备的4-(2-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图22是实施例11制备的4-(2-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图23是实施例12制备的4-苯基-6-(4-(三氟甲基)苯基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图24是实施例12制备的4-苯基-6-(4-(三氟甲基)苯基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图25是实施例13制备的6-(4-硝基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图26是实施例13制备的6-(4-硝基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图27是实施例14制备的4-苯基-6-苯乙烯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图28是实施例14制备的4-苯基-6-苯乙烯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图29是实施例15制备的4-苯基-6-(噻吩-2-基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图30是实施例15制备的4-苯基-6-(噻吩-2-基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图31是实施例16制备的6-苯基-4-(吡啶-4-基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图32是实施例16制备的6-苯基-4-(吡啶-4-基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图33是实施例17制备的6-苯基-4-(吡啶-3-基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图34是实施例17制备的6-苯基-4-(吡啶-3-基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图35是实施例18制备的6-苯基-4-(吡啶-2-基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图36是实施例18制备的6-苯基-4-(吡啶-2-基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图37是实施例19制备的4-苯基-6-丙基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图38是实施例19制备的4-苯基-6-丙基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图39是实施例20制备的6-异丙基-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图40是实施例20制备的6-异丙基-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图41是实施例21制备的6-环己基-4-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图42是实施例21制备的6-环己基-4-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图43是实施例22制备的4-(叔丁基)-6-苯基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图44是实施例22制备的4-(叔丁基)-6-苯基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图45是实施例23制备的6-(叔丁基)-4-(4-甲氧基苯基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图46是实施例23制备的6-(叔丁基)-4-(4-甲氧基苯基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图47是实施例24制备的4-(叔丁基)-6-(4-氯苯基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图48是实施例24制备的4-(叔丁基)-6-(4-氯苯基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图49是实施例25制备的4-(叔丁基)-6-(3-氯苯基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图50是实施例25制备的4-(叔丁基)-6-(3-氯苯基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图51是实施例26制备的6-(叔丁基)-4-(噻吩-2-基)-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图52是实施例26制备的6-(叔丁基)-4-(噻吩-2-基)-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图53是实施例27制备的4-(叔丁基)-6-丙基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图54是实施例27制备的4-(叔丁基)-6-丙基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图55是实施例28制备的4-(叔丁基)-6-环己基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图56是实施例28制备的4-(叔丁基)-6-环己基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图57是实施例29制备的6-(叔丁基)-4-异丙基-1,3,5-三嗪-2(1h)-酮的1hnmr谱图;
