一种蟾毒配基纳米晶体及其制备方法与流程
本发明属于医药技术领域,特别是涉及一种蟾毒配基纳米晶体及其制备方法。
背景技术:
蟾酥bufonisvenenum源于《本草衍义》,为我国传统中药。《中华人民共和国药典》(2015年版,一部)明确记载了蟾酥是其基源动物蟾蜍科动物中华大蟾蜍bufobufogargarzanscantor或黒眶蟾蜍bufomelanostictusschneider的耳后腺及皮肤腺分泌的白色浆液,经加工干燥而成。蟾酥又名蟾蜍眉酥《日华子本草》、蟾蜍眉脂《药性论》、癞蛤蟆浆《新疆药材》、蛤蟆《中药材手册》、蛤蟆酥《山东中药》,其性味辛、温,有毒;有解毒、止痛、开窍醒神、消肿等功能,主要用于治疗痈疽疗疮、咽喉肿痛、中暑神昏、腹痛吐泻等病症。
蟾酥含有多种化学成分,其中蟾酥甾类和吲哚生物碱类是其主要活性成分。研究表明,蟾酥抗癌的有效成分主要是蟾酥甾类成分中的蟾毒配基类化合物,其中蟾毒灵、华蟾毒配基和脂蟾毒配基是蟾毒配基中含量较高、活性较强的三个成分,不仅具有抗肿瘤作用,而且还能提高机体免疫力,减轻化疗的毒副反应,降低癌痛发生率,增强机体对放疗、化疗的耐受性,但其水溶性极差,因此需通过相应的制剂工艺提高药物的溶出速率或表观溶解度,达到提高其生物利用度的目的。
目前,为了提高蟾酥的生物利用度,研究者采用了诸多方法,如以下文献所述:
中国发明专利《蟾酥滴丸及其制备方法》(公开号cn1660141a)公开了一种蟾酥滴丸制剂,是利用表面活性剂为基质,与白花蛇舌草的有效成分一起制成固体分散剂,以加快药物的溶解而提高其生物利用度。但是,该滴丸制剂中所含蟾酥有效成分为蟾酥的乙醇提取物,成分较复杂,并不能很好的保证蟾酥临床应用的安全性。
中国发明专利《蟾酥提取物在制备治疗人脑胶质瘤药物中的应用》(公开号cn105687251a)公开了蟾毒灵、华蟾酥毒基和脂蟾毒配基的含量大于90%的蟾酥提取物在制备治疗人脑胶质瘤药物中的应用,其中涉及了蟾毒配基脂质纳米粒、蟾毒配基纳米脂质体、蟾毒配基亚微乳、蟾毒配基纳米囊、蟾毒配基纳米球、蟾毒配基纳米乳或蟾毒配基聚合物胶束,并与蟾毒配基溶液剂进行了比较,结果证实了蟾毒配基纳米制剂达到了增效减毒的目的,但这些制剂制备工艺复杂,载药量较低,大多稳定性较差。
中国发明专利《基于蟾酥提取物的抗脑胶质瘤药物及其制备方法》(公开号cn105477020a)公开了一种蟾毒配基亚微乳及蟾毒配基纳米粒制剂,取得了较理想的抗脑胶质瘤效果,但该蟾毒配基亚微乳和纳米粒均存在载药量低、制备工艺复杂等缺点。
中国发明专利《一种蟾酥靶向脂质体及其制备方法和应用》(公开号cn105561304a)公开了一种蟾酥靶向脂质体,该蟾酥靶向脂质体能够有针对性的对癌细胞进行抑制同时减小了其毒副作用,由于该脂质体为注射剂,制备工艺复杂且不能消除其在注射时蟾毒配基对血管的刺激性,导致患者顺应性差。
综上所述,上述现有技术方法还没有达到有效改善蟾毒配基口服生物利用度的目的,特别是实现三种成分同步而快速地溶出,目前仍需要一种优良的方法以进一步改善蟾毒配基的溶出速率而提高其生物利用度。
技术实现要素:
本发明的目的就在于克服上述现有技术的缺陷,提供一种可实现蟾毒配基中蟾毒灵、华蟾毒配基和脂蟾毒配基同步而快速溶出,从而提高蟾毒配基生物利用度,且稳定性好,制备工艺简单的蟾毒配基纳米晶体。
