一种固相萃取材料的制作方法
本发明属于固相萃取材料领域,尤其是固相微萃取材料。
背景技术:
固相萃取是一种基于化学分配/吸附机理的分离过程,具体是指在液体/萃取材料、气体/萃取材料的界面上,流体中的化合物会在萃取材料表面吸附,或者被萃取材料本体吸收,从而实现对流体中特定化合物的富集、分离、净化。固相萃取在化工分离、化学分析特别是样品前处理领域广泛使用。
固相微萃取是一种微型化的固相萃取技术,基本原理和固相萃取相同,主要用于水溶液样品中化合物的萃取富集。对于挥发性有机物(vocs)、半挥发性有机物(svocs),固相微萃取可以与热解析、气相色谱联用:固相微萃取结束后,把固相微萃取材料直接放入热解析装置中,高温直接把萃取的vocs或svocs脱附出来进入色谱,完成后续的分离分析。固相微萃取-热解析-气相色谱联用,把萃取富集、脱附解析、分离检测集中在一步完成,提高了分析通量。而且不使用有机溶剂,绿色环保。
萃取材料是固相微萃取方法的核心。分析化合物先从样品溶液中吸附、富集到萃取材料上,再通过热脱附或有机溶剂洗脱,进入到后续分析仪器。一般以相似相溶原则选择萃取材料,即萃取材料应与分析物性质匹配。目前,对于弱极性疏水化合物,聚二甲基硅氧烷(pdms)是性质较好、使用最普遍的萃取材料,对于极性有机化合物,聚丙烯酸酯(pa)、聚乙二醇(peg,carbowax,cw)是比较常见的商品化萃取相材料。另外,针对极性有机物萃取,科研报道的较好的萃取材料有聚酰亚胺(pi)、聚醚砜酮(ppesk)。
水在极性萃取材料pa、peg、pi、ppesk上的吸附量比较大,一般饱和吸附量在10%左右。如果微萃取相材料的质量为1毫克,则在萃取过程中,吸附的水有0.1毫克,而有机物的吸附量只有纳克级别。
固相微萃取上吸附的水,会降低后续的分离检测效率,甚至损毁仪器设备。在热解析-色谱联用过程中,热解析的高温瞬间使吸附的水完全气化,可能导致萃取相材料的碎裂,脱落;水汽进入气相色谱后,可能导致色谱固定相的溶胀、失效,降低色谱分离效率;水汽进入氢火焰离子化检测器(fid)后,会导致fid熄火;进入电子捕获检测器(ecd)、热导检测器(tcd)后,会极大缩短ecd、tcd检测器的寿命;进入质谱检测器(ms)后,可能导致电子发射灯丝(ei)的烧毁。
目前,对于水在极性萃取材料上吸附残留的相关研究很少,对抑制水吸附残留的萃取材料的报道更少。
技术实现要素:
本发明涉及一种固相萃取材料,特别是固相微萃取材料。本发明所述固相萃取材料为核壳结构,所述的核层为极性固相萃取材料,壳层为聚二甲基硅氧烷。
本发明另一方面提供所述固相萃取材料的制备方法,包括如下步骤:
(1)制备核层,包括核层薄膜和核层颗粒:
1a.制备核层薄膜:将固相萃取材料制备为0.1%~30%聚合物溶液;以浸渍法(dip-coating)、喷涂法(spray-coating)或旋涂法(spin-coating),在支撑载体表面形成0.01微米~1毫米的核层薄膜;并固化干燥;
1b.制备核层颗粒:将固相萃取材料制备为0.1%~50%聚合物溶液,以喷雾干燥、乳化、机械粉碎的方法制备0.1微米~1毫米颗粒,并固化干燥;
