偶氮色素组合物及其制造方法与流程
本发明涉及一种偶氮色素组合物及其制造方法。
背景技术:
偶氮色素化合物为可以使用于喷墨油墨、滤色器、染发剂(Hair dye)、升华型色素等各种用途的化合物。
例如,已知有包含特定结构的偶氮色素化合物(解离性偶氮染料)的染发剂组合物(例如,参考日本专利第4080947号公报)。
技术实现要素:
发明要解决的技术课题
然而,日本专利第4080947号公报中记载的偶氮色素化合物中,有时根据取代为色素骨架的取代基的种类或组合而结晶性非常高。因此有时上述偶氮色素化合物难以溶解于溶剂中,难以通过提纯(例如再结晶)获得高纯度的偶氮色素化合物。
本公开的目的在于提供一种可以获得高纯度的偶氮色素化合物的偶氮色素组合物及其制造方法。
用于解决技术课题的手段
用于实现上述课题的具体的方法为如下。
<1>一种偶氮色素组合物,其包含由式(I)表示的偶氮色素化合物和由式(II)表示的脲化合物。
[化学式1]
式(I)中,R11、R12、R13及R14分别独立地表示氢原子、卤素原子或脂肪族基,R15表示脂肪族基。
式(II)中,R21及R22分别独立地表示氢原子、脂肪族基或芳香族基,n表示0~2的整数。
<2>根据<1>所述的偶氮色素组合物,其中,R11、R12、R13及R14中的至少1个为卤素原子。
<3>根据<1>或<2>所述的偶氮色素组合物,其中,R21及R22分别独立地为脂肪族基。
<4>根据<1>~<3>中任一项所述的偶氮色素组合物,其中,R11、R12、R13及R14分别独立地为氢原子、卤素原子或碳原子数为1~3的烷基,且R11、R12、R13及R14中的至少1个是卤素原子,R15是碳原子数为1~3的烷基,R21及R22分别独立地为碳原子数为1~3的烷基。
<5>根据<1>~<4>中任一项所述的偶氮色素组合物,其中,R11及R13中的至少1个为卤素原子。
<6>根据<1>~<5>中任一项所述的偶氮色素组合物,其中,相对于偶氮色素组合物总量,由式(I)表示的偶氮色素化合物和由式(II)表示的脲化合物的总含量为95质量%以上。
<7>根据<1>~<6>中任一项所述的偶氮色素组合物,其中,由式(II)表示的脲化合物相对于由式(I)表示的偶氮色素化合物的含有质量比为0.5摩尔倍~1.5摩尔倍。
<8>一种偶氮色素组合物的制造方法,其为制造<1>~<7>中任一项所述的偶氮色素组合物的方法,该方法具有:准备工序,准备由式(I)表示的偶氮色素化合物的产物粗品;及析出工序,通过使产物粗品和由式(II)表示的脲化合物即第1溶剂在不同于第1溶剂的第2溶剂中接触而析出偶氮色素组合物。
<9>根据<8>所述的偶氮色素组合物的制造方法,其中,第2溶剂为选自包括芳香族烃类溶剂、酯类溶剂、酮类溶剂、腈类溶剂、醚类溶剂、脂肪族烃类溶剂及醇类溶剂的组中的至少1种。
<10>根据<8>或<9>所述的偶氮色素组合物的制造方法,其中,第2溶剂为选自包括甲苯、二甲苯、均三甲苯、乙基苯、乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酸乙酯、丙酮、甲乙酮、甲基异丁基酮、乙腈、丙腈、二异丙基醚、甲基叔丁基醚、四氢呋喃、二噁烷、己烷、庚烷、辛烷、环己烷、甲醇、异丙醇及乙二醇的组中的至少1种。
