一种纳米合金及其制备方法与流程
本发明涉及电化学催化领域,尤其涉及一种纳米合金及其制备方法。
背景技术
近年来,随着纳米合金材料的不断涌现和计算化学的不断发展,研究者们提出了一系列用以解释和预测合金特性的表面化学理论和相关数据图表,图表中可供检索的数据包括金属d带中心的位置以及合金内各元素原子的表面偏析的特性,且已在解释部分纳米合金催化剂性能上获得了初步的成功。利用金属-金属合金化的方法来提升催化性能,这样的方法已经在许多不同的体系中得到了有益的尝试。例如,pt-cu催化剂的orr活性相比于纯的pt催化剂有4倍以上的增强。在对金属协同作用的早期研究中,chu和jiang等人发现含有co/fe,ni/fe或者co/ni双金属的复合纳米材料的氧还原活性比任何一个单金属fe、co和ni形成的纳米材料要高。
目前研究纳米金属合金的方法已经很成熟,例如将pt与适当的贵金属(如ru,ir,pd,os,ag,au等)或者过渡金属(如cu,fe,co,ni等)形成纳米合金材料作为电化学催化剂近年来受到广泛的关注,基于对催化剂的组分、结构、尺寸以及表面进行了大量的研究和合成调控,电化学催化剂的催化活性和稳定性均有了显著的提高。由于合金的形状和结构各种各样,其对应的性能也有较大的差别,李亚栋和俞书洪等合成不同形状,高晶面指数的合金纳米粒子,具有很好的催化性能。但是合成小尺寸纳米、高指数晶面或核壳结构的零维纳米颗粒催化剂,形貌难以控制,制备工艺严苛,因其较高的表面能而具有通过聚集降低表面能的趋势,同时,在应用过程中,如在苛刻的电池运行条件下,纳米颗粒催化剂会溶解,碳支撑载体会被腐蚀,这种溶解和腐蚀在高电位或电池开关的情况下更为明显。
因此,现有技术还有待于改进和发展。
技术实现要素:
鉴于上述现有技术的不足,本发明的目的在于提供一种纳米合金及其制备方法,旨在解决现有的纳米合金的催化活性耐久性不足、制备工艺复杂的问题。
本发明的技术方案如下:
一种纳米合金的制备方法,其中,包括步骤:
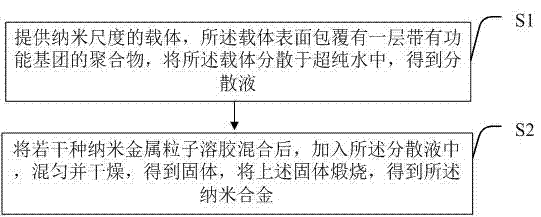
提供纳米尺度的载体,所述载体表面包覆有一层带有功能基团的聚合物,将所述载体分散于超纯水中,得到分散液;
将若干种纳米金属粒子溶胶混合后,加入所述分散液中,混匀并干燥,得到固体,将上述固体煅烧,得到所述纳米合金。
所述的纳米合金的制备方法,其中,所述载体的形状为纳米球、纳米线、纳米管、纳米棒或纳米片。
所述的纳米合金的制备方法,其中,所述载体为二氧化硅、ec-300j商业炭黑、中空碳球、碳纳米管、石墨烯或wo3纳米片。
所述的纳米合金的制备方法,其中,所述聚合物的功能基团为-sh、-nh2、-cooh、-oh和-conh-中的一种或多种。
所述的纳米合金的制备方法,其中,所述聚合物为聚多巴胺、聚乙烯亚胺、聚吡咯、聚苯胺或聚吡啶。
所述的纳米合金的制备方法,其中,所述纳米金属溶胶中纳米金属粒子的粒径为1-50nm。
所述的纳米合金的制备方法,其中,所述煅烧的温度为300~1500ºc、时间为1-100min。
所述的纳米合金的制备方法,其中,所述煅烧过程于惰性气体气氛下进行。
所述的纳米合金的制备方法,其中,所述纳米金属为pt、au、pd、ag、ti、ru、ir、os、cu、fe、co或ni。
一种纳米合金,其中,采用如上所述的制备方法制得。
有益效果:本发明在纳米尺度载体表面包覆一层带有与纳米金属配位的功能基团的聚合物,然后在载体上面负载不同纳米金属粒子,然后通过烧结使之熔合为合金,功能基团的存在增大了载体材料的比表面积,能够将更多的纳米金属粒子牢固地负载在载体表面,提高了金属的利用率及所制备的纳米合金的催化稳定性,而且合成工艺简单可控,便于对其催化活性形成机理进行探究,有利于纳米合金催化剂的推广应用。解决了现有的纳米合金的催化活性耐久性不足、制备工艺复杂的问题。
附图说明
图1为本发明所述纳米合金制备方法的较佳实施例流程图。
图2为本发明中实施例2合成催化剂的模拟图。
