一种纯α相碳化钼负载贵金属催化剂及其制备方法和应用与流程
本发明涉及碳化钼负载贵金属催化剂的制备及其应用,具体涉及一种纯α相碳化钼负载贵金属催化剂及其制备方法,以及其在水汽变换反应中的应用。
背景技术:
发明人所在课题组研究发现,在水汽变换反应中纯α相碳化钼催化剂(记为α-MoC1-x)的催化活性远远高于纯β相碳化钼催化剂(记为β-Mo2C);纯α相的金碳化钼催化剂(记为Au/α-MoC1-x)的催化活性高于α和β相混相金碳化钼催化剂(记为Au/MoCx)的催化活性,远高于纯β相的金碳化钼催化剂(记为Au/β-Mo2C)的催化活性。α-MoC1-x的合成目前为止只能通过氮化-碳化两步法才能实现,首先用氨气氮化MoO3得到γ-Mo2N,再以甲烷碳化γ-Mo2N将其转变为α-MoC1-x。这种合成方法的特点在于,碳化钼以与氮化钼呈相同晶型的形式生成,两者同为面心立方(fcc)晶型。该方法具有下述缺点:①碳化过程中,氮化钼的晶格氮与碳原子的交换需要在较高温度(700℃以上)进行,高温会加剧自由碳在颗粒表面聚合并生成积碳,大大降低了碳化钼的比表面积和表面催化活性位的暴露;②操作过程繁琐,制备周期长;③需要使用强烈刺激性气体NH3,不仅腐蚀设备也易发生危险。为克服上述氮化-碳化两步法合成α-MoC1-x及负载金属α-MoC1-x的缺陷,发明人所在课题组进行了一步碳化法合成负载金属α-MoC1-x催化剂的相关研究。结果表明,负载金属Ni、Co、Fe等到氧化钼前驱体中,得到碳化钼的晶型为β型;将一定量Cu、Pt、Au等金属引入前驱体后经碳化获得碳化钼的晶型为α型和β型的混晶。例如,发明人已公开的专利申请“一种金属改性的α型碳化钼催化剂的制备方法与其在低温水煤气变换反应中的应用”(专利公开号:CN102698783A)采用一步碳化法合成金或铜负载的α型碳化钼催化剂,从给出的XRD谱图中可以看出所获得碳化钼的晶型为α型和β型的混晶。迄今为止,一步碳化法(即无需先氮化仅经碳化过程)合成纯α相碳化钼负载金属催化剂未见报道。
技术实现要素:
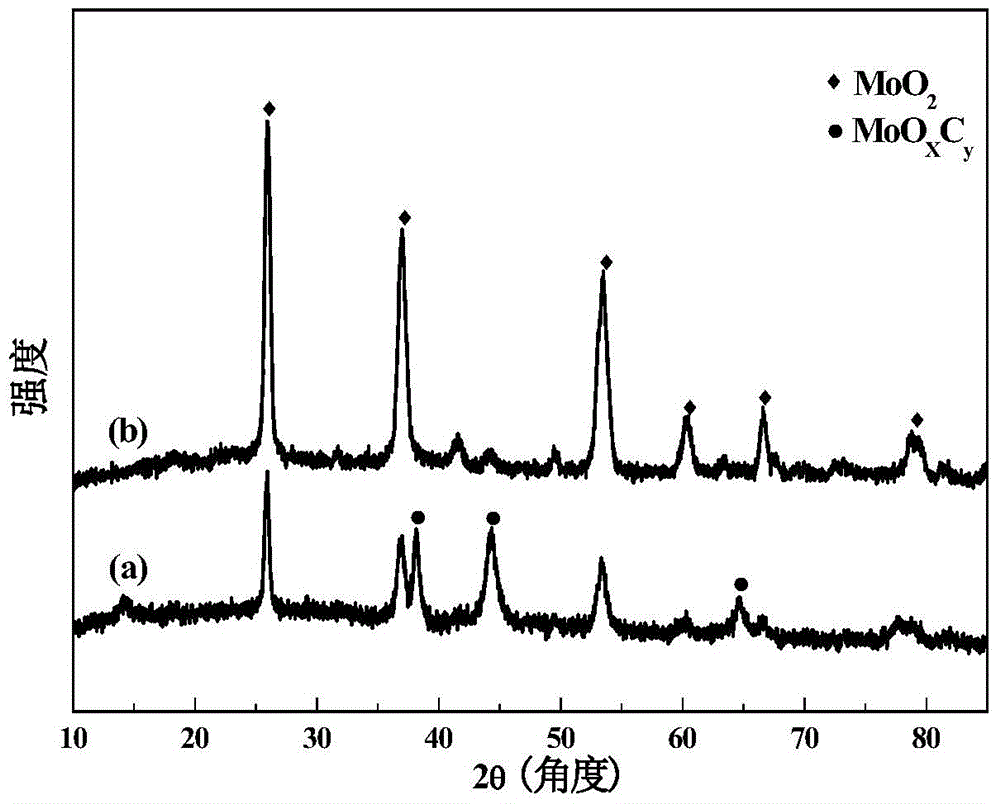
本发明解决的技术问题之一是提供一步碳化法合成纯α相碳化钼负载贵金属催化剂的制备方法。一种纯α相碳化钼负载贵金属催化剂的制备方法,所述制备方法包括如下步骤:(1)将贵金属盐溶液与仲钼酸铵水溶液混合,搅拌,经过滤、水洗后,沉淀物经干燥得到前驱体,前驱体中负载贵金属的质量百分比为0.5%-7%;(2)将步骤(1)所得前驱体进行非平衡等离子体处理,得到负载贵金属氧化钼;(3)将步骤(2)所得负载贵金属氧化钼在碳源气体下于580-800℃碳化。本发明所述制备方法利用非平衡等离子体处理过程代替传统的焙烧过程,经处理得到的负载贵金属氧化钼可经一步碳化过程直接获得纯α相碳化钼负载贵金属催化剂,省去了高污染的氮化过程。采用非平衡等离子体处理得到的负载贵金属氧化钼具有颗粒更小、金属分散性好、还原插碳过程在更低温度下发生的特点,从而更易形成MoOxCy中间物种,而不是MoO2中间物种(见附图1和2)。MoOxCy进一步碳化可形成具有面心立方结构的α-MoC1-x。同时,在碳化过程中,活性金属与碳化钼间的相互作用逐渐增强,活性金属进一步分散和锚定,最终形成晶型完整的α-MoC1-x负载贵金属催化剂。而未经等离子体处理的负载贵金属氧化钼经相同一步碳化过程后形成的是α型和β型碳化钼混合相负载贵金属催化剂(见对比例的结果)。优选地,所述贵金属盐为金或铂或钯所形成盐中的任何一种。