发明效果
根据本发明的方式,提供一种可以获得高纯度的偶氮色素化合物的偶氮色素组合物及其制造方法。
附图说明
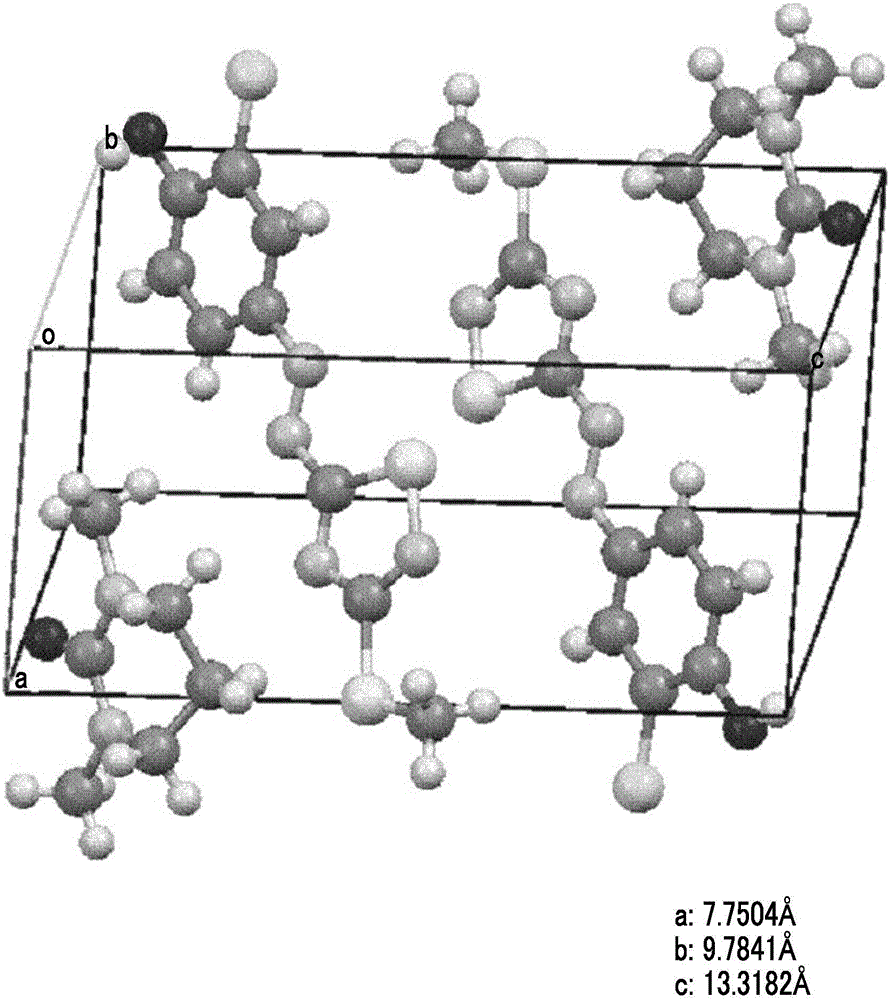
图1A是实施例1中的偶氮色素组合物(组合物24)的结构的概念图。
图1B是在图1A中示出的偶氮色素组合物(组合物24)的c轴投影图。
具体实施方式
以下,关于本公开的偶氮色素组合物及其制造方法详细地进行说明。
本说明书中,用“~”表示的数值范围表示将记载于“~”前后的数值作为下限值及上限值而包含的范围。
<偶氮色素组合物>
本公开的偶氮色素组合物包括:由下述式(I)表示的偶氮色素化合物(以下,也简称为“偶氮色素化合物”);及由下述式(II)表示的脲化合物(以下,也简称为“脲化合物”)。
[化学式2]
式(I)中,R11、R12、R13及R14分别独立地表示氢原子、卤素原子或脂肪族基,R15表示脂肪族基。
式(II)中,R21及R22分别独立地表示氢原子、脂肪族基或芳香族基,n表示0~2的整数。
偶氮色素化合物能够通过基于重氮化合物与偶联化合物的重氮偶联反应的方法等通常的合成方法合成。而且,通常被合成的偶氮色素化合物(产物粗品)中混入有杂质。作为将混入有杂质的偶氮色素化合物的产物粗品进行提纯的方法,可以举出使该产物粗品溶解于溶剂中进行提纯的方法(例如再结晶)。
然而,有时偶氮色素化合物的结晶性非常高,有时难以使其溶解于溶剂中。因此,有时通过提纯难以获得高纯度的偶氮色素化合物。
关于上述问题,本发明人等发现在包含由式(I)表示的偶氮色素化合物与由式(II)表示的脲化合物的组合的本公开的偶氮色素组合物中,在偶氮色素化合物的合成时混入的杂质的量得到减少。即,本发明人等发现在上述偶氮色素组合物中包含杂质少且纯度高的偶氮色素化合物。虽然该理由不明确,但认为与由式(I)表示的偶氮色素化合物具有酚性羟基、以及由式(II)表示的脲化合物为弱碱基有关联。
因此,根据本公开的偶氮色素组合物,能够获得高纯度的偶氮色素化合物。
关于由本公开的偶氮色素组合物获得高纯度的偶氮色素化合物(分离)的方法,将进行后述。
另外,在本说明书中,“偶氮色素化合物的产物粗品”是指包括作为纯物质的偶氮色素化合物和合成时混入的杂质的产物。
即,在“产物粗品”的概念中,不仅可以包括未提纯的产物(例如纯度低的产物),而且也可以包括已提纯的产物(例如纯度较高的产物)。
作为偶氮色素化合物的产物粗品中的偶氮色素化合物的含量(即,偶氮色素化合物的纯度),例如可以举出80.0质量%~99.0质量%,优选90.0质量%~99.0质量%,更优选95.0质量%~99.0质量%,尤其优选97.0质量%~98.5质量%。
以下,关于由式(I)表示的偶氮色素化合物及由式(II)表示的脲化合物进行说明。
(由式(I)表示的偶氮色素化合物)
本公开的偶氮色素组合物包括由下述式(I)表示的偶氮色素化合物。
[化学式3]
式(I)中,R11、R12、R13及R14分别独立地表示氢原子、卤素原子或脂肪族基,R15表示脂肪族基。
