一种抗硫抗水的低温脱硝复合分子筛催化剂及其制备方法与流程
本发明涉及一种抗硫抗水的低温脱硝复合分子筛催化剂及其制备方法,属于大气污染治理技术和环保催化材料领域。
背景技术:
我国的能源结构决定着我国是一个燃煤大国,随着我国的国民经济飞速发展,每年向大气中排放的大量氮氧化物,致使许多地区出现了酸雨、雾霾、微小颗粒污染等,对人体、环境、生态的危害以及对社会经济的破坏都十分巨大。近年来,我国氮氧化物排放快速增长,据环保部门公布的数据显示,2000年我国的氮氧化物排放总量为1210万吨,而2012年则达到了2338万吨,几乎翻了一番。据预测,如果不采取进一步控制措施,2030年我国氮氧化物排放将超过3500万吨。因此大力发展氮氧化物排放控制技术势在必行。
目前在烟气脱硝领域中,通常采用以nh3为还原剂的选择性催化还原(scr)工艺,商业用的催化剂主要是v2o5-wo3/tio2系列,该催化剂的操作温度为300℃~420℃,且价格昂贵。为了满足温度的需要,一般将催化床层布置与除尘器之前,但是这种布置方法一方面会引起催化剂的so2中毒及粉尘堵塞,另一方面需要较大的炉后空间;二是活性组分中的钒有毒性,对生态环境及身体健康都不利;此外,像钢铁厂烧结机和球团等设备排放的烟气温度均在200℃以下,不在此类中高温scr催化剂的活性温度窗口范围,因此,大力发展高抗硫性能的低温scr技术意义重大。
技术实现要素:
技术问题:本发明的目的是提供一种抗硫抗水的低温脱硝复合分子筛催化剂及其制备方法,该催化剂通过活性组分cu、mn及助剂ce、fe、co等金属的协同效应,显著的拓宽了催化剂脱硝反应温度温度,并且提高了催化剂的脱硝效率和抗硫抗水性能,同时该制备方法操作简单成本低、重复性高、制备的催化剂性能稳定。
技术内容:本发明提供了一种抗硫抗水的低温脱硝复合分子筛催化剂,该催化剂包含载体、活性组分和助剂,所述载体为菱沸石分子筛,所述活性组分为铜锰复合金属氧化物,所述助剂为ce、fe、co、mo或者cr中的一种;以载体的质量为基准,活性组分中铜元素和锰元素的质量分别为2%~10%,助剂的质量为1~10%。
其中:
所述的菱沸石分子筛为菱沸石分子筛h-sapo-34。
本发明还提供了一种如权利要求1所述的抗硫抗水的低温脱硝复合分子筛催化剂的制备方法,该方法包括以下步骤:
1)将载体菱沸石分子筛原粉末干燥后备用;
2)按比例称取所述活性组分中铜的可溶性盐溶解在去离子水中,充分搅拌均匀后,按比例加入活性组分中锰的可溶性盐和所述助剂的可溶性盐,充分搅拌制成混合溶液;
3)取步骤1)中干燥后的菱沸石分子筛原粉末溶于步骤2)制得的混合溶液中,充分搅拌浸渍后,将溶液升温并继续搅拌浸渍,待水分蒸干后,依次经烘干、研磨、筛选、煅烧后得到所述的抗硫抗水的低温脱硝复合型分子筛催化剂(cu-mn-x/sapo-34,其中x为ce、fe、co、mo、cr中的一种)。
其中:
步骤1)所述的干燥的温度为100~105℃,时间为25~30min。
步骤2)所述的铜的可溶性盐为cu(no3)2·3h2o或cu(ch3coo)2·h2o,锰的可溶性盐为硝酸锰或mn(ch3coo)2·4h2o),ce的可溶性盐为ce(no3)3·6h2o或ce(ch3coo)3·5h2o,co的可溶性盐为co(no3)2·6h2o或co(ch3coo)2·4h2o),fe的可溶性盐为fe(no3)3·9h2o,mo的可溶性盐为mo(no3)3·5h2o,cr的可溶性盐为cr(no3)3·9h2o。
步骤3)所述的充分搅拌浸渍后,将溶液升温并继续搅拌浸渍具体是指在水浴温度为20~30℃条件下,利用磁力搅拌器不间断的搅拌6~8h,之后将水浴温度升至75~85℃并继续搅拌浸渍2~4h。
所述的磁力搅拌器的搅拌转速为40~50r/s。
步骤3)中所述的烘干是指在温度为80~100℃的烘箱中烘8~12h;步骤3)中所述的煅烧的温度为450~550℃,时间为4~6h;步骤3)中所述的筛选的过筛目数为40~80目。
有益效果:与现有技术相比,本发明的抗硫抗水的低温脱硝复合型分子筛催化剂相对于现有的单一的铜基或锰基催化剂,由于两种活性组分和助剂的协同效果,有效的降低了催化剂的脱硝反应温度,提高了催化剂的热稳定性,抗硫抗水性,同时拓宽了催化剂脱硝反应的温度窗口。所制备的催化剂在微型固定床中模拟烟气条件下进行脱硝性能测试,发现该催化剂具有较低的起活温度,较高的脱硝效率和较宽的温度窗口。
附图说明
图1为本发明实例中制备的复合型分子筛催化剂的脱硝性能测试图;
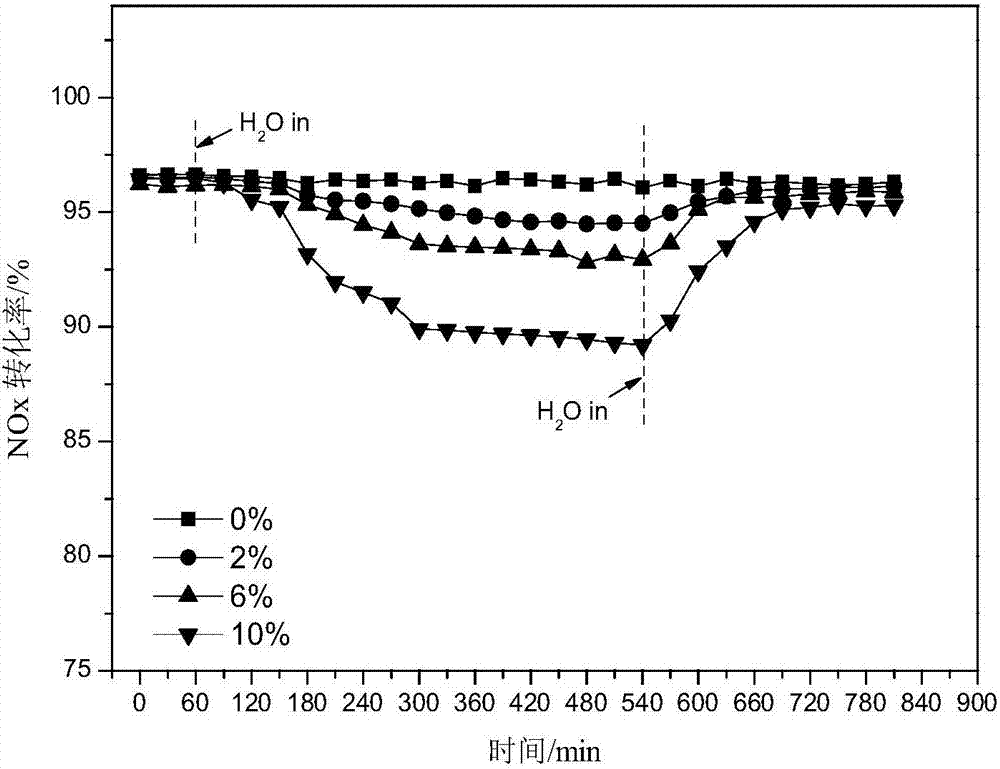
图2为本发明实例中制备的复合型分子筛催化剂的抗水性能测试图;
图3为本发明实例中制备的复合型分子筛催化剂的抗so2性能测试图;