图3为本发明中实施例2中得到纳米合金的投射电镜图。
图4为本发明中实施例2中得到纳米合金的粒径分布图。
图5为本发明中实施例2中得到纳米合金的能谱图。
图6为本发明中实施例7中得到纳米合金在0.1mol/l的koh溶液中的氧还原反应耐久性能测试图。
具体实施方式
本发明提供了一种纳米合金及其制备方法,为使本发明的目的、技术方案及效果更加清楚、明确,以下对本发明进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
所述的纳米合金的制备方法,如图1所示,包括步骤:
s1、提供纳米尺度的载体,所述载体表面包覆有一层带有功能基团的聚合物,将所述载体分散于超纯水中,得到分散液;
s2、将若干种纳米金属粒子溶胶混合后,加入所述分散液中,混匀并干燥,得到固体,将上述固体煅烧,得到所述纳米合金。
本发明先在纳米尺度的载体上包覆一层带有功能集团的聚合物,能够提高纳米金属离子与载体的结合力度,同时增加负载量,提高了纳米金属粒子的利用率及所制备的纳米合金的稳定性。
具体地,所述步骤s1中,选择纳米尺度的载体原料,分散在溶剂中,搅拌均匀,得到分散均匀的载体溶液;向所述载体溶液中加入带有功能集团的聚合物溶液,混合均匀,得到沉淀即为载体粒子,并将沉淀分离后洗涤干净后分散于超纯水中,得到分散液,备用。
所述溶剂可为甲醇、乙醇等有机溶剂,也可以为去离子水,载体原料加入到溶剂中,然后进行搅拌,为保证较好的分散效果,可以采用超声装置进行超声分散,超声20min-1h为宜,以获得分散均匀的纳米粒子溶液。
其中,加入的载体原料可以为球形二氧化硅、ec-300j商业炭黑、中空碳球的一种,也可以为纳米棒、纳米线、碳纳米管,还可以为二维材料如石墨烯、wo3纳米片的一种。采用纳米球或二维材料作为载体,不仅可以提高材料的比表面积,有利于以其为原料制备低成本、高性能,促进该项技术的推广应用。而在众多的载体中,选择二氧化硅、ec-300j商业炭黑、中空碳球、纳米棒、纳米线、碳纳米管、石墨烯、wo3纳米片,是因为上述载体较为常见、合成方法较为成熟易获得,有利于进一步降低成本。
较佳地,所述聚合物带有功能基团的可以为-sh、-nh2、-cooh、-oh、-conh-中的一种或多种,具体地,所述聚合物为聚多巴胺、聚乙烯亚胺、聚吡咯、聚苯胺或聚吡啶。所述聚合物要求不仅作为做为碳的前驱体,同时还能与金属离子形成配位。将聚合物加入到上述载体溶液中,包覆载体原料,为了保证包覆完全,载体原料与聚合物的质量比为0.5~2,一方面是为了保证加入金属纳米粒子后可以负载更多的金属;另一方面是防止煅烧后,聚合物转化为碳层的过程中,纳米金属脱落。
所述步骤s2中,预先制备纳米金属粒子溶胶并将两种以上纳米金属粒子溶解混合均匀后加入所述分散液中,混匀并真空干燥,得到固体进行煅烧即得到产物纳米合金。纳米金属溶胶中纳米金属粒子的粒径保证为1-50nm,所述纳米金属为纳米贵金属pt、au、pd、ag、ti、ru、ir或os,或者过渡金属cu、fe、co、ni等,其中选择熔合为合金的纳米金属,彼此的熔点要相近(相差不大于100℃),这样能够以与较低熔点金属的熔点温度相差不大的温度进行煅烧反应,较少金属损耗及损害,提供所制备的合金纯度及质量。采用真空干燥的方式干燥,能够使得分散液中的溶剂以适当的速度蒸发后转移至瓷舟中。
具体地,所述纳米金属溶胶利用还原剂还原金属盐溶液制备,具体可以利用柠檬酸钠还原法制备,然后将制备得到的纳米金属溶胶以1:1的摩尔质量比混合均匀,然后加入分散液中,室温下搅拌过夜后,得到负载纳米金属的载体,然后真空干燥,得到固体粉末,转移至干净的磁舟中,并将上述瓷舟置于管式加热炉中,在惰性气氛下进行煅烧,其中,煅烧温度是根据负载的纳米金属的熔点决定的,煅烧时间是根据负载的纳米金属熔合的程度决定的。较佳地,煅烧温度为300~1500ºc,煅烧时间为1-100min。