作为由R11~R14表示的卤素原子,可以举出氟原子、氯原子、溴原子或碘原子,优选氯原子或溴原子,更优选氯原子。
作为由R11~R14表示的脂肪族基,可以举出碳原子数为1~10的脂肪族基,优选碳原子数为1~6的脂肪族基,更优选碳原子数为1~3的脂肪族基。
并且,作为上述脂肪族基,优选烷基或烯基,更优选烷基。
作为由R11~R14表示的脂肪族基,优选碳原子数为1~10的烷基,更优选碳原子数为1~6的烷基,进一步优选碳原子数为1~3的烷基,进一步优选甲基或乙基,尤其优选甲基。
在由式(I)表示的偶氮色素化合物中,优选R11~R14中的至少1个为卤素原子,更优选R11及R13中的至少1个为卤素原子。
在R11~R14中的至少1个(更优选R11及R13中的至少1个)为卤素原子的情况下,偶氮色素化合物的溶解性提高。由此,由偶氮色素组合物获得的偶氮色素化合物的纯度进一步提高。并且,也有如下优点,即该情况下偶氮色素化合物的溶解性提高,由此制造本公开的偶氮色素组合物变得更加容易。
由R15表示的脂肪族基与由R11~R14表示的脂肪族基的含义相同,优选范围也相同。
由式(I)表示的偶氮色素化合物能够通过例如重氮盐与偶联化合物的重氮偶联反应等公知方法进行合成。作为由式(I)表示的偶氮色素化合物的合成方法,可以适当地参考例如日本专利第4080947号公报中记载的方法。
以下,示出由式(I)表示的偶氮色素化合物的具体例(例示化合物(I-1)~(I-13)),但本发明并不限定于这些具体例。
另外,在以下具体例中,“Me”表示甲基。
[化学式4]
(由式(II)表示的脲化合物)
本公开的偶氮色素组合物包括由下述式(II)表示的脲化合物。
作为由式(II)表示的脲化合物,优选为具有作为弱碱基性溶剂的功能的化合物。
[化学式5]
式(II)中,R21及R22分别独立地表示氢原子、脂肪族基或芳香族基,n表示0~2的整数。
由R21及R22表示的脂肪族基与由R11~R14表示的脂肪族基的含义相同,优选范围也相同。
作为由R21及R22表示的芳香族基,可以举出苯基或萘基,优选苯基。
从进一步提高由偶氮色素组合物获得的偶氮色素化合物的纯度的观点考虑,优选R21及R22分别独立地为氢原子或脂肪族基,更优选脂肪族基,进一步优选碳原子数为1~10(进一步优选碳原子数为1~6,进一步优选碳原子数为1~3)的烷基,尤其优选甲基或乙基。
n表示0~2的整数,优选0或1,尤其优选1。
另外,在由式(II)表示的脲化合物中,n为0的化合物为由下述式(II)-A表示的化合物,n为1的化合物为由下述式(II)-B表示的化合物,n为2的化合物为由下述式(II)-C表示的化合物。
[化学式6]
在式(II)-A、式(II)-B及式(II)-C中,R21与式(II)中的R21的含义相同,R22与式(II)中的R22的含义相同。
以下,示出由式(II)表示的脲化合物的具体例(例示化合物(II-1)~(II-4)),但本发明并不限定于这些具体例。
另外,在以下具体例中,“Me”及“Et”分别表示甲基及乙基。
[化学式7]
本公开的偶氮色素组合物可以包含由式(I)表示的偶氮色素化合物的基于由式(II)表示的脲化合物(溶剂)的溶剂化物,也可以包含上述偶氮色素化合物与上述脲化合物的盐。并且,本公开的偶氮色素组合物也可以包含上述溶剂化物及上述盐这两者。