图58是实施例29制备的6-(叔丁基)-4-异丙基-1,3,5-三嗪-2(1h)-酮的13cnmr谱图;
图59是实施例30制备的2-氯-4-(3-氯苯基)-6-苯基-1,3,5-三嗪的1hnmr谱图;
图60是实施例30制备的2-氯-4-(3-氯苯基)-6-苯基-1,3,5-三嗪的13cnmr谱图;
图61是实施例31制备的2-氯-4-苯基-6-(噻吩-2-基)-1,3,5-三嗪的1hnmr谱图;
图62是实施例31制备的2-氯-4-苯基-6-(噻吩-2-基)-1,3,5-三嗪的13cnmr谱图;
图63是实施例32制备的2-氯-4-异丙基-6-苯基-1,3,5-三嗪的1hnmr谱图;
图64是实施例32制备的2-氯-4-异丙基-6-苯基-1,3,5-三嗪的13cnmr谱图;
图65是实施例33制备的2-(叔丁基)-4-氯-6-丙基-1,3,5-三嗪的1hnmr谱图;
图66是实施例33制备的2-(叔丁基)-4-氯-6-丙基-1,3,5-三嗪的13cnmr谱图;
图67是实施例34制备的2-([1,1'-联苯]-3-基)-4-(3-氯苯基)-6-苯基-1,3,5-三嗪的1hnmr谱图;
图68是实施例34制备的2-([1,1'-联苯]-3-基)-4-(3-氯苯基)-6-苯基-1,3,5-三嗪的13cnmr谱图;
图69是实施例35制备的2-(3-氯苯基)-4-(二苯并[b,d]呋喃-4-基)-6-苯基-1,3,5-三嗪的1hnmr谱图;
图70是实施例35制备的2-(3-氯苯基)-4-(二苯并[b,d]呋喃-4-基)-6-苯基-1,3,5-三嗪的13cnmr谱图;
图71是实施例36制备的9-(4-(3-氯苯基)-6-苯基-1,3,5-三嗪-2-基)-9h-咔唑的1hnmr谱图;
图72是实施例36制备的9-(4-(3-氯苯基)-6-苯基-1,3,5-三嗪-2-基)-9h-咔唑的13cnmr谱图。
具体实施方式
下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
一种合成1,3,5-三嗪酮类化合物的方法,该反应通式如下,包括以下步骤:
步骤(1):在反应管中,加入反应底物酰胺,溶剂二甲苯,在常温搅拌下滴加草酰氯,60℃反应2h,然后加入脒升温至100-130℃,反应时间为12-24h;
步骤(2):反应结束后,将二甲苯旋干,用少量dmf将产物溶解后倒入热水中,趁热过滤得到固体1,3,5-三嗪酮类化合物。
所述酰胺为烷基酰胺、芳基酰胺和杂环芳基酰胺中的任意一种,所述酰胺的摩尔量为脒的摩尔量的1-5个当量。
所述脒为烷基脒、芳基脒和杂环芳脒中的任意一种。
一种合成含有三个不同取代基的2,4,6-三取代-1,3,5-三嗪类化合物的方法,该反应通式如下,包括以下步骤:
步骤(1):在反应管中,加入反应底物酰胺,溶剂二甲苯,在常温搅拌下滴加草酰氯,60℃反应2h,然后加入脒升温至100-130℃,反应时间为12-24h;
步骤(2):反应结束后,将二甲苯旋干,用少量dmf将产物溶解后倒入热水中,趁热过滤得到固体1,3,5-三嗪酮类化合物。
所述酰胺为烷基酰胺、芳基酰胺和杂环芳基酰胺中的任意一种,所述酰胺的摩尔量为脒的摩尔量的1-5个当量。
所述脒为烷基脒、芳基脒和杂环芳脒中的任意一种;
步骤(3):在反应管中,加入反应底物1,3,5-三嗪酮类化合物、pocl3、三乙胺、乙腈,90℃反应12h,反应结束后,恢复至室温,加入二氯甲烷萃取有机产物,然后浓缩有机溶剂,所得粗品进行柱层析纯化,得到2-氯-4,6-二取代1,3,5-三嗪类化合物,三氯氧磷、三乙胺、1,3,5-三嗪酮类化合物三者的摩尔比为3:3:1;
步骤(4):将2-氯-4,6-二取代1,3,5-三嗪类化合物与各种亲核试剂偶联,实现对2-氯-4,6-二取代1,3,5-三嗪中氯原子的取代,从而制备得到含有三个不同取代基的2,4,6-三取代-1,3,5-三嗪类化合物;根据实际需求,可以选择胺类、硼酸类、醇类和有机锡试剂等亲核底物实现对2-氯-4,6-二取代1,3,5-三嗪类化合物中氯原子的取代。
实施例1
结构式如下的三嗪酮的制备方法
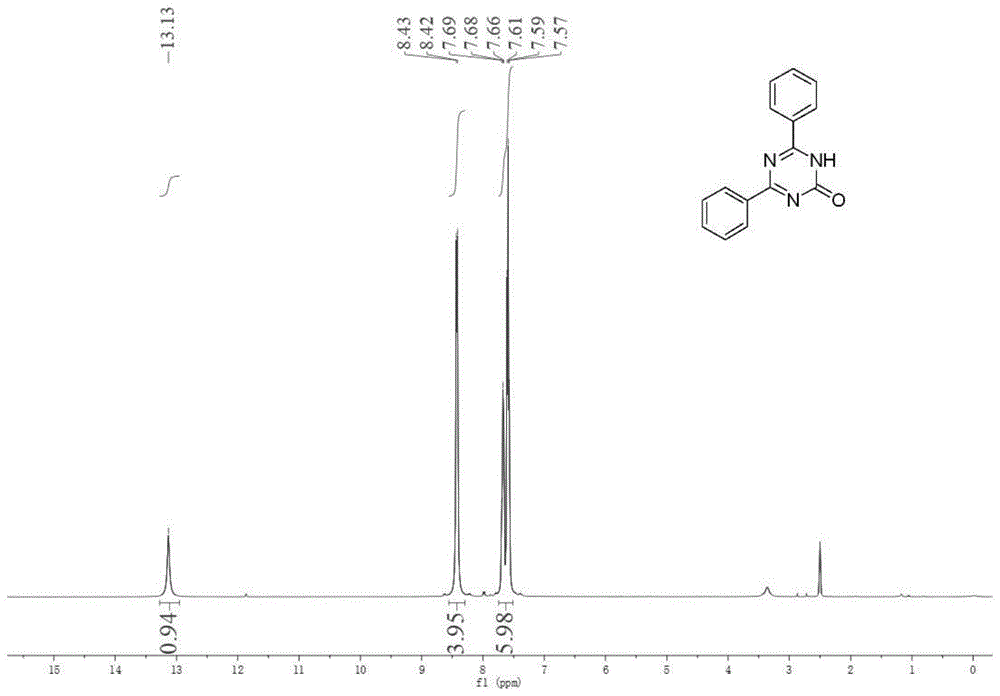
在25ml的schlenk管中,投入1.4mmol(169mg)苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体120mg。产品经nmr确定结构为4,6-二苯基-1,3,5-三嗪-2(1h)-酮,收率为80%。目标产物表征数据:whitesolid,mp260-265℃.1hnmr(400mhz,dmso-d6)δ13.15(s,1h),8.44(d,j=7.8hz,4h),7.69(t,j=7.3hz,2h),7.61(t,j=7.5hz,4h).13cnmr(101mhz,dmso-d6)δ133.32,128.86,128.85;lrms(ei):m/zcalcdforc15h11n3o[m]+,249;found,249.
