一种确定表面活性剂溶解吸附行为的方法及系统与流程
本发明涉及半导体制造领域,特别涉及一种确定表面活性剂溶解吸附行为的方法及系统。
背景技术:
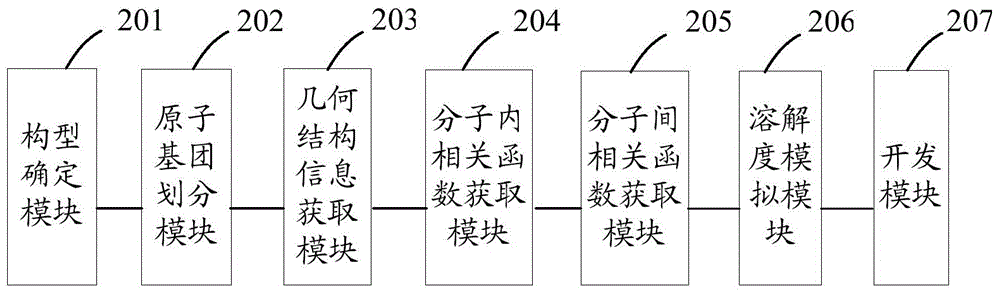
:表面活性剂(ComplexingAgent,CA),是指加入少量能使其溶液体系的界面状态发生明显变化的物质。在集成电路化学机械研磨(ChemicalMechanicalPlanarization,CMP)工艺过程中,研磨作用导致大量不同种类和尺寸的颗粒吸附在晶圆表面,无法去除。CA作为研磨液的必要组分可以降低研磨液和疏水性薄膜间的表面张力,使研磨液和疏水性薄膜更紧密贴合,减少和控制晶圆疏水性薄膜表面的残留物和研磨粒子等缺陷,可以改善化学机械研磨效果;此外,CA还具有较低的临界胶束浓度,容易使研磨颗粒更分散,显著地稳定研磨液中的研磨颗粒,提高研磨液各组分的稳定性,降低研磨表面的清洗难度等。因此,开发出高性能CA以满足CMP工艺需求在半导体制造领域具有重要意义。在高分子CA开发过程中,其溶解吸附行为是衡量CA结构和性质的重要指标,尤其小分子在CA中的溶解度对CA的结构设计、实验合成、过程与本体相特征的最终控制具有重要指导意义。为了获取溶解吸附数据,实验测量仍旧是目前采用的重要方法,然而,实验虽能提供直接的测量数据,但并不能提供各种条件所需的研究和应用数据,更不能预测新体系或新条件下的相关参数。现有技术可以通过分子模拟获得和实验几乎一致的溶解参数,然而分子模拟计算周期较长,这在一定程度上影响实验开发效率,导致CA开发成本较高。技术实现要素:本发明提供了一种确定表面活性剂溶解吸附行为的方法及系统,通过建立一种简洁高效、准确可靠的模拟方法和系统,解决计算机分子模拟确定CA 吸附行为的计算周期长,影响实验开发效率的问题,达到降低CA开发成本的目的。本发明提供了一种确定表面活性剂溶解吸附行为的方法,包括:确定高分子表面活性剂的分子构型,并将高分子链的单体原子划入同一原子基团;根据高分子表面活性剂分子的几何结构信息,获取高分子表面活性剂的原子基团间分子内相关函数;根据原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂的分子间相关函数;根据分子间相关函数获取小分子在高分子表面活性剂中的溶解度;根据溶解度辅助开发高分子表面活性剂。优选的,所述表面活性剂分子的构型为多点半自由链。优选的,所述根据高分子表面活性剂分子的几何结构信息,获取高分子表面活性剂的原子基团间分子内相关函数包括:将各原子以联合原子基团的方式转换为各原子基团作用点;根据高分子表面活性剂分子的几何结构信息,通过生成矩阵法获取高分子表面活性剂的原子基团间分子内相关函数。优选的,所述根据已获取的原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂的分子间相关函数包括:确定各原子基团间的作用力场;根据PY近似及各原子基团间的作用力场确定相关函数近似方程;根据相关函数近似方程及原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂的分子间相关函数。优选的,所述确定各原子基团间的作用力场还包括:根据平均力位能与高分子表面活性剂的分子内、分子间作用的相关性,修正各原子基团间的作用力场。一种确定表面活性剂溶解吸附行为的系统,包括:构型确定模块,用于确定高分子表面活性剂的分子构型;原子基团划分模块,用于将高分子链的单体原子划入同一原子基团;几何结构信息获取模块,用于获取高分子表面活性剂分子的几何结构信息;分子内相关函数获取模块,用于根据高分子表面活性剂分子的几何结构信息,计算高分子表面活性剂的分子内相关函数;分子间相关函数获取模块,用于根据原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂的分子间相关函数;溶解度模拟模块,用于根据分子间相关函数获取小分子在高分子表面活性剂中的溶解度;开发模块,用于根据溶解度辅助开发高分子表面活性剂。