(2)壳层的制备:将聚二甲基硅氧烷与交联剂按质量比1:2~50混溶,然后用烃类溶剂按照体积比稀释到0.1~50%;浸泡步骤(1)制备的核层薄膜或核层颗粒;然后取出固化烘干。
所述方法制备的本发明的固相萃取材料对于弱极性、非极性的小分子有机物(logp≥0),如酚类、硝基苯类、多环芳烃、有机磷、有机氯等化合物,壳层导致的萃取效率下降,不大于30%。对于强极性的小分子化合物(logp<0),吸附量会减少,其中,水在核层极性固相萃取材料上的吸附会减少80%以上。本发明材料制备方法简单,弱极性、非极性的小分子萃取效率基本维持不变,同时大幅度降低水的吸附残留,延长固相萃取材料的使用寿命,提高后续的色谱分离与质谱检测的效率。
基于此,本发明进一步提供所述的材料在固相萃取或固相微萃取中的应用。
特征性地,所述的固相萃取材料可以萃取富集疏水有机小分子化合物(logp≥0);也可以抑制极性亲水小分子化合物(logp<0)、特别是水的吸附。
在所述的应用中,还可包括萃取完成后对所述固相萃取材料的回收利用的步骤。具体的方式,包括使用有机溶剂解析、高温加热解析,把萃取的化合物脱附下来,用于后续分析。
附图说明
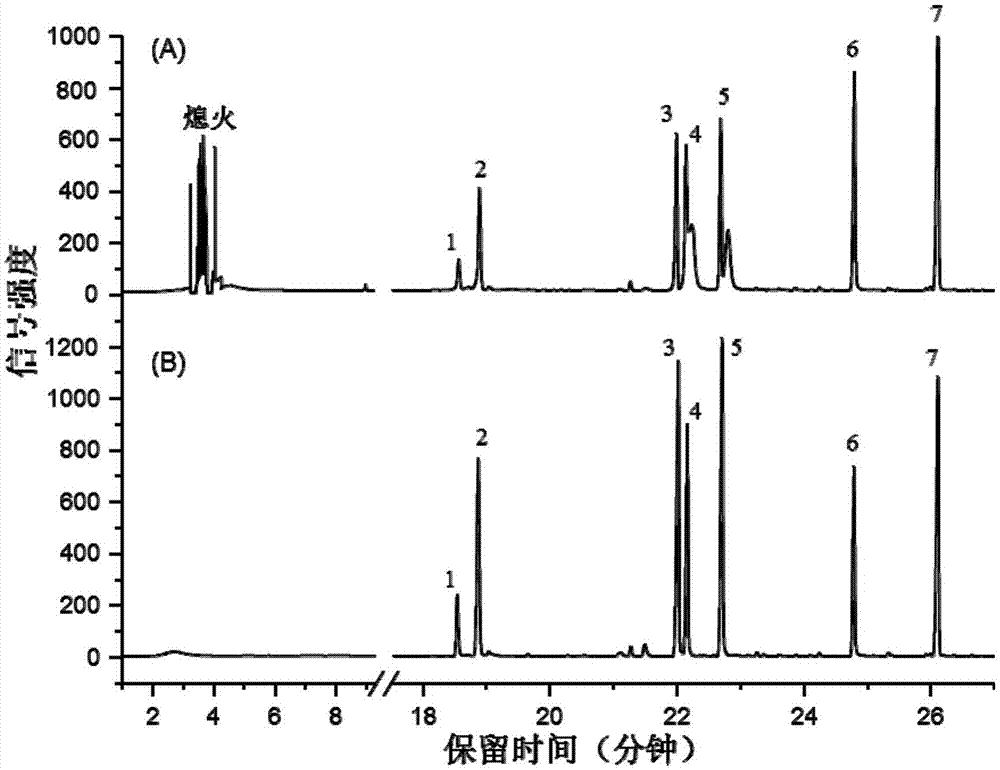
附图1是有机酚萃取测试试验中,经热解析脱附联用气相色谱检测的谱图,其中:(a)是pi萃取搅拌棒的测试结果;(b)是pdms/pi萃取搅拌棒的测试结果。
附图2是硝基苯类化合物萃取测试试验中,经热解析脱附联用气相色谱检测的谱图,其中:(a)是pi萃取搅拌棒的测试结果;(b)是pdms/pi萃取搅拌棒的测试结果。
具体实施方式
本发明提供一种固相萃取材料,所述材料为核壳结构,所述的核层为极性固相萃取材料,壳层为聚二甲基硅氧烷。在本说明书中,具有核壳结构的固相萃取材料不约束于具体的外形、可以是颗粒状或薄膜状材料。在任何形态的材料中,所述的聚二甲基硅氧烷壳层的厚度为1纳米~10微米。
具体的实施方式中,所述的极性固相萃取材料选自聚酰亚胺(pi)、聚苯并咪唑(pbi)、聚醚砜酮(ppesk)、聚乙二醇(peg)、聚丙烯酸酯(paa)、聚二乙烯苯(pdvb)、石墨化炭黑(gcb)、碳分子筛及其衍生物或组合物。优选选自聚酰亚胺、聚醚砜酮、聚苯并咪唑、聚乙二醇和聚丙烯酸酯。
进一步,本发明提供上述固相萃取材料的制备方法,所述方法包括核层制备和壳层制备的步骤。
具体的实施方式中,核层的制备可根据具体需要选择方法,来制备薄膜状核层或颗粒状核层:
1a.制备核层薄膜:将固相萃取材料制备为0.1%~30%聚合物溶液;以浸渍法(dip-coating)、喷涂法(spray-coating)或旋涂法(spin-coating),在支撑载体表面形成0.01微米~1毫米的核层薄膜;并固化干燥;其中所述聚合物溶液的溶剂选自二甲基亚砜、二乙基亚砜、氮甲基吡咯烷酮及其混合体系;
1b.制备核层颗粒:将固相萃取材料制备为0.1%~50%聚合物溶液,以喷雾干燥、乳化、机械粉碎的方法制备0.1微米~1毫米颗粒,并固化干燥;其中所述聚合物溶液的溶剂选自二甲基亚砜、二乙基亚砜、氮甲基吡咯烷酮及其混合体系。