实施例2
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(270mg)4-甲基苯甲酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得黄色固体125mg。产品经nmr确定结构为4-苯基-6-(对甲苯基)-1,3,5-三嗪-2(1h)-酮,收率为79%。目标产物表征数据:yellowsolid,mp225-234℃.1hnmr(400mhz,dmso)δ13.08(s,1h),8.39(dd,j=39.7,7.5hz,4h),7.65(dt,j=14.7,7.1hz,3h),7.40(d,j=7.8hz,2h),2.42(s,3h).13cnmr(101mhz,dmso)δ143.74,133.13,129.38,128.73,21.18.hrms(dart)calcdforc16h13n3o([m+h]+):264.1131,found:264.1128.
实施例3
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(211mg)4-甲氧基苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体118mg。产品经nmr确定结构为6-(4-甲氧基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮,收率为70%。目标产物表征数据:whitesolid,mp290-295℃.1hnmr(400mhz,dmso-d6)δ13.01(s,1h),8.69–8.30(m,4h),7.69(m,3h),7.36–7.06(m,2h),3.93(s,3h);13cnmr(101mhz,dmso-d6)δ163.65,133.21,130.98,128.85,128.82,114.36,55.71;hrms(dart)calcdforc16h13n3o2([m+h]+):280.1081,found:280.1080.
实施例4
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入3.4mmol(412mg)苯甲酰胺、5.1mmol(429ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(103mg)4-羟基卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体124mg。产品经nmr确定结构为6-(4-羟基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮,收率为78%。目标产物表征数据:whitesolid,mp305-309℃.1hnmr(400mhz,dmso)δ12.88(s,1h),10.51(s,1h),8.39(dd,j=30.0,7.7hz,4h),7.61(dt,j=14.8,7.1hz,3h),6.97(d,j=8.6hz,2h).13cnmr(101mhz,dmso)δ145.51,131.06,128.69,115.64.hrms(dart)calcdforc15h11n3o2([m+h]+):266.0924,found:266.0941.
实施例5
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(278mg)4-氟苯甲酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应18小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得黄色固体125mg。产品经nmr确定结构为6-(4-氟苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮,收率为78%。目标产物表征数据:yellowsolid,mp216-220℃.1hnmr(400mhz,dmso)δ13.14(s,1h),8.58–8.29(m,4h),7.63(dt,j=14.9,7.2hz,3h),7.40(t,j=8.6hz,2h).13cnmr(101mhz,dmso)δ166.46,163.96,133.31,131.60,131.51,128.76,128.69,115.93,115.71.hrms(dart)calcdforc15h10fn3o([m+h]+):268.0881,found:268.0878.
实施例6
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入0.7mmol(108mg)4-氯苯甲酰胺、1mmol(84ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应20小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体145mg。产品经nmr确定结构为6-(4-氯苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮,收率为85%。目标产物表征数据:whitesolid,mp240-248℃.1hnmr(400mhz,dmso-d6)δ13.21(s,1h),8.43(m,4h),7.65(m,5h);13cnmr(101mhz,dmso-d6)δ138.17,133.48,130.67,128.98,128.90,128.80;hrms(dart)calcdforc15h10cln3o([m+h]+):284.0585,found:284.0585.
实施例7
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(310mg)3-氯苯甲酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体124mg。产品经nmr确定结构为4-(3-氯苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮,收率为73%。目标产物表征数据:whitesolid,mp248-252℃.1hnmr(400mhz,dmso-d6)δ13.21(s,1h),8.49–8.31(m,4h),7.74–7.64(m,2h),7.59(td,j=7.7,3.0hz,3h).13cnmr(101mhz,dmso)δ133.59,133.42,132.73,130.66,128.81,128.73,128.22,127.36.hrms(dart)calcdforc15h10cln3o([m+h]+):284.0585,found:284.0605.
实施例8
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(310mg)2-氯苯甲酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应24小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得红色固体83mg。产品经nmr确定结构为4-(2-氯苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮,收率为49%。目标产物表征:redsolid,mp218-223℃.1hnmr(400mhz,dmso)δ13.35(s,1h),8.36(d,j=7.5hz,2h),7.82(d,j=7.2hz,1h),7.59(ddd,j=21.7,13.5,5.5hz,6h).13cnmr(101mhz,dmso)δ133.25,132.57,131.36,131.16,130.18,128.92,128.78,127.36.hrms(dart)calcdforc15h10cln3o([m+h]+):284.0585,found:284.0583.
实施例9
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(280mg)3-溴苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得黄色固体177mg。产品经nmr确定结构为4-(3-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮,收率为90%。目标产物表征:yellowsolid,mp235-251℃.1hnmr(400mhz,dmso)δ13.21(s,1h),8.51(s,1h),8.38(d,j=6.6hz,3h),7.83(d,j=7.8hz,1h),7.73–7.40(m,4h).13cnmr(101mhz,dmso)δ135.59,133.40,131.11,130.87,128.80,128.72,127.72,122.00.hrms(dart)calcdforc15h10brn3o([m+h]+):328.0080,found:328.0078.