优选的,所述分子内相关函数获取模块用于根据高分子表面活性剂分子的几何结构信息,获取高分子表面活性剂的原子基团间分子内相关函数包括:转换单元,用于将各原子作用点以联合原子基团的方式转换为各原子基团作用点;原子基团间分子内相关函数获取单元,用于根据高分子表面活性剂分子的几何结构信息,通过生成矩阵法获取高分子表面活性剂的原子基团间分子内相关函数。优选的,所述分子间相关函数获取模块包括:作用力场确定单元,用于确定各原子基团间的作用力场;相关函数近似方程确定模块,用于根据PY近似及各原子基团间的作用力场确定相关函数近似方程;分子间相关函数获取单元,用于根据相关函数近似方程及原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂分子间相关函数。优选的,所述作用力场确定单元还包括:结构因子获取子单元,用于根据分子间相关函数获取高分子CA的结构因子;平均力位能获取子单元,用于根据结构因子、直接相关函数获取平均力位能;修正子单元,用于根据平均力位能与高分子表面活性剂的分子内、分子间作用的相关性,修正各原子基团间的作用力场。优选的,所述溶解度模拟模块包括:化学位获取单元,用于根据分子间相关函数获取小分子在高分子CA中的低压超额化学位和/或高压超额化学位;溶解度模拟单元,用于根据低压超额化学位和/或高压超额化学位模拟小分子在高分子CA中的溶解度。本发明提供的一种确定表面活性剂溶解吸附行为的方法及系统,首先确定CA的构型,并将高分子链的单体原子划入同一原子基团,以原子基团简化高分子构型,然后根据高分子CA的几何结构信息计算高分子CA的分子内相关函数,并根据分子内相关函数,通过PRISM理论模型获取高分子CA的分子间相关函数,以此预测小分子在高分子CA中的溶解度。由于采用了PRISM理论模型,并以原子基团作为后续模拟的单元,使得该方法较分子模拟复杂度显著降低,不但能保持较高的模拟精度,而且能显著提高模拟速度,是速度和精度的有效结合,能够满足模拟高分子CA溶解吸附行为的精度和速度的要求。附图说明为了更清楚地说明本申请实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明中记载的一些实施例,对于本领域普通技术人员来讲,还可以根据这些附图获得其他的附图。图1为根据本发明实施例确定高分子表面活性剂溶解吸附行为方法的一种流程图;图2为根据本发明实施例确定高分子表面活性剂溶解吸附行为系统的一种结构示意图。具体实施方式下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自 始至终相同或类似的标号表示相同或类似的参数或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能解释为对本发明的限制。为了更好地理解本发明,下面首先对现有技术中确定CA溶解吸附行为做简单说明。吸附行为是由于多相界面上一定表面能的存在,促使表面原子趋于吸引物质以降低表面能态,吸附力可分为基于范德华力的弱吸附,即物理吸附,和基于化学键的强吸附,即化学吸附。小分子与CA之间通过物理吸附使小分子溶解在CA中,溶解度在一定程度上反映了小分子在CA中的溶解能力,也说明了CA对小分子的吸附强弱,可以理解为CA对小分子的吸附能力,也就是小分子的溶解能力,即溶解度在一定程度上反映了小分子在CA中的溶解能力,也说明了CA对小分子的吸附强弱。