在一些具体的实施方式中,核层颗粒材料可以通过直接购买市售商品获得,也可以用于本发明。此处可举例但不限于聚二乙烯苯(pdvb)、石墨化炭黑(gcb)、碳分子筛颗粒。
在所制备的核层基础上,进一步制备壳涂层,壳层的制备方法为:将聚二甲基硅氧烷与交联剂按质量比1:2~50混溶,然后用烃类溶剂按照体积比稀释到0.1~50%;浸泡步骤(1)制备的核层薄膜或核层颗粒;然后取出固化烘干。具体实施方式中,所述的交联剂是道康宁的sylgard184。所述的烃类溶剂选自正己烷、环己烷、正戊烷、苯、甲苯或其混合物。
下面以具体实施例的方式对本发明做进一步说明,但不应当理解为对本发明的任意形式的限定。
实施例1
制备ppesk萃取搅拌棒
20%ppesk(大连聚合物新材料公司)充分溶解在二甲基乙酰胺溶剂中,将石英棒(长40mm、直径2.5mm)分别用乙醇、水清洗,并在100℃烘干。提拉法制备ppesk萃取材料:将石英棒插入ppesk溶液,匀速提起,然后直接浸入纯净水中,进行相转化。24小时后取出,空气中晾干。在20ml/min氮气吹扫下,高温老化ppesk萃取材料:40℃维持30min、5℃/min升到100℃维持30min、5℃/min升到200℃维持30min、5℃/min升到280℃维持300min,自然降到室温。
实施例2
制备pi萃取搅拌棒
10%pi(p84,evonic)充分溶解在二甲基甲酰胺溶剂中,其他操作条件同
实施例1。
实施例3
制备pdms/ppesk、pdms/pi萃取搅拌棒
按质量比10:1配制pdmssylgard184溶液,混溶后,用正己烷稀释到0.5%、1%、2%、5%、10%、20%、50%v/v浓度。将实施例1制备的ppesk萃取搅拌棒、实施例2制备的pi萃取搅拌棒,分别浸入相应浓度的pdmssylgard184溶液中,浸泡10分钟后取出,空气中晾干,在烘箱中60℃交联固化pdms。然后再在氮气保护下,老化pdms/ppesk、pdms/pi萃取搅拌棒。老化程序:40℃维持30min、5℃/min升到100℃维持30min、5℃/min升到200℃维持30min、5℃/min升到280℃维持300min,自然降到室温。
实施例4
测量表面接触角
经检测:pi萃取棒自身,水的接触角67°±5°;实施例3所制备的0.5%,1%,2%,5%,10%,20%,50%v/vpdms/ppesk萃取棒的水的接触角112°±10;这表明pdms已经涂覆到ppesk表面,且浓度低至0.5%的pdms,就可以有效涂覆pi材料外表面。
实施例5
观测pdms涂层微观形貌
电子扫描显微镜观察实施例3所制得的材料截面,发现:0.5%,1%,2%,5%,10%,20%pdms涂层厚度小于1微米,而50%pdms涂层厚度小于5-10微米。
电子扫描显微镜观察材料表面,发现:pi萃取棒表面有密集的纳米孔,随着pdms浓度的增加,pdms涂层逐渐封闭了表面的纳米孔,在10%pdms涂层时,表面纳米孔基本消失,但仍可见表面微米级的起伏;在50%dms涂层时,表面起伏降至纳米范围。
实施例6
测定pi萃取搅拌棒、pdms/pi萃取搅拌棒的吸水量