作为上述偶氮色素化合物的基于上述脲化合物(溶剂)的溶剂化物,可以举出下述溶剂化物1。下述溶剂化物1为上述脲化合物一个分子相对于上述偶氮色素化合物一个分子进行溶剂化的一种溶剂化物的例子。
作为上述偶氮色素化合物与上述脲化合物的盐,可以举出下述盐1。
[化学式8]
在上述溶剂化物1中,R11、R12、R13、R14、R15、R21、R22及n与式(I)及式(II)中的R11、R12、R13、R14、R15、R21、R22及n的含义相同。
在上述盐1中,R11、R12、R13、R14、R15、R21、R22及n与式(I)及式(II)中的R11、R12、R13、R14、R15、R21、R22及n的含义相同。
如上述盐的例子(例如盐1)所示,在本公开的偶氮色素组合物中,由式(I)表示的偶氮色素化合物中,其一部分或全部也可以被离子化。同样地,在本公开的偶氮色素组合物中,由式(II)表示的脲化合物中,其一部分或全部也可以被离子化。
相对于偶氮色素组合物的总量,本公开的偶氮色素组合物中的由式(I)表示的偶氮色素化合物和由式(II)表示的脲化合物的总含量为优选95质量%以上,更优选98质量%以上,尤其优选99质量%以上。
若上述总含量为95质量%以上,则能够获得更高纯度的偶氮色素化合物。
上述总含量的上限理想的是100质量%,但作为上限也可以举出99.9质量%。
并且,在本公开的偶氮色素组合物中,由式(II)表示的脲化合物相对于由式(I)表示的偶氮色素化合物的含有质量比(含有质量比〔由式(II)表示的脲化合物/由式(I)表示的偶氮色素化合物〕)优选0.5摩尔倍~1.5摩尔倍,更优选0.7摩尔倍~1.3摩尔倍,进一步优选0.8摩尔倍~1.2摩尔倍,进一步优选0.9摩尔倍~1.1摩尔倍。
并且,从进一步提高由偶氮色素组合物获得的偶氮色素化合物的纯度的观点考虑,本发明的偶氮色素组合物中的式(I)及式(II)中的各基团的优选的组合为以下组合。
上述优选组合为如下,
R11、R12、R13及R14分别独立地为氢原子、卤素原子或碳原子数为1~10的烷基(优选碳原子数为1~6的烷基,更优选碳原子数为1~3的烷基),且R11、R12、R13及R14中的至少1个(优选R11及R13中的至少1个)为卤素原子,
R15是碳原子数为1~10的烷基(优选碳原子数为1~6的烷基,更优选碳原子数为1~3的烷基),
R21及R22分别独立地为碳原子数为1~10的烷基(优选碳原子数为1~6的烷基,更优选碳原子数为1~3的烷基)的组合。
以下,示出本公开的偶氮色素组合物中的、式(I)中的各基团及式(II)中的各基团的组合的具体例(组合物1~40),但本发明并不限定于以下具体例。
[化学式9]
作为由本公开的偶氮色素组合物获得(分离)偶氮色素化合物的方法,可以使用通常的方法,但例如可以举出对上述偶氮色素组合物进行酸处理的方法、使上述偶氮色素组合物通过离子交换膜树脂的方法等。
作为使用于酸处理的酸,可以举出盐酸、硫酸、磷酸等无机酸,甲磺酸、三氟乙酸等有机酸。
并且,在酸处理中使用酸水溶液的情况下,酸水溶液中的酸的浓度优选1质量%~10质量%,更优选3质量%~6质量%。