优选地,所述步骤(2)中非平衡等离子体处理的具体过程为,将步骤(1)所得前驱体放入非平衡等离子体反应器中,在氧气气氛或氧气与氩气混合气氛下,室温在两电极上施加频率为2-6kHz,输入功率为5-10W的正弦交流电压,大气压下处理30-60min。优选地,所述非平衡等离子体的放电形式为介质阻挡放电。优选地,所述步骤(3)中的碳源气体为甲烷与氢气的混合气。进一步优选地,甲烷与氢气的混合气中甲烷的体积分数为20%。优选地,所述步骤(3)中的碳化温度为590-700℃。本发明解决的技术问题之二是提供利用上述制备方法制备的纯α相碳化钼负载贵金属催化剂。本发明解决的技术问题之三是提供上述纯α相碳化钼负载贵金属催化剂在水汽变换反应中的应用。所述纯α相碳化钼负载贵金属催化剂在进行水汽变换反应前的预处理过程如下,以CH4与H2混合气对催化剂进行预处理,混合气中CH4的体积分数为10-20%,预处理温度为500-700℃,时间为1-3小时。优选地,所述水汽变换反应的条件为,质量空速90000-100000ml/g/h,反应气氛中H2的体积分数20-50%,反应温度120-200℃。本发明中的纯α相碳化钼负载贵金属催化剂对水汽变换反应具有优异的催化性能;在上述富氢气氛、高空速、低温下仍具有优异的催化性能,实验证明,在9%CO/30%H2O/39%H2/6%CO2/16%N2的富氢反应气氛中,90000ml/g/h空速下,0.5%Au/α-MoC1-x催化剂在150℃时,单位Au上的反应速率为2.30molco/molAu/s。本发明的有益效果如下:(1)本发明利用非平衡等离子体处理过程代替传统的焙烧过程,经处理得到的负载贵金属氧化钼可经一步碳化过程直接获得纯α相碳化钼负载贵金属催化剂,省去了高污染的氮化过程。(2)本发明方法制备的催化剂具有优异的水汽变换性能,实验证明,在9%CO/30%H2O/39%H2/6%CO2/16%N2的富氢反应气氛中,90000ml/g/h空速下,0.5%Au/α-MoC1-x催化剂在150℃时,单位Au上的反应速率为2.30molco/molAu/s。与目前文献报道的结果相比,本发明中0.5%Au/α-MoC1-x催化剂在150℃具有最高的TOF(见表1)。表1本发明制得的0.5%Au/α-MoC1-x与文献中已有催化剂在水汽变换反应中的反应速率对比结果a2%CO/10%H2O/He;b11%CO/21%H2O/43%H2/6%CO2/19%N2;c10.5%CO/21%H2O/He附图说明本发明附图8幅,图1为实施例2所得3%Au/MoO3-plasma的电镜照片;图2为实施例1所得0.5%Au/MoO3-plasma(a)与对比例1所得0.5%Au/MoO3-500℃(b)在碳化过程中所形成的中间物体的XRD对比图;图3为实施例2所得3%Au/α-MoC1-x的电镜照片;图4为实施例1所得0.5%Au/α-MoC1-x(a)和对比例1所得0.5%Au/MoCx(b)的XRD对比图;图5为实施例5所得1%Pt/α-MoC1-x(a)和对比例2所得1%Pt/MoCx(b)的XRD对比图;图6为实施例6所得1%Pd/α-MoC1-x(a)和对比例3所得1%Pd/MoCx(b)的XRD对比图;图7为实施例1所得0.5%Au/α-MoC1-x(a)和对比例1所得0.5%Au/MoCx(b)与商用催化剂Cu-Zn-Al催化水汽变换反应的活性对比图;图8为实施例5所得1%Pt/α-MoC1-x(a)和对比例2所得1%Pt/MoCx(b)与商用催化剂Cu-Zn-Al催化水汽变换反应的活性对比图。