所述惰性保护气可以为氮气或氩气,可以避免催化剂在高温煅烧过程中发生氧化等副反应,其中,优选氩气作为惰性保护气,且在试验开始前,预先向管式炉中通氩气至少20min,形成保护气氛围,防止催化剂在升温过程中被氧化。所述加热过程采用程序升温方式进行,以2~10ºc/min的速率升温到300~1500ºc,保温1-100min。优选的,煅烧温度在500~800ºc较为合适,煅烧时间为10min。高温煅烧结束后,进行程序降温处理,200ºc前以5-10ºc/min降温,当管式炉温度低于50ºc时,取出瓷舟。优选的,降温速率10ºc/min效果较好。
本发明还提供了一种纳米合金,其中,采用如上所述的制备方法制得。
下面通过实施例对本发明进行详细说明。
以下实施例中,使用的纳米金属颗粒的制备方法,以纳米au、pt为例。
实施例1
利用柠檬酸钠还原法分别制备粒径3nm左右的pt、au纳米粒子:
将1.0ml1%haucl4·3h2o加入90ml超纯水中,搅拌1min后,加入2ml38.8mm柠檬酸钠,搅拌1min,最后快速加入1ml0.075%nabh4,搅拌5min既得橙红色au纳米溶胶;
向100ml圆底烧瓶中加入47ml超纯水、2ml0.1m的柠檬酸钠和1ml0.1mh2ptcl6.6h2o。油浴80ºc,机械搅拌,恒温1h。然后加入微量的nabh4,搅拌30min即得到深棕色pt纳米溶胶;
向100ml圆底烧瓶中加入32.5ml超纯水、1.64mgpvp和7.5ml4mmh2pdcl4.6h2o搅拌均匀,几分钟后加入10ml无水乙醇。油浴100℃,机械搅拌,回流3h,冷却至室温,即得到pd纳米溶胶,放置冰箱待用;
50ml圆底烧瓶加入50mmol.l-1氯化镍的乙二醇溶液3ml,一定体积的pd纳米溶胶与ni2+摩尔比为1:40,加入1ml水合肼和一定体积的乙二醇,保证反应液的体积为30ml,搅拌均匀至于60°c油浴下加热即得ni纳米粒子溶胶;
将0.017gagno3与0.029gc6h5na3o7均同时加入35°c恒温的400ml蒸馏水中,得到agno3与c6h5na3o7浓度均为0.25mm的混合溶液,将混合溶液置于磁力搅拌器上剧烈搅拌,迅速加入12ml10mmnabh4的水溶液中。加入水溶液后,混合溶液立即呈现土黄色,旋即颜色变深,类似于墨绿色,随即又变回澄清透明的土黄色,并保持颜色恒定不变,即得ag纳米粒子溶胶,放置冰箱过夜,以待备用。
实施例2
称取50mgsio2纳米粒子于烧瓶中,加入2ml去离子水,充分溶解分散;之后,称取100mg多巴胺盐酸盐于烧瓶中,加入50mltris-hcl缓冲溶液,充分溶解后加入上述sio2纳米粒子,室温下机械搅拌5h,得到sio2@pda纳米粒子载体,将沉淀离心分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
将实施例1所制备合成的金属pt、au纳米溶胶以摩尔比1:1加入到上述分散液中,机械搅拌过夜,最后将得到的沉淀离心分离,并用超纯水洗涤沉淀,然后将样品置于真空干燥箱中,干燥过夜,得到sio2@pda@pt-au纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在900ºc的氩气氛围下高温炭化10min,即得sio2@c@pt/au双金属纳米合金。
其中,sio2@c@pt/au双金属纳米合金的催化合成模拟如图2所示。
将上述实施例2制备的sio2@c@pt/au纳米合金进行形貌表征,结果分布如图3和4所示,图3为纳米合金的投射电镜图,图4为合成的纳米合金粒径分布图,通过对比,烧结后的金属粒子粒径明显大于未烧结的粒子。
为了进一步表征pt、au纳米粒子是否熔合,对烧结后的纳米合金颗粒及进行能谱表征,结果如图5所示,根据图5可知,pt、au粒子已完全熔合为合金。
实施例3