作为上述酸处理的具体的方法可以举出:在含有固体偶氮色素组合物的液体中添加酸的方法;在含有酸的溶液中添加固体偶氮色素组合物的方法;及用含有酸的溶液清洗固体偶氮色素组合物的方法等。
酸处理也可以在加热条件下(例如,在液温为10℃~80℃的条件下,优选在液温为30℃~60℃的条件下)进行。
在上述酸处理之后,通过过滤等,从溶液中分离偶氮色素化合物,并干燥分离出的偶氮色素化合物,由此能够获得纯度高的偶氮色素化合物。
<偶氮色素组合物的制造方法>
本公开的偶氮色素组合物的制造方法(以下,也称作“本公开的制造方法”)为制造上述本公开的偶氮色素组合物的方法,该方法具有:准备工序,准备由式(I)表示的偶氮色素化合物的产物粗品;及析出工序,使产物粗品和由式(II)表示的脲化合物即第1溶剂在不同于第1溶剂的第2溶剂中接触,从而析出上述偶氮色素组合物。
根据本公开的偶氮色素组合物的制造方法,能够制造可以获得高纯度的偶氮色素化合物的偶氮色素组合物。
虽然发挥该效果的作用机理不明确,但可以如下进行推测。
即,可以认为在上述析出工序中包含于产物粗品中的杂质溶解于第2溶剂,且产物粗品中的偶氮色素化合物与作为第1溶剂的脲化合物相互作用,从而析出偶氮色素组合物。因此可以认为在所析出的偶氮色素组合物中含有杂质的量得到减少且纯度高的偶氮色素化合物。
而且,可以认为通过对偶氮色素组合物实施酸处理等,能够从偶氮色素组合物分离纯度高的上述偶氮色素化合物。
作为上述相互作用,可以举出脲化合物在偶氮色素化合物中进行溶剂化的作用、形成偶氮色素化合物和脲化合物的盐的作用等。
(准备工序)
准备工序为准备由式(I)表示的偶氮色素化合物的产物粗品的工序。
准备工序并不限定于只准备预先被合成的产物粗品的工序,也可以是合成产物粗品的工序。
作为合成产物粗品的方法,例如可以举出日本专利第4080947号公报中所记载的公知的方法。
(析出工序)
析出工序为通过使偶氮色素化合物的产物粗品和脲化合物即第1溶剂在不同于第1溶剂的第2溶剂中接触而析出偶氮色素组合物的工序。
作为偶氮色素化合物的产物粗品与脲化合物即第1溶剂接触的具体的方式,可以举出以下3种方式。
(方式1)是首先将产物粗品添加到第2溶剂中而制作溶液,接着,在该溶液中添加脲化合物(第1溶剂)的方式。
(方式2)是首先将脲化合物(第1溶剂)添加到第2溶剂中而制作溶液,接着,在该溶液中添加产物粗品的方式。
(方式3)是对第2溶剂同时添加产物粗品及脲化合物(第1溶剂)的方式。
上述3种方式中,从更有效地析出偶氮色素组合物的观点考虑,优选方式1。
并且,在析出工序中,脲化合物(第1溶剂)相对于产物粗品的质量比〔脲化合物(第1溶剂)/产物粗品〕优选0.5~10.0,更优选0.8~5.0,尤其优选1.0~3.0。
并且,在析出工序中,产物粗品及脲化合物(第1溶剂)的合计相对于第2溶剂的浓度〔(产物粗品+脲化合物(第1溶剂))/第2溶剂〕优选50g/L~700g/L,更优选100g/L~500g/L。
并且,析出工序中的产物粗品与脲化合物(第1溶剂)的接触时间优选3分钟~3小时,更优选10分钟~2小时,尤其优选15分钟~1小时。