具体实施方式下述非限定性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。实施例1(1)将6.92g仲钼酸铵溶于40ml去离子水中,室温下磁力搅拌,使其完全溶解;然后将5.93ml浓度为0.02428mmol/ml的氯金酸溶液逐滴加入仲钼酸铵溶液中,室温避光磁力搅拌4-5小时,过滤得到白色沉淀物,水洗后于80℃水浴蒸干,110℃干燥过夜,得到负载质量百分比0.5%Au的前驱体;(2)将负载质量百分比0.5%Au的前驱体置于介质阻挡放电(DBD)非平衡等离子体反应器中,以氧气(100ml/min)为工作气体,采用非平衡等离子体进行处理,两电极上施加6000v的交流电压,处理时间为30min,获得负载质量百分比0.5%Au的氧化钼(记为0.5%Au/MoO3-plasma);(3)取1.2g0.5%Au/MoO3-plasma样品置于石英反应器中,在CH4与H2的混合气氛(CH4的体积分数为20%)中进行程序升温碳化,先由室温升至300℃,升温速率为5℃/min,再由300℃缓慢升至700℃,升温速率为1℃/min,在700℃下恒温2h,再迅速冷却至室温,得到负载质量百分比0.5%Au的纯α相碳化钼催化剂(记为0.5%Au/α-MoC1-x)。(4)水汽变换反应在内径6mm的石英管固定床反应器中进行,实验所需各路气体流量均有质量流量计调节和控制,混合后流入反应器。称取200mg的0.5%Au/α-MoC1-x置于石英管中,以CH4的体积分数为15%的CH4与H2混合气对催化剂于590℃进行预处理2h,然后于下述条件进行活性评价,反应气氛为:按体积分数计,9%CO/30%H2O/39%H2/6%CO2/16%N2,气体质量空速为90000ml/g/h,反应温度为150-400℃。150℃时,CO的转化率为58%,单位Au上的反应速率为2.30molco/molAu/s。实施例2本实施例的步骤及工艺条件等与实施例1均相同,区别仅在于下述两点,①称取36.5ml氯金酸溶液制备得到负载质量百分比3%Au的纯α相碳化钼催化剂(记为3%Au/α-MoC1-x);②对其进行活性评价,反应气氛为:按体积分数计,11%CO/26%H2O/26%H2/7%CO2/30%N2,气体质量空速为90000ml/g/h,反应温度为120-400℃。150℃时,CO的转化率为82%。实施例3本实施例的步骤及工艺条件等与实施例1均相同,区别仅在于下述两点,①称取62.2ml氯金酸溶液制备得到负载质量百分比5%Au的纯α相碳化钼催化剂(记为5%Au/α-MoC1-x);②对其进行活性评价,反应气氛为:按体积分数计,11%CO/26%H2O/26%H2/7%CO2/30%N2,气体质量空速为90000ml/g/h,反应温度为120-400℃。150℃时,CO的转化率为86%。实施例4本实施例的步骤及工艺条件等与实施例1均相同,区别仅在于下述两点,①称取88.9ml氯金酸溶液制备得到负载质量百分比7%Au的纯α相碳化钼催化剂(记为7%Au/α-MoC1-x);②对其进行活性评价,反应气氛为:按体积分数计,11%CO/26%H2O/26%H2/7%CO2/30%N2,气体质量空速为90000ml/g/h,反应温度为120-400℃。150℃时,CO的转化率为87%。