首先,称取50mg碳纳米管cnt至于烧瓶中,加入2ml乙醇,充分溶解分散。之后,称取50mg聚乙烯吡咯烷酮pvp于烧瓶中,充分溶解后加入上述cnt溶液,室温下机械搅拌,得到cnt@pvp纳米粒子载体,将沉淀离心分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
然后将实施例1所制备合成的金属pt、pd纳米溶胶以摩尔比1:1加入到上述分散液中,机械搅拌过夜,最后将得到的沉淀离心分离,沉淀用超纯水洗涤,然后将样品置于真空干燥箱中,干燥过夜,得到cnt@pvp@pt-pd纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在900ºc的氩气氛围下高温炭化30min,即得cnt@c@pt/pd双金属纳米合金。
实施例4
首先,称取50mg纳米银线agnw至于烧瓶中,加入2ml去离子水,充分溶解分散。之后,称取80mg聚吡咯,充分溶解后加入上述agnw,室温下机械搅拌,得到agnw@pda纳米粒子载体,将沉淀离心分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
然后将实施例1所制备合成的金属au、pd纳米溶胶以摩尔比1:1加入到上述分散液中,机械搅拌过夜,最后将得到的沉淀离心分离,沉淀用超纯水洗涤,然后将样品置于真空干燥箱中,干燥过夜,得到agnw@pda@au-pd纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在900ºc的氩气氛围下高温炭化10min,即得agnw@c@au/pd双金属纳米合金。
实施例5
首先,称取50mgec-300j商用炭黑于烧瓶中,加入100ml去离子水,充分溶解,超声分散形成均一溶液;再称取100mg多巴胺盐酸盐于烧瓶中,加入50mltris-hcl缓冲溶液,充分溶解后加入上述溶液中,室温下机械搅拌,得到c@pda纳米粒子载体,将沉淀抽滤洗涤分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
然后将实施例1所制备合成的金属pt、ni纳米溶胶以摩尔比1:1加入到上述分散液中,搅拌过夜,最后将得到的沉淀抽滤洗涤分离,用超纯水洗涤4遍,然后将样品置于真空干燥箱中,干燥过夜,得到c@pda@pt-ni纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在700ºc的氩气氛围下高温炭化90min。即得c@pt/ni双金属纳米合金。
实施例6
首先,称取50mg碳纳米管cnt于烧瓶中,加入100ml乙醇,充分溶解,超声分散形成均一溶液;再称取50mg聚吡咯ppy,充分溶解后加入上述cnt溶液中,室温下机械搅拌,得到cnt@ppy纳米粒子载体,沉淀抽滤洗涤分离,用超纯水洗涤4遍,重新分散于超纯水中,得到载体分散液;
然后将实施例1所制备合成的金属ag、pd纳米溶胶以摩尔比1:1加入到上述分散液中,搅拌过夜,最后将得到的沉淀离心洗涤分离,用超纯水洗涤4遍,然后将样品置于真空干燥箱中,干燥过夜,得到cnt@ppy@ag-pd纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在500ºc的氮气氛围下高温炭化70min,即得cnt@c@ag/pd双金属纳米合金。
将上述所制备的cnt@c@ag/pd双金属纳米合金在0.1mol/l的koh溶液中进行氧还原反应耐久性能测试,其结果如图6所示,根据图6可知,所制备的cnt@c@ag/pd双金属纳米合金催化活性对碱的耐久性能良好。
实施例7
首先,称取50mgec-300j商用炭黑于烧瓶中,加入100ml乙醇,充分溶解,超声分散形成均一溶液;再称取100mg多巴胺盐酸盐于烧瓶中,加入50mltris-hcl缓冲溶液,充分溶解后加入上述ec-300j商用炭黑溶液中,室温下机械搅拌5h,得到c@pda纳米粒子载体,沉淀抽滤洗涤分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