实施例10
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(280mg)4-溴苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体151mg。产品经nmr确定结构为4-(4-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮,收率为77%。目标产物表征:whitesolid,mp300-306℃.1hnmr(400mhz,dmso)δ13.21(s,1h),8.39(dd,j=19.4,8.0hz,4h),7.80(d,j=8.4hz,2h),7.71(t,j=7.2hz,1h),7.61(t,j=7.6hz,2h).13cnmr(101mhz,dmso)δ133.39,131.83,130.71,128.80,128.70,127.21.hrms(dart)calcdforc15h10brn3o([m+h]+):328.0080,found:328.0083.
实施例11
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入3.4mmol(680mg)3-溴苯甲酰胺、5.1mmol(429ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应24小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体100mg。产品经nmr确定结构为4-(2-溴苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮,收率为51%。目标产物表征数据:whitesolid,mp218-223℃.1hnmr(400mhz,dmso)δ13.34(s,1h),8.36(d,j=7.3hz,2h),7.88–7.73(m,2h),7.73–7.45(m,5h).13cnmr(101mhz,dmso)δ133.25,132.58,131.01,128.94,128.80,127.82,120.37.hrms(dart)calcdforc15h10brn3o([m+h]+):328.0080,found:328.0080.
实施例12
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(265mg)4-三氟甲基苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体171mg。产品经nmr确定结构为4-苯基-6-(4-(三氟甲基)苯基)-1,3,5-三嗪-2(1h)-酮,收率为90%。目标产物表征数据:whitesolid,mp230-238℃.1hnmr(400mhz,dmso-d6)δ13.30(s,1h),8.48(dd,j=75.1,8.3hz,4h),7.90(d,j=8.4hz,2h),7.77–7.47(m,3h).13cnmr(101mhz,dmso)δ137.98,133.44,132.86,132.54,132.23,131.91,129.48,128.76,128.67,127.92,125.56,125.52,125.48,125.45,125.21,122.50,119.79.hrms(dart)calcdforc16h10f3n3o([m+h]+):318.0849,found:318.0847.
实施例13
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(232mg)4-硝基苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体147mg。产品经nmr确定结构为6-(4-硝基苯基)-4-苯基-1,3,5-三嗪-2(1h)-酮,收率为83%。目标产物表征数据:whitesolid,mp295-300℃.1hnmr(400mhz,dmso)δ13.34(s,1h),8.58(d,j=8.9hz,2h),8.34(dd,j=15.7,8.2hz,4h),7.69(t,j=7.3hz,1h),7.58(t,j=7.6hz,2h).13cnmr(101mhz,dmso)δ149.89,133.59,130.02,128.81,128.67,123.64.hrms(dart)calcdforc15h10n4o3([m+h]+):295.0826,found:295.0827.
实施例14
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(294mg)肉桂酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至120℃,反应24小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得黄色固体129mg。产品经nmr确定结构为4-苯基-6-苯乙烯基-1,3,5-三嗪-2(1h)-酮,收率为78%。目标产物表征数据:yellowsolid,mp127-130℃.1hnmr(400mhz,dmso)δ12.87(s,1h),8.46(d,j=7.5hz,2h),8.32(d,j=15.9hz,1h),7.86–7.72(m,2h),7.71–7.46(m,6h),7.00(d,j=15.9hz,1h).13cnmr(101mhz,dmso)δ171.07,166.14,143.52,135.33,134.64,133.03,129.46,129.15,128.87,128.69,120.87.hrms(dart)calcdforc17h13n3o([m+h]+):276.1131,found:276.1134.
实施例15
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(254mg)2-噻吩酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至120℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得黄色固体92mg。产品经nmr确定结构为4-苯基-6-(噻吩-2-基)-1,3,5-三嗪-2(1h)-酮,收率为60%。目标产物表征数据:yellowsolid,mp248-253℃.1hnmr(400mhz,dmso)δ13.10(s,1h),8.32(t,j=18.1hz,3h),8.03(s,1h),7.64(dt,j=15.1,7.2hz,3h),7.44–7.20(m,1h).13cnmr(101mhz,dmso)δ134.78,133.34,132.67,129.06,128.79,128.57.hrms(dart)calcdforc13h9n3os([m+h]+):256.0539,found:256.0541.
实施例16
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(242mg)苯甲酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)4-吡啶甲眯盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体108mg。产品经nmr确定结构为6-苯基-4-(吡啶-4-基)-1,3,5-三嗪-2(1h)-酮,收率为72%。目标产物表征数据:whitesolid,mp230-236℃.1hnmr(400mhz,dmso-d6)δ13.41(s,1h),8.83(d,j=5.0hz,2h),8.40(d,j=7.7hz,2h),8.26(d,j=5.0hz,2h),7.79–7.45(m,3h).13cnmr(101mhz,dmso)δ150.59,133.62,128.89,128.79,122.12.hrms(dart)calcdforc14h10n4o([m+h]+):251.0927,found:251.0928.