图4为本发明实例中制备的复合型分子筛催化剂中毒前后的sem图;其中:a为未改性新鲜的催化剂的sem图、b为未改性的催化剂so2中毒后的sem图、c为2%ce改性催化剂so2中毒后的sem图、d为2%co改性催化剂so2中毒后的sem图、e为2%fe改性催化剂so2中毒后的sem图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。
实施例1:
(1)称取8g菱沸石分子筛(h-sapo-34)原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中100℃干燥25min备用;
(2)按比例称取0.3775gcu(no3)2·3h2o粉末,溶解在30ml的去离子水中,充分搅拌均匀后,加入2.7412g浓度为50%的硝酸锰溶液,充分搅拌制成含不同比例的两种活性组分的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在30℃的水浴、磁力搅拌器上不间断的搅拌8h,充分的浸渍后,将其升温至75℃并继续搅拌浸渍2h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在100℃下干燥8h,然后置于管式炉中,在450℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(2)-mn(6)/sapo-34(450)催化剂,该式表示以sapo-34为载体,cu的质量为载体的2%,mn的质量为载体的6%,煅烧温度为450℃。
实施例2:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中103℃干燥30min备用;
(2)按比例称取0.3775gcu(no3)2·3h2o粉末,溶解在35ml的去离子水中,充分搅拌均匀后,加入0.9091g浓度为50%的硝酸锰溶液和0.1549gce(no3)3·6h2o粉末,充分搅拌制成含不同比例的两种活性组分和含铈助剂的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在20℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至85℃并继续搅拌浸渍4h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在80℃下干燥12h,然后置于管式炉中,在500℃的高温有氧环境中煅烧6h;
(1)(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(2)-mn(2)-ce(1)/sapo-34(500)催化剂,该式表示以sapo-34为载体,cu的质量为载体的2%,mn的质量为载体的2%,ce的质量为载体的1%,煅烧温度为450℃。
实施例3:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中105℃干燥30min备用;
(2)按比例称取0.3775gcu(no3)2·3h2o粉末,溶解在40ml的去离子水中,充分搅拌均匀后,加入2.7412g浓度为50%的硝酸锰溶液和0.3099gce(no3)3·6h2o粉末,充分搅拌制成含不同比例的两种活性组分和含铈助剂的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在20℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至85℃并继续搅拌浸渍4h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在80℃下干燥12h,然后置于管式炉中,在550℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(2)-mn(6)-ce(2)/sapo-34(550)催化剂。
实施例4:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中105℃干燥30min备用;
(2)按比例称取1.8875gcu(no3)2·3h2o粉末,溶解在50ml的去离子水中,充分搅拌均匀后,加入4.5455g浓度为50%的硝酸锰溶液和1.5504gce(no3)3·6h2o粉末,充分搅拌制成含不同比例的两种活性组分和含铈助剂的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在20℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至85℃并继续搅拌浸渍4h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在80℃下干燥12h,然后置于管式炉中,在450℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(10)-mn(10)-ce(10)/sapo-34(450)催化剂。