小分子在CA中的溶解度对CA开发具有重要指导意义,现实中,如何选取一种结构稳定、分散性好、易于吸附在晶圆或研磨粒子表面,同时又容易吸附其他研磨杂质小分子的CA,是开发CA需要考虑的重要因素。为了获取溶解吸附数据,实验测量仍旧是目前采用的重要方法,例如吸附量和等温线测定等,但是实验测量不能预测新体系或新条件下的溶解吸附数据。现有技术可以通过模拟获得和实验几乎一致的溶解参数,例如分子动力学(MolecularDynamics,MD)模拟小分子的溶解度。MD模拟法是通过牛顿经典力学计算许多分子在空间中的轨迹,模拟系统随时间推进的微观过程,通过统计方法得到系统的平衡参数或运输性质,如溶解度参数。以下为一种可行的MD模拟步骤:首先,选取要研究的系统及其边界,选取系统内粒子间的作用势能模型;然后,设定系统中粒子的初始位置和初始动量;建立模型,计算粒子间作用力及各粒子的速度和位置;依据相关的统计理论模型,模拟体系达到平衡后各宏观参数。由于MD通过模拟粒子之间的作用力及各粒子运动状态来获得热力学性能,受模拟能力限制,MD模拟方法不能模拟大量分子,只能模拟有限空间中的有限个数分子,而为了提高模拟精度,需要模拟大量分子,使得模拟过程中计算量巨大, 模拟周期很长。不同于MD模拟方法,本发明提供的一种确定表面活性剂溶解吸附行为的方法及系统,在确定CA的构型后,将高分子链的单体原子划入同一原子基团,以原子基团为模拟的作用点,能大大降低后续模拟过程中的计算量;然后根据CA分子的几何结构信息,获取CA的分子内相关函数,并根据PRISM理论模型获取分子间相关函数,这样就保证了分子间相关函数的精确性;所述分子间相关函数可以用于模拟小分子在CA中的溶解度,因此,本发明提供的方法能快速、准确的模拟CA的溶解吸附行为,并可根据其辅助开发高分子表面活性剂。为了更好的理解本发明的技术方案和技术效果,以下将结合流程示意图对具体的实施例进行详细的描述。如图1所示,本发明方法包括以下步骤:步骤S01,确定高分子表面活性剂的分子构型,并将高分子链的单体原子划入同一原子基团。在本发明实施例中,表面活性剂分子的构型可以为多点半自由链。一般地,用于CMP体系的高分子CA可以简化为链状高分子聚合物,常见的聚合物链有自由连接链、多点半自由链、旋转异构链等。多点半自由链在描述真实高分子体系过程中具有很高的模拟精度,因此,本实施例主要以多点半自由链为示例。为了减少后续模拟过程的计算量,往往需要对高分子链构型进行简化,在本实施例中,通过采用联合原子基团的方式,将高分子链的单体原子划入同一原子基团,并将原子基团作为后续模拟过程中的作用点,从而在不失模拟精度的前提下,减小模拟复杂度。其中,所述单体指有聚合能力的低分子,通过单体间的聚合反应可以合成聚合物,聚合物一般由单体结构重复构成。例如,一个C原子链接两个H原子,可将这三个单体内原子联合为一个原子基团。步骤S02,根据高分子表面活性剂分子的几何结构信息,获取高分子表面活性剂的原子基团间分子内相关函数。在本实施例中,由于根据CA分子的实际几何结构信息,例如键长、键角、键能等,计算CA的分子内相关函数。进一步的,为了保证该相关函数的精确度,可以采用生成矩阵法来获取CA的分子内相关函数。在一个具体实施例中,获取CA分子的键长、键角、键能等几何结构信息参数,根据上述几何结构参数,通过生成矩阵法获取分子内相关函数ω(r)。所述生成矩阵法是高分子链构象统计理论中求解高分子均方末端矩、回转半径等结构特征的数学工具。步骤S03,根据原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂分子间相关函数。在本实施例中,PRISM理论模型,主要是建立高分子CA分子内和分子间的相关函数的内在关联,通过PRISM理论模型获取CA分子间相关函数g(r),具体的数学表达式如式(1)所示:H^(k)=Ω^(k)C^(k)[Ω^(k)+H^(k)]---(1)]]>其中,ρα为α作用点的数密度,为分子内相关函数ω(r)的傅里叶变换形式。