实施例17
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(170mg)苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)3-吡啶甲眯盐酸盐,搅拌下加热升温至130℃,反应12小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得黄色固体78mg。产品经nmr确定结构为6-苯基-4-(吡啶-3-基)-1,3,5-三嗪-2(1h)-酮,收率为52%。目标产物表征数据:yellowsolid,mp235-240℃.1hnmr(400mhz,dmso)δ13.31(s,1h),9.55(d,j=1.7hz,1h),8.84(dd,j=4.8,1.6hz,1h),8.73(dt,j=8.0,1.9hz,1h),8.49–8.39(m,2h),7.66(dq,j=13.5,7.5hz,4h).13cnmr(101mhz,dmso)δ153.33,149.79,136.26,133.49,128.84,128.79,123.79.hrms(dart)calcdforc14h10n4o([m+h]+):251.0927,found:251.0927.
实施例18
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(170mg)苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)2-吡啶甲眯盐酸盐,搅拌下加热升温至130℃,反应24小时后停止反应,冷却,真空旋蒸除去二甲苯,固体用dmf溶解后倒入热水中,趁热过滤,得白色固体63mg。产品经nmr确定结构为6-苯基-4-(吡啶-2-基)-1,3,5-三嗪-2(1h)-酮,收率为42%。目标产物表征数据:whitesolid(63mg,42%yield);mp210-215℃.1hnmr(400mhz,dmso)δ12.87(s,1h),8.84(d,j=4.3hz,1h),8.62(d,j=7.8hz,1h),8.49(d,j=7.4hz,2h),8.14(t,j=7.1hz,1h),7.77(dd,j=7.0,5.0hz,1h),7.68(t,j=7.3hz,1h),7.59(t,j=7.5hz,2h).13cnmr(101mhz,dmso)δ149.59,138.12,133.10,129.05,128.72,128.12,124.05.hrms(dart)calcdforc14h10n4o([m+h]+):251.0927,found:251.0927.
实施例19
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(174mg)丁酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至100℃反应12h。反应结束后,反应液旋蒸过柱得到白色固体97mg,收率75%。产品经nmr确定结构为4-苯基-6-丙基-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid(hexane/ea=3:1asaneluent);mp194-198℃.1hnmr(400mhz,cdcl3)δ13.69(s,1h),8.55(d,j=7.6hz,2h),7.60(t,j=7.3hz,1h),7.50(t,j=7.5hz,2h),2.82(t,j=7.6hz,2h),2.05–1.87(m,2h),1.08(t,j=7.4hz,3h).13cnmr(101mhz,cdcl3)δ174.70,172.10,159.81,134.91,133.46,129.96,128.57,36.94,20.18,13.59.hrms(dart)calcdforc12h13n3o([m+h]+):216.1131,found:216.1185.
实施例20
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(174mg)异丁酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至100℃反应12h。反应结束后,反应液旋蒸过柱得到白色固体92mg,收率71%。产品经nmr确定结构为6-异丙基-4-苯基-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid(hexane/ea=3:1asaneluent);mp120-126℃.1hnmr(400mhz,cdcl3)δ13.59(s,1h),8.56(d,j=7.5hz,2h),7.59(t,j=7.2hz,1h),7.50(t,j=7.3hz,2h),3.17–3.01(m,1h),1.45(d,j=6.9hz,6h).13cnmr(101mhz,cdcl3)δ176.42,174.83,159.81,135.05,133.44,130.00,128.54,34.54,20.22.hrms(dart)calcdforc12h13n3o([m+h]+):216.1131,found:216.1161.
实施例21
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入3.4mmol(432mg)环己基甲酰胺、5.1mmol(429ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(94mg)卞脒盐酸盐,搅拌下加热升温至100℃反应12h。反应结束后,反应液旋蒸过柱得到白色固体130mg,收率85%。产品经nmr确定结构为6-环己基-4-苯基-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid(hexane/ea=3:1asaneluent);mp215-220℃.1hnmr(400mhz,cdcl3)δ13.45(s,1h),8.55(d,j=7.3hz,2h),7.55(dt,j=37.8,7.1hz,3h),2.78(tt,j=11.5,3.3hz,1h),2.04(d,j=11.6hz,2h),1.82(dt,j=23.9,12.4hz,5h),1.57–1.31(m,3h).13cnmr(101mhz,cdcl3)δ175.60,159.79,135.24,133.48,130.11,128.64,43.87,30.38,25.61,25.54;hrms(dart)calcdforc15h17n3o([m+na]+):278.1264,found:278.1254.
实施例22
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2.7mmol(327mg)苯甲酰胺、4mmol(336ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应12h。反应结束后,反应液旋蒸过柱得到白色固体100mg,收率73%。产品经nmr确定结构为4-(叔丁基)-6-苯基-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid(hexane/ea=3:1asaneluent);mp180-190℃.(hexane/ea=3:1asaneluent)1hnmr(400mhz,cdcl3)δ13.00(s,1h),8.57(d,j=7.6hz,2h),7.60(t,j=7.3hz,1h),7.51(t,j=7.4hz,2h),1.54(s,9h);13cnmr(101mhz,cdcl3)δ178.06,174.41,159.48,135.20,133.43,129.99,128.60,38.45,28.06;hrms(dart)calcdforc13h15n3o([m+na]+):252.1107,found:252.1104.