实施例5:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中100℃干燥25min备用;
(2)按比例称取0.3775gcu(no3)2·3h2o粉末,溶解在30ml的去离子水中,充分搅拌均匀后,加入0.9091g浓度为50%的硝酸锰溶液和0.3607gfe(no3)3·9h2o,充分搅拌制成含不同比例的两种活性组分和含助剂fe的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在30℃的水浴、磁力搅拌器上不间断的搅拌8h,充分的浸渍后,将其升温至75℃并继续搅拌浸渍2h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在100℃下干燥8h,然后置于管式炉中,在450℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(2)-mn(2)-fe(1)/sapo-34(450)催化剂。
实施例6:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中105℃干燥25min备用;
(2)按比例称取1.8875gcu(no3)2·3h2o粉末,溶解在50ml的去离子水中,充分搅拌均匀后,加入4.5455g浓度为50%的硝酸锰溶液和3.6071gfe(no3)3·9h2o,充分搅拌制成含不同比例的两种活性组分和含助剂fe的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在30℃的水浴、磁力搅拌器上不间断的搅拌8h,充分的浸渍后,将其升温至75℃并继续搅拌浸渍2h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在100℃下干燥8h,然后置于管式炉中,在550℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(10)-mn(10)-fe(10)/sapo-34(550)催化剂。
实施例7:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中100℃干燥27min备用;
(2)按比例称取0.3775gcu(no3)2·3h2o粉末,溶解在30ml的去离子水中,充分搅拌均匀后,加入0.9091g浓度为50%的硝酸锰溶液和0.2469gco(no3)2·6h2o,充分搅拌制成含不同比例的两种活性组分和含助剂co的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在25℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至80℃并继续搅拌浸渍3h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在90℃下干燥10h,然后置于管式炉中,在450℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(2)-mn(2)-co(1)/sapo-34(450)催化剂。
实施例8:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中105℃干燥27min备用;
(2)按比例称取1.8875gcu(no3)2·3h2o粉末,溶解在50ml的去离子水中,充分搅拌均匀后,加入4.5455g浓度为50%的硝酸锰溶液和2.4661gco(no3)2·6h2o,充分搅拌制成含不同比例的两种活性组分和含助剂co的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在25℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至80℃并继续搅拌浸渍3h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在90℃下干燥10h,然后置于管式炉中,在550℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(10)-mn(10)-co(10)/sapo-34(550)催化剂。