实施例5本实施例的步骤及工艺条件等与实施例1均相同,区别仅在于下述两点,①将氯金酸改为氯铂酸,称取9.47ml浓度为0.0386mol/l的氯铂酸溶液制备得到负载质量百分比1%Pt的纯α相碳化钼催化剂(记为1%Pt/α-MoC1-x),②对其进行活性评价,反应气氛为:按体积分数计,11%CO/26%H2O/26%H2/7%CO2/30%N2,气体质量空速为90000ml/g/h,反应温度为150-400℃。200℃时,CO的转化率为80%。实施例6本实施例的步骤及工艺条件等与实施例1均相同,区别仅在于下述两点,①将氯金酸改为硝酸钯,称取0.207g硝酸钯溶于40ml去离子水中配成溶液制备得到负载质量百分比1%Pd的纯α相碳化钼催化剂(记为1%Pd/α-MoC1-x);②对其进行活性评价,反应气氛为:按体积分数计,11%CO/26%H2O/26%H2/7%CO2/30%N2,气体质量空速为90000ml/g/h,反应温度为150-400℃。200℃时,CO的转化率为83%。对比例1(1)将6.92g仲钼酸铵溶于40ml去离子水中,室温下磁力搅拌,使其完全溶解;然后将5.93ml浓度为0.02428mmol/ml的氯金酸溶液逐滴加入仲钼酸铵溶液中,室温避光磁力搅拌4-5小时,过滤得到白色沉淀物,水洗后于80℃水浴蒸干,110℃干燥过夜;500℃空气气氛下焙烧4小时,得到负载质量百分比0.5%Au的氧化钼(记为0.5%Au/MoO3-500℃);(2)取1.2g0.5%Au/MoO3-500℃样品置于石英反应器中,在CH4与H2的混合气氛(CH4的体积分数为20%)中进行程序升温碳化,先由室温升至300℃,升温速率为5℃/min,再由300℃缓慢升至700℃,升温速率为1℃/min,在700℃下恒温2h,再迅速冷却至室温,得到传统方法制备的负载质量百分比0.5%Au的α和β相混相碳化钼催化剂(记为0.5%Au/MoCx)。(3)水汽变换反应在内径6mm的石英管固定床反应器中进行,实验所需各路气体流量均有质量流量计调节和控制,混合后流入反应器。称取200mg的0.5%Au/MoCx催化剂置于石英管中,以CH4的体积分数为15%的CH4与H2混合气对催化剂于590℃进行预处理2h,然后于下述条件进行活性评价,反应气氛为:按体积分数计,9%CO/30%H2O/39%H2/6%CO2/16%N2,气体空速为90000ml/g/h,反应温度为150-400℃。150℃时,CO的转化率为18%。对比例2本对比例的步骤及工艺条件等与对比例1均相同,区别仅在于下述两点,①称取9.47ml浓度为0.0386mol/l的氯铂酸溶液制备得到负载质量百分比1%Pt的α和β相混相碳化钼催化剂(记为1%Pt/MoCx);②对其进行活性评价,反应气氛为:按体积分数计,11%CO/26%H2O/26%H2/7%CO2/30%N2,气体空速为90000ml/g/h,反应温度为150-400℃。200℃时,CO的转化率为65%。对比例3本对比例的步骤及工艺条件等与对比例1均相同,区别仅在于下述两点,①称取0.207g硝酸钯于40ml去离子水中配成溶液制备得到负载质量百分比1%Pd的α和β相混相碳化钼催化剂(记为1%Pd/MoCx);②对其进行活性评价,反应气氛为:按体积分数计,11%CO/26%H2O/26%H2/7%CO2/30%N2,气体空速为90000ml/g/h,反应温度为150-400℃。200℃时,CO的转化率为62%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1