实施例23
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入0.7mmol(109mg)4-甲氧基苯甲酰胺、1mmol(84ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应12h。反应结束后,反应液旋蒸过柱得到白色固体116mg,收率75%。产品经nmr确定结构为6-(叔丁基)-4-(4-甲氧基苯基)-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid,(hexane/ea=3:1asaneluent);mp235-240℃1hnmr(400mhz,dmso)δ12.44(s,1h),8.32(d,j=8.8hz,2h),7.09(d,j=8.9hz,2h),3.85(s,3h),1.34(s,9h).13cnmr(101mhz,dmso)δ163.23,156.21,130.93,114.03,55.47,37.75,27.39.hrms(dart)calcdforc14h17n3o2([m+h]+):260.1394,found:260.1376.
实施例24
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(218mg)4-氯苯甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应12h。反应结束后,反应液旋蒸过柱得到白色固体137mg,收率87%。产品经nmr确定结构为4-(叔丁基)-6-(4-氯苯基)-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid,(hexane/ea=3:1asaneluent);mp190-194℃.1hnmr(400mhz,dmso)δ12.61(s,1h),8.34(d,j=8.6hz,2h),7.61(d,j=8.6hz,2h),1.35(s,9h).13cnmr(101mhz,dmso)δ137.76,130.61,128.82,38.89,27.41;hrms(dart)calcdforc13h14cln3o([m+na]+):286.0718,found:286.0708.
实施例25
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(312mg)3-氯苯甲酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应12h。反应结束后,反应液旋蒸过柱得到白色固体122mg,收率77%。产品经nmr确定结构为4-(叔丁基)-6-(3-氯苯基)-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid,(hexane/ea=3:1asaneluent);mp220-224℃.1hnmr(400mhz,dmso)δ12.65(s,1h),8.27(d,j=1.9hz,2h),7.60(dt,j=11.9,8.1hz,2h),1.34(s,9h).13cnmr(101mhz,dmso)δ177.94,170.35,156.08,137.47,133.44,132.50,130.58,128.22,127.39,37.64,27.29.hrms(dart)calcdforc13h14cln3o([m+h]+):264.0898,found:264.0899.
实施例26
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(284mg)2-噻吩酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应24h。反应结束后,反应液旋蒸过柱得到白色固体81mg,收率57%。产品经nmr确定结构为6-(叔丁基)-4-(噻吩-2-基)-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid,(hexane/ea=3:1asaneluent);mp255-260℃.1hnmr(400mhz,dmso)δ12.44(s,1h),8.31–7.64(m,2h),7.26(dd,j=4.8,3.9hz,1h),1.33(s,9h).13cnmr(101mhz,dmso)δ155.81,134.46,132.51,128.82,37.45,27.23.hrms(dart)calcdforc11h13n3os([m+h]+):236.0852,found:236.0874.
实施例27
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入3.4mmol(296mg)丁酰胺、5.1mmol(429ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应24h。反应结束后,反应液旋蒸过柱得到白色固体102mg,收率87%。产品经nmr确定结构为4-(叔丁基)-6-丙基-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid,(hexane/ea=3:1asaneluent);mp70-73℃.1hnmr(400mhz,cdcl3)δ13.57(s,0.65h),12.59(s,0.25h),2.68(t,j=7.5hz,2h),1.90–1.74(m,2h),1.33(s,9h),0.99(t,j=7.4hz,3h).13cnmr(101mhz,cdcl3)δ190.00,170.90,159.37,39.88,36.38,28.14,19.82,13.21.hrms(dart)calcdforc10h17n3o([m+h]+):196.1444,found:196.1474.
实施例28
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入1.4mmol(178mg)环己基甲酰胺、2.1mmol(176ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应24h。反应结束后,反应液旋蒸过柱得到白色固体71mg,收率50%。产品经nmr确定结构为4-(叔丁基)-6-环己基-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:whitesolid,(hexane/ea=3:1asaneluent);mp115-119℃.1hnmr(400mhz,cdcl3)δ13.23(s,0.59h),12.59(s,0.42h),2.64(tt,j=11.5,3.4hz,1h),1.93(d,j=11.9hz,2h),1.82(dd,j=10.0,3.0hz,2h),1.65(dt,j=34.9,11.8hz,3h),1.35(s,9h),1.26(dt,j=25.7,14.3hz,3h).13cnmr(101mhz,cdcl3)δ185.72,179.94,154.65,44.25,39.61,30.15,30.05,28.07,25.38,25.26,25.24.hrms(dart)calcdforc13h21n3o([m+h]+):236.1757,found:236.1756.