实施例9:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中100℃干燥29min备用;
(2)按比例称取0.3775gcu(no3)2·3h2o粉末,溶解在30ml的去离子水中,充分搅拌均匀后,加入0.9091g浓度为50%的硝酸锰溶液和0.2287gmo(no3)2·5h2o,充分搅拌制成含不同比例的两种活性组分和含助剂mo的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在28℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至85℃并继续搅拌浸渍4h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在80℃下干燥12h,然后置于管式炉中,在450℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(2)-mn(2)-mo(1)/sapo-34(450)催化剂。
实施例10:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中105℃干燥29min备用;
(2)按比例称取1.8875gcu(no3)2·3h2o粉末,溶解在50ml的去离子水中,充分搅拌均匀后,加入4.5455g浓度为50%的硝酸锰溶液和2.2865gmo(no3)2·5h2o,充分搅拌制成含不同比例的两种活性组分和含助剂mo的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在28℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至85℃并继续搅拌浸渍4h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在80℃下干燥12h,然后置于管式炉中,在550℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(10)-mn(10)-mo(10)/sapo-34(550)催化剂。
实施例11:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中100℃干燥30min备用;
(2)按比例称取0.3775gcu(no3)2·3h2o粉末,溶解在30ml的去离子水中,充分搅拌均匀后,加入0.9091g浓度为50%的硝酸锰溶液和0.3847gcr(no3)3·9h2o,充分搅拌制成含不同比例的两种活性组分和含助剂cr的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在20℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至85℃并继续搅拌浸渍4h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在80℃下干燥12h,然后置于管式炉中,在450℃的高温有氧环境中煅烧6h;
(4)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(2)-mn(2)-cr(1)/sapo-34(450)催化剂。
实施例12:
(1)称取8g菱沸石分子筛h-sapo-34原粉末,将其放入刚玉材质的方舟中,一起置于鼓风式干燥箱中105℃干燥30min备用;
(2)按比例称取1.8875gcu(no3)2·3h2o粉末,溶解在50ml的去离子水中,充分搅拌均匀后,加入4.5455g浓度为50%的硝酸锰溶液和3.8465gcr(no3)3·9h2o,充分搅拌制成含不同比例的两种活性组分和含助剂cr的混合溶液。
(3)取步骤(1)中干燥后的h-sapo-34分子筛原粉末5g溶于步骤(2)制得的混合溶液,在20℃的水浴、磁力搅拌器上不间断的搅拌6h,充分的浸渍后,将其升温至85℃并继续搅拌浸渍4h,待水分蒸干后,将蒸发结晶的产物放入烘箱中在80℃下干燥12h,然后置于管式炉中,在550℃的高温有氧环境中煅烧6h;
(5)将煅烧后的沉淀物研磨筛选至粒径为40~80目,即得到了cu(10)-mn(10)-cr(10)/sapo-34(550)催化剂。
实施例13:催化剂活性测定及抗硫抗水性能测试