取实施例2中制备的pi萃取搅拌棒,以及实施例3中的制备的0.5%pdms/pi萃取搅拌棒,萃取相尺寸相同:萃取相厚度60微米,长度2cm。萃取条件:10ml纯水溶液中,40℃,搅拌速度600rpm,萃取时间30min。萃取完成后直接放入热解析器中,260℃高温加热7min,热解析脱附的水蒸汽直接进入agilent7890b,进行后续分离分析。
色谱分离条件:hp-plot/q色谱柱,30m×320μm×20.0μm,载气为氦气(99.999%),恒流模式下3ml/min;进样口300℃。柱温:40℃保持9min,以8℃/min的升温速率,升温至200℃保持5min;tcd检测器。
色谱定量分析表明:pi萃取搅拌棒吸附1.4毫克水,0.5%pdms/pi吸附水量只有0.098毫克,吸水量下降了93%;5%pdms/pi吸附水量为0.042毫克,吸水量下降了97%。
实施例7
pdms/pi萃取搅拌棒,萃取酚类化合物。
检测材料为实施例2中制备的pi萃取搅拌棒,以及实施例3中的制备的0.5%pdms/pi萃取搅拌棒。有机酚标准样:苯酚、2-氯酚、2-硝基酚、2、4-二甲基酚、2、4-二氯酚、4-氯-3-甲基酚、2、4、6-三氯酚。萃取相尺寸相同:萃取相厚度60微米,长度2cm。萃取条件:100ppb、10ml有机酚水溶液样品,40℃,搅拌速度600rpm,ph3,萃取时间30min。萃取完成后直接放入热解析器中,260℃高温加热7min,热解析脱附的酚类化合物直接进入agilent7890b,进行分离分析。
色谱分离条件:agilentdb-5色谱柱,30m×250μm×1.0μm,载气为氦气(99.999%),恒流模式下3ml/min;进样口300℃。柱温:40℃保持9min,以8℃/min的升温速率,升温至280℃保持5min;fid温度为300℃,h2流速38ml/min,空气流速400ml/min。
色谱分析谱图如附图1所示。如图1中的(a),pi萃取棒吸附大量水,并在热解析中气化产生大量水蒸汽,导致fid熄火多次;苯酚、2-氯酚、2-硝基酚的色谱峰高偏低;2、4-二甲基酚、2、4-二氯酚色谱峰分裂。而0.5%pdms/pi萃取棒吸附水少,少量水汽没有导致fid熄火,也没有降低色谱分离效率,色谱峰尖锐对称、没有分裂,如图1中的(b)。
实施例8
pdms/pi萃取搅拌棒,萃取硝基苯化合物。
检测材料为实施例2中制备的pi萃取搅拌棒,以及实施例3中的制备的0.5%pdms/pi萃取搅拌棒。硝基苯标准样:硝基苯、2-硝基苯、2-硝基甲苯、3-硝基甲苯、4-硝基苯、2.4-二硝基苯、2.6-二硝基苯。萃取条件:20ppb、10ml硝基苯类水溶液样品,40℃,搅拌速度600rpm,ph7;萃取时间30min。萃取完成后直接放入热解析器中,250℃高温加热7min,热解析脱附的硝基苯类化合物直接进入agilent7890b色谱柱,进行分离分析。色谱分离条件与实施例7相同。
色谱分析谱图如附图2所示。如图2中的(a),pi萃取棒吸附大量水,导致硝基苯、2-硝基苯、2-硝基甲苯、3-硝基甲苯的色谱峰高偏低,而且4-硝基甲苯左右色谱基线波动很大。而5%pdms/pi萃取棒吸附水少,色谱峰尖锐、基线平稳,如图2中的(b)。
- 还没有人留言评论。精彩留言会获得点赞!