并且,在析出工序中,包含第1溶剂、第2溶剂及产物粗品的混合溶液的液温优选30℃~80℃,更优选40℃~70℃,尤其优选40℃~60℃。
第2溶剂为不同于第1溶剂(即,脲化合物)的溶剂,其中,优选为溶解在偶氮色素化合物的合成时混入的杂质且析出偶氮色素组合物的溶剂。
作为第2溶剂,具体而言,可以举出:甲苯、二甲苯、均三甲苯、乙基苯等芳香族烃类溶剂;乙酸乙酯、乙酸丙酯、乙酸丁酯、丙酸乙酯等酯类溶剂;丙酮、甲乙酮、甲基异丁基酮等酮类溶剂;乙腈、丙腈等腈类溶剂;二异丙基醚、甲基叔丁基醚、四氢呋喃、二噁烷等醚类溶剂;己烷、庚烷、辛烷、环己烷等脂肪族烃类溶剂;乙醇、甲醇、异丙醇、乙二醇等醇类溶剂等。
作为第2溶剂,优选芳香族烃类溶剂、酯类溶剂、酮类溶剂、腈类溶剂或醚类溶剂,更优选芳香族烃类溶剂(优选甲苯)、酯类溶剂(优选乙酸乙酯)或酮类溶剂(优选丙酮),尤其优选芳香族烃类溶剂(优选甲苯)或酯类溶剂(优选乙酸乙酯)。
上述本公开的偶氮色素组合物的制造方法,作为偶氮色素化合物的提纯处理的一部分而优选。
即,在通过上述制造方法制造偶氮色素组合物之后,实施所述酸处理等(另外,根据需要,过滤、干燥等处理),从而可以获得纯度高的偶氮色素化合物(提纯物)。
实施例
以下,通过实施例对本发明更具体地进行说明,但本发明并不限定于以下实施例。
以下,只要无特别说明,则“%”是指“质量%”。
并且,“HPLC”是指高效液相色谱法(High performance liquid chromatogra phy)。
并且,化合物的纯度(%)是指通过HPLC而测定出的HPLC面积比(%)(HPLC area%)。
〔实施例1〕
<偶氮色素组合的制作>
制作出含有偶氮色素化合物即例示化合物(I-4)和脲化合物即例示化合物(II-3)的偶氮色素组合物(所述组合物24)。
以下示出详细内容。
[化学式10]
首先,合成了上述例示化合物(I-4)的产物粗品(纯度97.5%)。例示化合物(I-4)为橙色偶氮色素化合物。
上述产物粗品的合成是参考日本专利第4080947号公报的0174~0176段落进行的。
详细而言,将5-氨基-3-甲硫基-1,2,4-噻二唑(12.0mmol)添加到磷酸(50mL)中,将所得到的混合物在冰水中进行了冷却并搅拌。将亚硝酸钠晶体(1.0g)添加到上述混合物中,并搅拌1小时,由此制备出重氮溶液。将2-氯苯酚(10.0mmol)添加到甲醇(60ml)中,将所得到的混合物在冰水中进行冷却并搅拌之后,缓慢地添加上述所制备的重氮溶液。将所得到的溶液在冷却条件下搅拌1小时,进而,在室温下搅拌1小时,在所得到的反应液中添加水(150ml)及乙酸乙酯(80ml),提取有机层。然后,用饱和盐水(60ml)将有机层清洗2次,将清洗过的有机层在减压下进行浓缩,接着,用硅胶柱层析法进行提纯,得到了例示化合物(I-4)的产物粗品(纯度97.5%)。
使上述所得到的例示化合物(I-4)的产物粗品5.0g(纯度97.5%)悬浮于作为第2溶剂的甲苯40mL中,接着,将液温升温至50℃,由此使上述产物粗品溶解而得到了溶液。