本发明的另一目的是提供上述蟾毒配基纳米晶体的制备工艺。
为实现上述发明目的所采取的技术方案为:
一种蟾毒配基纳米晶体,其特征在于该蟾毒配基纳米晶体是将蟾毒配基、电荷稳定剂、空间稳定剂和纯化水混合后经纳米化处理后所得,其载药量为20%~90%,粒径分布范围为30~1000nm。
所述蟾毒配基纳米晶体中蟾毒配基的总浓度为1mg/ml~500mg/ml;电荷稳定剂的质量体积浓度为0.01%~2.0%;空间立体稳定剂的质量体积浓度为0.01%~5.0%。
所述电荷稳定剂的质量体积浓度为0.10%~0.22%;空间立体稳定剂的质量体积浓度为0.10%~0.30%。
所述蟾毒配基是由蟾毒灵、华蟾毒配基或/和脂蟾毒配基组成的无定形态或微晶态的物质,且其总纯度为20~98%,其中由蟾毒灵、华蟾毒配基和脂蟾毒配基三种物质组成时,蟾毒灵占4~15%,华蟾毒配基占6~30%,脂蟾毒配基占10~53%。
所述电荷稳定剂为十二烷基硫酸钠、泊洛沙姆188、泊洛沙姆407、大豆卵磷脂、海藻糖和壳聚糖中的一种或几种。
所述空间稳定剂为羟丙基甲基纤维素e5、羧甲基纤维素钠、交联羧甲基纤维素钠、聚维酮k30、羟丙基纤维素和交联羧甲基淀粉钠中的一种或几种。
所述蟾毒配基纳米晶体的粒径范围为30~200nm,电位值范围为-58~30mv。
所述蟾毒配基纳米晶体经表面修饰或者包衣、干燥处理后,与适宜药用辅料混制成药剂学上可接受的各种剂型。
所述表面装饰是指利用α-生育酚琥珀酸聚乙二醇酯、壳聚糖及其衍生物以物理和/或化学的方式改变蟾毒配基纳米晶体的表面结构和状态。
所述剂型为注射剂、片剂、口服液、微丸或者胶囊剂。
所述纳米化处理是指采用湿法研磨法或者高压均质法。
上述蟾毒配基纳米晶体的制备方法,其特征在于其工艺步骤为:
1)将电荷稳定剂、空间稳定剂和纯化水充分混合,形成稳定剂溶液a;
2)将蟾毒配基加入到上述稳定剂溶液a中,通过磁力搅拌使药物混悬于其中,得蟾毒配基粗混悬液b;
3)将上述所得蟾毒配基粗混悬液b转移至纳米研磨机/高压均质机中进行研磨/均质而得到蟾毒配基纳米晶体。
所述纳米研磨机为循环式纳米研磨机,所用研磨珠的粒度为0.2~0.8mm,材质为玻璃、钇稳定的氧化锆或聚苯乙烯衍生物的聚合物。
所述研磨过程中,研磨时间控制在15~60min,研磨转速控制在1000~6000rpm。
所述高压均质机为超高压纳米均质机,均质压力为50~200mpa、均质温度为5~60℃,均质次数为2~20次。
本发明具有以下技术优势:
1、本发明采用电荷稳定剂、空间稳定剂与蟾毒配基混合制备蟾毒配基纳米晶体,充分利用电荷稳定剂所具有的1)提供电荷斥力或与药物分子以氢键等方式结合而减弱药物粒子的聚集,2)增加药物的溶解度,3)抑制药物在分散、破碎过程中产生泡沫,从而增加药物与药物、药物与研磨腔壁、药物与研磨介质间的碰撞作用等特性;以及空间稳定剂所具有的1)增大空间位阻以防止药物粒子聚集,2)在一定时间内可维持药物分子以过饱和状态存在,3)加快由该纳米晶体所制备的其他固体制剂的崩解以利于药物快速溶出等特性,将由表面活性剂或聚合物构成的电荷稳定剂及空间稳定剂以物理吸附或氢键作用等方式包覆于蟾毒配基粒子表面,在阻止药物聚集的同时避免了药物分子与外界环境的接触,最终实现了制备效率和产率的提高,蟾毒配基中三种成分的快速而同步地溶出,蟾毒配基纳米晶体物理或/及化学稳定性的提高等等优势,实验表明:蟾毒配基纳米晶体在人工肠液中三种成分可按最初的质量比同步溶出,且溶出速率比蟾毒配基普通混悬剂提高了约1~10倍;在室温和低温条件下储存两个月,纳米晶体粒径和药物含量均无明显变化。