实施例29
结构式如下的三嗪酮的制备方法
在25ml的schlenk管中,投入2mmol(174mg)异丁酰胺、3mmol(252ul)草酰氯、3ml二甲苯;搅拌下加热升温至60℃,反应2小时后停止反应,加入0.6mmol(82mg)叔丁基胍盐酸盐,搅拌下加热升温至100℃反应24h。反应结束后,反应液旋蒸过柱得到黄色固体73mg,收率62%。产品经nmr确定结构为6-(叔丁基)-4-异丙基-1,3,5-三嗪-2(1h)-酮,目标产物表征数据:yellowsolid,(hexane/ea=3:1asaneluent);mp118-123℃.1hnmr(400mhz,cdcl3)δ13.04(d,j=248.5hz,1h),2.89(hept,j=6.9hz,1h),1.29(s,9h),1.25(d,j=6.9hz,6h).13cnmr(101mhz,cdcl3)δ159.46,28.20,26.86,20.23,18.85.hrms(dart)calcdforc10h17n3o([m+h]+):196.1444,found:196.1442.
实施例30
结构式如下的2-氯-4-(3-氯苯基)-6-苯基-1,3,5-三嗪的制备方法
在25ml的schlenk管中,投入4-(3-氯苯基)-6-苯基-1,3,5-三嗪-2(1h)-酮(57mg,0.2mmol),pocl3(56ul,0.6mmol),三乙胺(83ul,0.6mmol)和乙腈(2ml),搅拌下加热升温至90℃反应12小时。反应结束后,反应液旋蒸过柱得到白色固体42mg,收率70%。产品经nmr确定结构为2-氯-4-(3-氯苯基)-6-苯基-1,3,5-三嗪,目标产物表征数据:whitesolid,(hexane/dcm=6:1asaneluent);mp145-148℃.1hnmr(400mhz,cdcl3)δ8.64–8.54(m,3h),8.53–8.45(m,1h),7.67–7.42(m,5h).13cnmr(101mhz,cdcl3)δ173.55,172.28,172.16,136.18,135.10,134.06,133.79,133.44,130.09,129.47,129.25,128.89,127.45.hrms(dart)calcdforc15h9cl2n3([m+h]+):302.0246,found:302.0252.
实施例31
结构式如下的2-氯-4-苯基-6-(噻吩-2-基)-1,3,5-三嗪的制备方法
在25ml的schlenk管中,投入6-苯基-4-(噻吩-2-基)-1,3,5-三嗪-2(1h)-酮(51mg,0.2mmol),pocl3(56ul,0.6mmol),三乙胺(83ul,0.6mmol)和乙腈(2ml),搅拌下加热升温至90℃反应12小时,反应结束后,反应液旋蒸过柱得到白色固体41mg,收率75%。产品经nmr确定结构为2-氯-4-苯基-6-(噻吩-2-基)-1,3,5-三嗪,目标产物表征数据:whitesolid,(hexane/dcm=6:1asaneluent);mp115-117℃.1hnmr(400mhz,cdcl3)δ8.62–8.54(m,2h),8.30(dd,j=3.8,1.2hz,1h),7.70(dd,j=5.0,1.2hz,1h),7.66–7.59(m,1h),7.58–7.49(m,2h),7.23(dd,j=4.9,3.8hz,1h).13cnmr(101mhz,cdcl3)δ173.24,171.77,169.29,139.95,134.10,134.01,133.60,133.22,129.39,128.86,128.81.
实施例32
结构式如下的2-氯-4-异丙基-6-苯基-1,3,5-三嗪的制备方法
在25ml的schlenk管中,投入4-异丙基-6-苯基-1,3,5-三嗪-2(1h)-酮(51mg,0.2mmol),pocl3(56ul,0.6mmol),三乙胺(83ul,0.6mmol)和乙腈(2ml),搅拌下加热升温至90℃反应12小时,反应结束后,反应液旋蒸过柱得到黄色固体37mg,收率78%。产品经nmr确定结构为2-氯-4-异丙基-6-苯基-1,3,5-三嗪,目标产物表征数据:yellowsolid,(hexane/dcm=6:1asaneluent);mp50-53℃.1hnmr(400mhz,cdcl3)δ8.46(dd,j=8.4,1.3hz,2h),7.53(ddd,j=6.6,3.9,1.3hz,1h),7.48–7.40(m,2h),3.11(hept,j=6.9hz,1h),1.34(d,j=6.9hz,6h).13cnmr(101mhz,cdcl3)δ186.26,173.19,171.68,134.30,133.48,129.38,128.80,37.39,20.89.hrms(dart)calcdforc12h12cl3n3([m+h]+):234.0793,found:234.0790.
实施例33
结构式如下的2-(叔丁基)-4-氯-6-丙基-1,3,5-三嗪的制备方法
在25ml的schlenk管中,投入6-(叔丁基)-4-丙基-1,3,5-三嗪-2(1h)-酮(39mg,0.2mmol),pocl3(56ul,0.6mmol),三乙胺(83ul,0.6mmol)和乙腈(2ml),搅拌下加热升温至90℃反应12小时,反应结束后,反应液旋蒸过柱得到黄色液体29mg,收率67%,产品经nmr确定结构为2-(叔丁基)-4-氯-6-丙基-1,3,5-三嗪,目标产物表征数据:yellowliquid,(hexane/dcm=6:1asaneluent);1hnmr(400mhz,cdcl3)δ2.83–2.72(m,2h),1.83–1.71(m,2h),1.30(s,9h),0.93(t,j=7.4hz,3h).13cnmr(101mhz,cdcl3)δ188.01,181.45,171.00,40.56,39.68,28.72,20.94,13.72.hrms(dart)calcdforc10h16cln3([m+h]+):214.1106,found:214.1102.