在上述溶液的液温维持在50℃的状态下,对上述溶液,经5分钟滴加DMPU(N,N’-二甲基丙烯基脲;作为第1溶剂的例示化合物(II-3))9.0g,加热搅拌30分钟的结果,产生了紫红色的析出物。接着,将包含上述析出物的反应液冷却至液温成为25℃。
接着,从上述冷却后的反应液滤取析出物,并干燥所滤取的析出物,由此得到了紫红色固体5.8g(产率80%)。
通过NMR、气相色谱分析及X射线结构分析对所得到的固体进行分析的结果,确认到该固体为包含例示化合物(I-4)和例示化合物(II-3)的偶氮色素组合物(所述组合物24)。
并且,确认到例示化合物(I-4)和例示化合物(II-3)的总含量相对于上述固体(偶氮色素组合物)的总量为95质量%以上。
并且,确认到在上述固体(偶氮色素组合物)中,例示化合物(II-3)相对于例示化合物(I-4)的合计质量比〔例示化合物(II-3)/例示化合物(I-4)〕为1.0摩尔倍。
以下示出偶氮色素组合物(组合物24)的NMR测定结果。
-偶氮色素组合物(组合物24)的NMR测定结果-
1HNMR(400MHz,DMSO-d6):δ=8.04(s,1H),7.93(dd,1H),7.22(d,1H),3.19(t,4H),2.76(s,6H),2.52(S,3H)2.51(s,1H),1.88(m,2H)
图1A是通过X射线结构分析而求出的偶氮色素组合物(组合物24)的结构的概念图,图1B是图1A中示出的偶氮色素组合物(组合物24)的c轴投影图。
<偶氮色素化合物的纯度的测定>
通过对上述紫红色固体(组合物24)进行酸处理而得到了例示化合物(I-4)的提纯物。以下,示出详细的内容。
在三口烧瓶中放入50g的组合物24、250mL的乙腈及300mL的丙酮,在室温下进行搅拌而使它们悬浮。对所得到的悬浮液,在室温下,添加25.2g的三乙胺而使其溶解,将内部温度升温至50℃而得到了溶液。
另外,将浓盐酸30.6g及水315mL进行混合,制备出盐酸浓度为3.3质量%的稀盐酸。
对上述升温至50℃的溶液,在50℃下,经1小时滴加所得到的稀盐酸。将所得到的混合物搅拌30分钟之后,将内部温度冷却至20℃。从冷却后的混合物滤取析出物,用水100mL及甲醇100mL对所滤取的析出物进行了清洗。将清洗后的析出物在50℃下经24小时进行干燥,得到了例示化合物(I-4)的提纯物27g(产率78%)。
接着,通过下述测定条件下的HPLC而测定出上述提纯物中的例示化合物(I-4)的纯度。其结果,确认到上述提纯物中的例示化合物(I-4)的纯度为99.5%,相对于原料即产物粗品中的例示化合物(I-4)的纯度(97.5%)提高。
-HPLC测定条件-
A溶液:超纯水、0.1%三乙胺、0.1%乙酸
B溶液:乙腈、0.1%三乙胺、0.1%乙酸
梯度条件:0分钟(体积比〔A溶液/B溶液〕=70/30)→60分钟(体积比〔A溶液/B溶液〕=10/90)
色谱柱:TOSOH CORPORATION制TSKgel(注册商标)ODS-80Ts、色谱柱尺寸4.6mm×150mm
流速:1mL/min
色谱柱温度:40℃
检测波长:254nm
装置:Shimadzu Corporation制造的HPLC装置(装置名称:LC2010)
〔比较例1〕