然后将实施例1所制备合成的金属au、pd纳米溶胶以摩尔比1:1加入到上述溶液中,机械搅拌过夜,最后将得到的沉淀抽滤分离洗涤,沉淀用超纯水洗涤,然后将样品置于真空干燥箱中,干燥过夜,得到c@pda@au-pd纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在600ºc的氩气氛围下高温炭化10min,即得c@au/pd双金属纳米合金。
实施例8
首先,称取50mgwo3纳米片于烧瓶中,加入2ml去离子水,充分溶解分散;之后,称取60mg多巴胺盐酸盐于烧瓶中,加入30mltris-hcl缓冲溶液,充分溶解后加入上述wo3纳米溶液,室温下机械搅拌5h,得到wo3@pda纳米载体,沉淀离心分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
然后将实施例1所制备合成的金属pt、pd和ag纳米粒子以摩尔比1:1:1加入到上述分散液中,机械搅拌过夜,最后将得到的沉淀离心分离,沉淀用超纯水洗涤,然后将样品置于真空干燥箱中,干燥过夜,得到wo3@pda@pt-pd-ag纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在600ºc的氮气氛围下高温炭化20min,即得wo3@c@pt/pd/ag三元金属纳米合金。
实施例9
首先,称取50mgec-300j商用炭黑于烧瓶中,加入100ml去离子水,充分溶解,超声分散形成均一溶液;再称取100mgpvp于烧瓶中,充分溶解后加入上述ec-300j商用炭黑溶液中,室温下机械搅拌,得到c@pvp纳米粒子载体,沉淀抽滤洗涤分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
然后将实施例1所制备合成的金属pd、au和ni纳米粒子以摩尔比1:1:1加入到上述溶液中,机械搅拌过夜,最后将得到的沉淀抽滤分离洗涤。沉淀用超纯水洗涤,然后将样品置于真空干燥箱中,干燥过夜,得到c@pvp@pd-au-ni纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在700ºc的氩气氛围下高温炭化30min,即得c@pd/au/ni三元金属纳米合金。
实施例10
首先,称取50mg碳纳米管cnt于烧瓶中,加入100ml乙醇,充分溶解,超声分散形成均一溶液;再称取50mg聚吡咯于烧瓶中,充分溶解后加入上述cnt溶液中,室温下机械搅拌,得到cnt@ppy纳米粒子载体,将沉淀抽滤洗涤分离,用超纯水洗涤4遍,重新分散于超纯水中,得到分散液;
然后将实施例1所制备合成的金属ag、au、ni纳米粒子以摩尔比1:1:1加入到上述溶液中,搅拌过夜,最后将得到的沉淀抽滤洗涤分离,用超纯水洗涤4遍,然后将样品置于真空干燥箱中,干燥过夜,得到cnt@ppy@ag-au-ni纳米粒子;
最后,将干燥后的样品粉末转移到瓷舟中,置于管式炉中,在1000ºc的氮气氛围下高温炭化60min。即得cnt@c@ag/au/ni三元金属纳米合金。
综上所述,本发明提供的纳米合金的制备方法,在纳米尺度载体表面包覆一层带有与纳米金属配位的功能基团的聚合物,然后在载体上面负载不同纳米金属粒子,然后通过烧结使之熔合为合金,功能基团的存在增大了载体材料的比表面积,能够将更多的纳米金属粒子牢固地负载在载体表面,提高了金属的利用率及所制备的纳米合金的催化稳定性,而且合成工艺简单可控,便于对其催化活性形成机理进行探究,有利于纳米合金催化剂的推广应用。解决了现有的纳米合金的催化活性耐久性不足、制备工艺复杂的问题。
应当理解的是,本发明的应用不限于上述的举例,对本领域普通技术人员来说,可以根据上述说明加以改进或变换,所有这些改进和变换都应属于本发明所附权利要求的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!