分子间相关函数g(r)和总相关函数h(r)之间的关系为:g(r)=h(r)+1。该理论模型需要用到近似方程,常见的近似方程有超网链近似、PY近似及平均球近似等。在一个具体实施例中,对分子内相关函数ω(r)进行傅里叶变换,可以获得k空间分子内任意两原子基团α、γ之间相关函数的表达式,如式(2)所示:ω^αγ(k)=sin(Bαγk)Bαγke-Aαγ2k2---(2)]]>其中,Aαγ2=<rαγ2>(1-Cαγ)/6;Bαγ2=Cαγ<rαγ2>;Cαγ2=12(5-3<rαγ4><rαγ2>2).]]>和分别为二阶矩和四阶矩,可以通过生成矩阵法获得。PRISM理论模型如式(1)所示,可以通过快速傅里叶变换结合迭代算法求解。其中,为k空间的原子基团间直接相关函数,其和C(r)可通过傅里叶变换进行相互转换。近似方程是PRISM理论模型的构成部分,本实施例采用PY近似,相应的C(r)数学表达式如式(3)所示:Cαγ(r)=(1+hαγ(r)-Cαγ(r))(e-uαγ(r)/kBT-1)---(3)]]>其中,Cαγ(r)为α原子基团与γ原子基团间的直接相关函数,uαγ(r)为α原子基团与γ原子基团间的作用力场,kB为波尔滋曼常数,T为绝对温度。此外,本实施例还可以采用其他近似,例如超网链近似、平均球近似等,从而可以比对不同近似方法对高分子CA的溶解吸附行为的影响。S04,根据分子间相关函数获取小分子在高分子表面活性剂中的溶解度。在本发明实施例中,由于小分子在高分子表面活性剂中的溶解度和化学位相关,而化学位可通过分子间相关函数获取,因此,可以先通过分子间相关函数计算化学位,然后再根据化学位模拟小分子在高分子表面活性剂中的溶解度。需要说明的是,化学位包括:低压超额化学位和高压超额化学位,其适用的环境不同,以下以研磨液高分子CA为例,模拟小分子在高分子CA中的溶解度。由于研磨液的工作环境为低压,相应的采用低压超额化学位模拟小分子在高分子CA中的溶解度。具体如下所示:低压小分子溶解度的计算公式如式(4)所示:S=1kBTe-1kBT(μref+μatt)---(4)]]>其中,μref和μatt分别为参考项及吸引项化学位,S为溶解度。μref和μatt的数学表达式分别如式(5)、式(6)所示:μref=kBTπ2Σα∈A,γ∈Bdαργ∫01[(dγ+λdα)2gαγhs(λ)(dγ+λdα2)]dλ---(5)]]>μatt=kBT4πΣα∈A,γ∈Bργ∫σαγ∞[r2uαγ(r)gαγatt(r)]dr---(6)]]>其中,dα为α作用点硬球直径,σαγ为α与γ作用点间软球直径,ργ为γ作用点分子数密度,和分别为两作用点间硬球项和吸引项分子间相关函数。A代表小分子,B代表CA。对于高压超额化学位,具体数学公式如式(7)所示:S=1kBTe-1kBT(μref+μatt-lnz)---(7)]]>其中,z为小分子气相逸度,而参考项μref和吸引项μatt的数学表达式与低 压小分子溶解度中表示不同,分别如式(8)、式(9)所示:μref=kBTπ2{Σα∈A,γ∈Bdαργ∫01[(dγ+λdα)2gαγhs(λ)(dγ+λdα2)]dλ+Σα∈A,α′∈A(dα+dα′)ρα′∫01[(λdα′+λdα)2gαα′hs(λ)(λdα′+λdα2)]dλ}---(8)]]>μatt=kBT4π{Σα∈A,γ∈Bργ∫σαγ∞[r2uαγ(r)gαγatt(r)]dr+Σα∈A,α′∈Aρα′∫σαα′∞[r2uαα′(r)gαα′att(r)]dr}---(9)]]>进一步的,可以通过平均力位能Wαγ(r)对原子基团间作用力场uαγ(r)进行修正,以提高PRISM理论模型的自洽性,进而提高模拟的精确度。