实施例34
结构式如下的2-([1,1'-联苯]-3-基)-4-(3-氯苯基)-6-苯基-1,3,5-三嗪的制备方法
在25ml的schlenk管中投入实施例30得到的2-氯-4-(3-氯苯基)-6-苯基-1,3,5-三嗪(150mg,0.5mmol),并依次加入联苯-3-硼酸(198mg,1mmol),pd(pph3)2cl2(18mg,5mol%),k2co3(276mg,2mmol),甲苯(5ml)和h2o(0.5ml),随后,将反应在氮气氛围下于110℃搅拌12小时。反应结束后,反应液旋蒸过柱得到白色固体189mg,收率90%。产品经nmr确定结构为2-([1,1'-联苯]-3-基)-4-(3-氯苯基)-6-苯基-1,3,5-三嗪,目标产物表征数据:whitesolid,(hexane/dcm=4:1asaneluent);mp145-150℃.1hnmr(400mhz,cdcl3)δ8.95(t,j=1.6hz,1h),8.79–8.68(m,4h),8.68–8.59(m,1h),7.85–7.78(m,1h),7.77–7.70(m,2h),7.68–7.47(m,8h),7.43(t,j=7.4hz,1h).13cnmr(101mhz,cdcl3)δ171.83,171.79,170.54,141.81,140.82,138.12,136.52,135.92,134.86,132.75,132.45,131.47,129.92,129.15,129.06,128.94,128.71,127.93,127.66,127.38,127.12.hrms(dart)calcdforc27h18cln3([m+h]+):420.1262,found:420.1266.
实施例35
结构式如下的2-(3-氯苯基)-4-(二苯并[b,d]呋喃-4-基)-6-苯基-1,3,5-三嗪的制备方法
在25ml的schlenk管中投入实施例30得到的2-氯-4-(3-氯苯基)-6-苯基-1,3,5-三嗪(150mg,0.5mmol),并依次加入二苯并呋喃-4-硼酸(212mg,1mmol),pd(pph3)2cl2(18mg,5mol%),k2co3(276mg,2mmol),甲苯(5ml)和h2o(0.5ml)。随后,将反应在氮气氛围下于110℃搅拌12小时。反应结束后,反应液旋蒸过柱得到白色固体206mg,收率95%。产品经nmr确定结构为2-(3-氯苯基)-4-(二苯并[b,d]呋喃-4-基)-6-苯基-1,3,5-三嗪,目标产物表征数据:whitesolid,(hexane/dcm=4:1asaneluent);mp185-190℃.1hnmr(400mhz,cdcl3)δ8.90–8.83(m,3h),8.80(dd,j=7.8,1.2hz,1h),8.75(dt,j=7.6,1.3hz,1h),8.20(dd,j=7.6,1.3hz,1h),8.03(d,j=7.6hz,1h),7.79(d,j=8.2hz,1h),7.69–7.51(m,7h),7.42(t,j=7.5hz,1h).13cnmr(101mhz,cdcl3)δ171.87,170.75,170.55,156.66,155.60,138.17,136.01,134.89,132.76,132.45,129.92,129.24,129.15,128.92,128.72,127.65,127.12,126.35,124.81,123.41,123.08,122.75,121.46,120.58,112.10.hrms(dart)calcdforc27h16cln3o([m+k]+):472.0613,found:472.0663.
实施例36
结构式如下的9-(4-(3-氯苯基)-6-苯基-1,3,5-三嗪-2-基)-9h-咔唑的制备方法
在25ml的schlenk管中投入咔唑(117mg,0.7mmol),nah(112mg,2.8mmol),dmf(5ml),将反应在氮气氛围下室温搅拌1h。随后,向schlenk管中投入实施例30得到的2-氯-4-(3-氯苯基)-6-苯基-1,3,5-三嗪(301mg,1mmol),室温搅拌12h。反应结束后,向混合物中加入冰水,过滤并用dcm洗涤,得到白色固体287mg,收率95%。产品经nmr确定结构为9-(4-(3-氯苯基)-6-苯基-1,3,5-三嗪-2-基)-9h-咔唑,目标产物表征数据:whitesolid;mp200-205℃.1hnmr(400mhz,cdcl3)δ9.11(d,j=8.4hz,2h),8.78–8.67(m,3h),8.62(dt,j=7.6,1.3hz,1h),8.08(d,j=7.2hz,2h),7.70–7.51(m,7h),7.50–7.40(m,2h).13cnmr(101mhz,cdcl3)δ172.52,171.17,165.08,139.01,138.11,135.96,134.94,132.85,132.54,130.03,129.15,129.13,128.82,127.10,127.02,126.71,123.41,119.66,117.75.hrms(dart)calcdforc27h17cln4([m+h]+):433.1215,found:433.1212.
不同反应条件下合成1,3,5-三嗪酮类化合物的反应收率如表1所示:
表1
以上所述的仅是本发明的一些实施方式。对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!