使实施例1中所合成的例示化合物(I-4)的产物粗品5.0g(纯度97.5%)悬浮于甲苯40mL中,接着,将液温升温至50℃,由此溶解产物粗品而得到了溶液。
将上述溶液在液温50℃下搅拌1小时,接着,冷却至液温25℃的结果,产生了橙色析出物。滤取析出物,并干燥所滤取的析出物,由此得到了橙色固体4.7g。
对上述橙色固体,通过与实施例1相同条件的HPLC而测定出上述橙色固体中的例示化合物(I-4)的纯度。其结果,确认到上述橙色固体中的例示化合物(I-4)的纯度为97.6%,相对于原料即产物粗品中的例示化合物(I-4)的纯度(97.5%)几乎没有变化。
〔比较例2〕
在比较例1中将甲苯变更为THF(四氢呋喃),除此以外,进行与比较例1相同的操作,得到了橙色固体4.4g。
对上述橙色固体,通过与实施例1相同条件的HPLC而测定出上述橙色固体中的例示化合物(I-4)的纯度。其结果,确认到橙色固体中的例示化合物(I-4)的纯度为98.1%,相对于原料即产物粗品中的例示化合物(I-4)的纯度(97.5%)几乎没有变化。
〔比较例3〕
使实施例1中所合成的例示化合物(I-4)的产物粗品5.0g(纯度97.5%)悬浮于甲苯40mL中,接着,将液温升温至50℃,由此溶解上述产物粗品而得到了溶液。
在将上述溶液的液温维持在50℃的状态下,对上述溶液添加三乙胺7.1g的结果,未确认到析出物。
将液温维持在50℃的状态下,在添加三乙胺之后的溶液中滴加5%盐酸30mL的结果,产生了橙色析出物。接着,将包含析出物的反应液冷却至液温成为25℃。接着,滤取析出物并干燥所滤取的析出物,由此得到了橙色固体4.8g。
对上述橙色固体进行与实施例1相同条件下的HPLC,由此测定出上述橙色固体中的例示化合物(I-4)的纯度。其结果,确认到橙色固体中的例示化合物(I-4)的纯度为97.5%,相对于原料即产物粗品中的例示化合物(I-4)的纯度(97.5%)没有变化。
〔比较例4〕
在比较例3中将三乙胺变更为N,N-二甲基乙酰胺,除此以外,进行与比较例3相同的操作,得到了橙色固体4.5g。
对所得到的橙色固体,以与比较例3相同的方式进行HPLC,由此测定出上述橙色固体中的例示化合物(I-4)的纯度。其结果,确认到橙色组合物中的例示化合物(I-4)的纯度为98.3%,相对于原料即产物粗品中的例示化合物(I-4)的纯度(97.5%)几乎没有变化。
〔比较例5~10〕
在比较例3中将三乙胺变更为吡啶(比较例5)、N,N-二乙基苯胺(比较例6)、N,N-二甲基甲酰胺(比较例7)、N-甲基-2-吡咯烷酮(比较例8)、N-乙基-2-吡咯烷酮(比较例9)或四甲基脲(比较例10),除此以外,进行了与比较例3相同的操作。
其结果,在比较例5~10中均确认到滤取及干燥后固体中的例示化合物(I-4)的纯度相对于原料即产物粗品中的例示化合物(I-4)的纯度几乎没有变化。
2014年9月2日申请的日本专利申请2014-178029公开的全部内容,通过参考而援用于本说明书中。
在本说明书中记载的所有文献、专利申请及技术标准中,通过参考而援用各文献、专利申请及技术标准的情况与通过具体且分别记载的情况程度相同地,通过参考而援用于本说明书中。
- 还没有人留言评论。精彩留言会获得点赞!