其中,平均力位能Wαγ(r)是介质诱导产生的、能影响高分子链分子间相互作用的势能,Wαγ(r)与结构因子及分子内、分子间作用相关,其数学表达式如式(10)所示:Wαγ(r)=-kBTΣijC^αi(k)ρS^ij(k)C^jγ(k)---(10)]]>其中,ρ为:作用点的数密度,kB为波尔滋曼常数,T为绝对温度,为结构因子。高分子CA的结构因子的数学表达式如式(11)所示:S^(k)=Ω^(k)+ρ~h^(k)=(I-ρΩ^(k)C^(k))-1Ω^(k)---(11)]]>其中,ρ为作用点的数密度,为分子内相关函数,是ω(r)的傅里叶变换形式,为k空间的原子基团间直接相关函数,其和C(r)可通过傅里叶变换进行相互转换。通过高分子CA的结构因子计算出平均力位能,然后可以通过平均力位能Wαγ(r)对原子基团间作用力场uαγ(r)进行修正,以达到提高PRISM理论模型的自洽性,进而提高模拟的精确度。S05,根据溶解度辅助高分子表面活性剂开发。在本发明实施例中,可以根据S04步骤获得的小分子溶解度预测其溶解吸附行为,并据此辅助高分子表面活性剂的开发,例如,在碱性研磨液的开发过程中,由于氧化剂双氧水不太稳定,很容易发生分解,生成水和氧气,而氧气在研磨液中的溶解在一定程度上会影响研磨液的性质以及研磨过程中的效果。一般地,在研磨液中添加磷酸等可以抑制双氧水的分解。事实上,活性剂的添加在一定程度上也会吸附小分子及其他杂质分子,从而可以降低此类杂质分子对研磨液结构和性质的影响。比如现在有三种待 选择的高分子CA,首先固定模拟过程中CA的密度参数和温度参数,通过更改氧气的密度参数,分别模拟氧气在三种CA中的溶解度参数,选择溶解度参数最大的一种,比如是CA1;然后轻微改变CA的温度参数,模拟温度变化对以上三种CA对氧气溶解度参数的影响,选择出影响最大的一种,比如是CA2;从密度变化及温度变化中比对二者对氧气溶解度的影响,选择出吸附能力最强的一种CA(比如CA1)作为备选CA,或者根据一定权重选择CA1与CA2的混合组分作为研磨液CA的添加剂,然后进行相关CMP实验以优化CA的开发。此外,还可以通过对比S04步骤获得的当前CA的小分子溶解度与现实中各领域所需CA的小分子溶解度,然后采用当前CA在溶解度相近领域进行实验,验证模拟的结果,可以大大降低高分子CA的开发周期。本发明实施例提供的确定表面活性剂溶解吸附行为的方法,通过把高分子CA的高分子链的单体原子划入同一原子基团,并以原子基团为作用点,能有效降低后续模拟过程中的计算时间,并且根据高分子CA的几何结构信息获取原子基团间分子内相关函数,通过PRISM理论模型获取高分子CA的分子间相关函数,保证了据此进行模拟获得的溶解度的精确度,可以满足实际应用中模拟获取小分子在高分子CA中溶解度的要求,因此可以根据该获取方法辅助高分子表面活性剂的开发。相应地,本发明还提供了与上述方法对应的确定表面活性剂溶解吸附行为的系统,如图2所示,包括:构型确定模块201,用于确定高分子表面活性剂的分子构型;原子基团划分模块202,用于将高分子链的单体原子划入同一原子基团;几何结构信息获取模块203,用于获取高分子表面活性剂分子的几何结构信息;分子内相关函数获取模块204,用于根据高分子表面活性剂分子的几何结构信息,计算高分子表面活性剂的分子内相关函数;分子间相关函数获取模块205,用于根据已获得的原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂分子间相关函数;溶解度模拟模块206,用于根据分子间相关函数获取小分子在高分子表面活性剂中的溶解度;开发模块207,用于根据溶解度辅助高分子表面活性剂的开发。