分别取上述实施例所得催化剂压片筛分,取40~60目的催化剂颗粒装入固定床反应器进行催化剂活性测试。活性检测条件如下:反应系统温度为90℃~350℃,反应压力为常压,原料气空速为15000h-1,原料气体积含量:no:350×10-6,nh3:350×10-6,o2:3%,载气:n2,气体总流量为500ml/min,各路气体经过质量流量计后逐步混合最后进入空气预混器充分混合;反应器为内径10mm的不锈钢管,带温控系统的三段加热立式管式炉提供反应温度条件;用气袋在采样口采集烟气后由testo350烟气分析仪进行分析。
催化剂的活性用no的转化率来评价:
其中,noin、noout分别表示固定床反应器入口和出口no的浓度,所有的数据均在脱硝反应稳定后读取。活性测试结果见附图1,从图中可以看出,助剂的添加均能提高复合分子筛催化剂的脱硝活性,其中ce的添加表现最为明显,cu-mn-ce/sapo-34多金属复合型分子筛催化剂在温度120℃时,no转化率就可以达到82%以上,温度在180℃~300℃之间均能高达90%以上,活性最高可达99.6%。
催化剂抗水抗硫性能测试结果见附图2和附图3,制备新鲜的催化剂进行活性测试待反应稳定1h后,通入不同浓度的水蒸气或so2浓度,进行毒化实验8h后停止通入水和so2。附图2是催化剂在不同浓度的水蒸气环境下脱硝活性的变化情况,从图中可以看出,通入水蒸气后催化剂的脱硝活性随着水浓度的增加而有所下降,但是当停止通入水蒸气后催化剂的活性基本上均能恢复到初始水平,表明催化剂抗水性能较好;附图3是经添加ce、co、fe等几种不同助剂的复合分子筛催化剂抗硫性能测试结果,从图中可以看出,未添加助剂的cu-mn/sapo-34分子筛在500ppm的so2中毒化8h后,催化剂的活性由95%降至65%,添加助剂ce、fe、co后催化剂的抗硫性能明显有所提升,其中ce的添加最为明显,cu-mn-ce/sapo-34毒化8h后催化剂的活性仅由95%降至82%左右,停止通入so2后活性能够恢复并稳定在85%左右。
实施例14:bet比表面积和孔结构分析
使用美国micromeritics公司asap2020m全自动比表面积及微孔孔隙分析仪测定。通过催化剂对氮气的吸附和脱附测量催化剂的比表面积,在吸附过程中利用液氮制造低温环境,在最大程度上使氮气充满每一个微孔孔隙,然后在接近零度的温度下使催化剂进行脱附,将孔隙中的氮气排出。利用氢气作载气,氮气作吸附气,液氮温度(77k)下n2被吸附,以a12o3做参比,比表面积使用brunauer-emmett-teller(bet)方法计算,孔容和孔径使用barrett-joyner-halenda(bjh)方法计算。
表1复合型分子筛催化剂的bet比表面积及孔结构分析
表1是对ce、co、fe三种不同过渡金属助剂添加的催化剂so2中毒前后进行bet表征。从表中可以看出,添加助剂后催化剂的比表面积、孔容均有所减少,孔径有所增加。可能是由于这些助剂覆盖在催化剂表面或进入分子筛孔道所致。对比改性后的催化剂中毒前后理化性能参数的变化,可以看出改性催化剂so2中毒前后比表面积减少量的大小依次是fe>co>ce。由ce改性后的催化剂比表面积减少的最小,仅仅由423.14m2/mg减少至390.30m2/mg,而未添加助剂改性的催化剂so2中毒后比表面积由456.58m2/mg降至331.47m2/mg,so2中毒后催化剂bet比表面积减少主要原因是生成的硫酸铵盐附着在催化剂表面。表明添加适量的ce和co能够很好的提高催化剂抗so2中毒性能。
实施例15:扫描电镜(sem)联用x射线能谱(eds)分析
催化剂表面活性组分晶体形貌、孔隙大小、团聚程度等都会直接影响到催化剂的性能,扫描电镜(sem)可以清楚地反映这些微观特性,x射线能谱(eds)可以对表面晶体元素组成进行定性和定量分析。本文中扫描电镜sem联用eds是采用荷兰fei公司生产的sirion-50型扫描电子显微镜,其分辨率为150ev。
表2本发明实例中制备的复合型分子筛催化剂中毒亲后eds分析
图4和表2是对未改性的新鲜催化剂、未改性so2中毒后的催化剂及经ce、co、fe改性so2中毒后的催化剂进行了sem联用eds分析,从图中可以明显看出未改性的催化剂so2中毒后催化剂表面发生团聚严重,有许多亮白色的物质,结合eds分析和前文的xrd、tg-dtg分析可知该物质是硫酸铵盐类物质。改性后的催化剂so2中毒后催化剂表面明显比未改性的催化剂so2中毒后的表面光滑许多,也没有出现大量的团聚现象。ce改性后的催化剂so2中毒后表面s元素的质量含量明显低于其它几种催化剂,说明经ce改性后的催化剂能够减少硫酸铵盐类物质在催化剂表面沉积,可能是由于ce能够降低了nh4+和so4-2之间的键能,使(nh4)2so4热稳定性降低,更容易分解,从而降低了硫酸铵盐在催化剂表面的堆积。
- 还没有人留言评论。精彩留言会获得点赞!