在实际应用中,由于分子内相关函数的精度在PRISM理论模型中具有重要意义,因此要求所述分子内相关函数获取模块204获取的参数精度较高,为了提高分子内相关函数的计算精度,在本发明实施例中,所述分子内相关函数获取模块204包括:转换单元,用于将各原子作用点以联合原子基团的方式转换为各原子基团作用点。由于后续的模拟过程以原子基团为作用点,能进一步有效降低后续模拟复杂度;原子基团间分子内相关函数获取单元,用于根据高分子表面活性剂分子的几何结构信息,通过生成矩阵法获得高分子表面活性剂的原子基团间分子内相关函数。需要说明的是,PRISM理论模型需要根据近似方程以获取分子间相关函数,因此所述分子间相关函数获取模块205可以包括:作用力场确定单元,用于确定各原子基团间的作用力场;相关函数近似方程获取单元,用于根据PY近似及各原子基团间的作用力场确定相关函数近似方程;分子间相关函数获取单元,用于根据相关函数近似方程及原子基团间分子内相关函数,通过PRISM理论模型获取高分子表面活性剂分子间相关函数。进一步的,为了提高系统的精确度,可以对系统进行修正,例如,所述作用力场确定单元还可以包括:结构因子获取子单元,用于根据分子间相关函数获取高分子CA结构因子;平均力位能获取子单元,用于根据结构因子、直接相关函数获取平均力位能;修正子单元,用于根据平均力位能与高分子表面活性剂的分子内、分子间作用的相关性,修正各原子基团间的作用力场。在实际应用中,由于小分子在高分子表面活性剂中的溶解度可以通过小分子在CA中的化学位获得,而小分子在CA中的化学位和分子间相关函数具有关联性,因此,溶解度模拟模块206可以包括:化学位获取单元,用于根据分子间相关函数获取小分子在高分子CA中的低压超额化学位和/或高压超额化学位;溶解度模拟单元,用于根据低压超额化学位和/或高压超额化学位模拟小分子在高分子CA中的溶解度。本发实施例明提供的确定表面活性剂溶解吸附行为的系统,通过原子基团划分模块202简化高分子模型,然后根据几何结构信息获取模块203,来获取高分子相关信息,并通过分子内相关函数获取模块204,获取高分子CA的分子内相关函数,可根据其通过PRISM理论模型获取高分子CA分子间相关函数。由于以原子基团作为作用点,使得根据分子间相关函数获取小分子在高分子CA中溶解度的复杂度较传统分子模拟的复杂度显著降低,这样,该系统不但能保持较高的模拟精度,而且能大大提高模拟速度,是速度和精度的有效结合,可以满足实际应用中高分子CA的开发需求,准确快速的模拟小分子在当前CA中的溶解度。当然,在实际应用中,该系统还可进一步包括:存储模块(未图示),用于保存任意模块和/或任意单元获得的参数,比如:小分子在当前高分子CA中的溶解度、分子内相关函数等。这样,可以根据已有的信息,以降低高分子CA的开发周期。本说明书中的各个实施例均采用递进的方式描述,各个实施例之间相同相似的部分互相参见即可。尤其,对于系统实施例而言,由于其基本相似于方法实施例,所以描述得比较简单,相关之处参见方法实施例的部分说明即可。以上所描述的系统实施例仅仅是示意性的,其中所述作为分离部件说明的单元可以是或者也可以不是物理上分开的,作为单元显示的部件可以是或者也可以不是物理单元,即可以位于一个地方,或者也可以分布到多个仿真窗口上。可以根据实际的需要选择其中的部分或者全部模块来实现本实施例方案的目的。本领域普通技术人员在不付出创造性劳动的情况下,即可以理解并实施。以上对本发明实施例进行了详细介绍,本文中应用了具体实施方式对本发 明进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及系统;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1