2、本发明采用纳米研磨技术对蟾毒配基进行处理,研磨过程中,药物粒子之间以及药物粒子与研磨介质、器壁间在高速剪切力、撞击力、摩擦力等作用下,将碰撞机械能转化为混悬液分子势能,使得药物粒径减小,比表面积增大,达到粒度均一的纳米晶体;同时通过药物与稳定剂的共研磨改善了难溶性药物蟾毒配基的体外溶出特性,并能充分的防止药物粒子的聚集,保证了混悬液的稳定性。
3、本发明采用高压均质机对蟾毒配基进行处理,均质过程中,药物颗粒在空穴、湍流、剪切、撞击力等共同作用下被粉碎成纳米颗粒,稳定剂同时也受此作用形成单分子界面,进而对药物纳米颗粒进行包裹,有效阻止了药物颗粒的聚集,采用此法制得的纳米晶体粒径均匀,稳定性好。
4、本发明采用的制备工艺简单,制得的纳米晶体载药量高,稳定性好,可批量化生产。
综上所述,本发明的蟾毒配基纳米晶体同步提高了蟾毒配基中三种成分的溶出速率,载药量高,稳定性好,为蟾毒配基的其他制剂提供了良好的中间体,且制备工艺简单、高效、安全,适应工业化生产。
附图说明
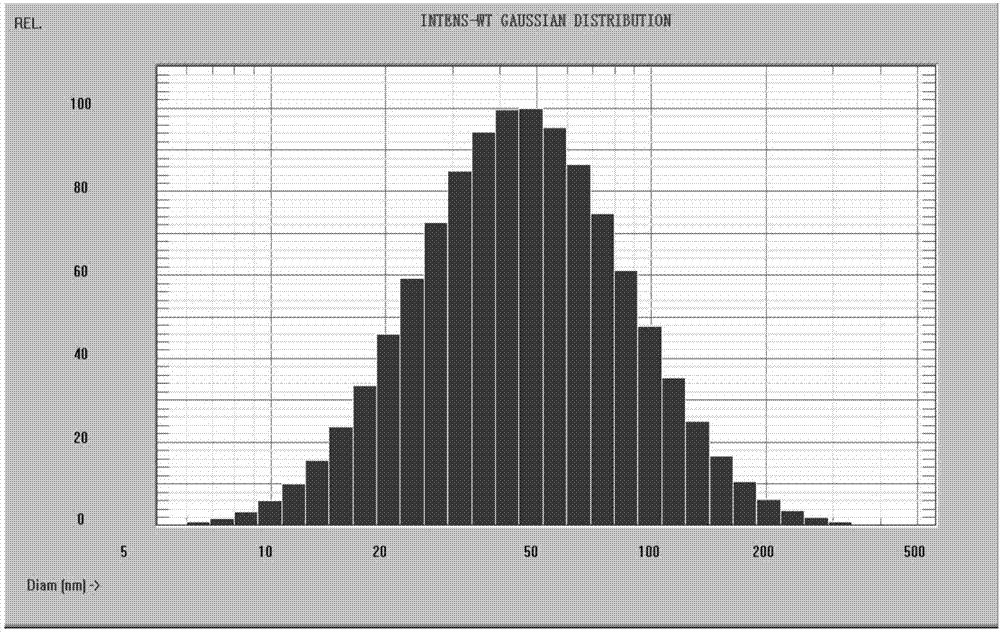
图1为本发明蟾毒配基纳米晶体的粒径分布图。
图2为本发明蟾毒配基纳米晶体的透射电镜图。
图3为本发明蟾毒配基纳米晶体在人工肠液中(ph=6.8)的溶出曲线图。
图4为本发明蟾毒配基纳米晶体在纯化水中的溶出曲线图。
图5为本发明蟾毒配基纳米晶体在醋酸缓冲液中(ph=4.5)的溶出曲线图。
图6为本发明蟾毒配基纳米晶体的红外光谱图(a为十二烷基硫酸钠,b为交联羧甲基纤维素钠,c为蟾毒配基原料药,d为物理混合物,e为纳米晶体)。
图7为本发明蟾毒配基纳米晶体的粉末x射线衍射图(a为十二烷基硫酸钠,b为交联羧甲淀粉钠,c为蟾毒配基原料药,d为物理混合物,e为纳米晶体)。
具体实施方法
下面用实例予以说明本发明,应该理解的是,实例是用于说明本发明而不是对本发明的限制。本发明的范围与核心内容依据权利要求书加以确定。
下列实施例中,蟾毒配基是由蟾毒灵、华蟾毒配基和脂蟾毒配基三种成分组成的无定形态或微晶态的混合物,总纯度为20~98%,其中蟾毒灵约占4~15%,华蟾毒配基约占6~30%,脂蟾毒配基约占10~53%。
实施例1:一种蟾毒配基纳米晶体的制备
称取十二烷基硫酸钠0.08g,交联羧甲基纤维素钠0.05g,加入50ml蒸馏水中,使其充分溶解溶胀,在磁力搅拌下加入0.5g蟾毒配基原料药,磁力搅拌30分钟,得粗混悬液。然后将此粗混悬液转移至纳米研磨机中进行研磨。研磨条件为:钇稳定的氧化锆研磨珠粒径为0.6-0.8mm,搅拌轴转速为2000rpm。30分钟后取样,测得粒径为30.2nm,电位为-47.9mv。
实施例2:一种蟾毒配基纳米晶体的制备
称取泊洛沙姆1880.05g,羟丙甲纤维素0.5g,加入50ml蒸馏水中,使其充分溶解溶胀,在磁力搅拌下加入0.8g蟾毒配基原料药,搅拌30分钟,得粗混悬液。然后将此粗混悬液转移至纳米研磨机中进行研磨。研磨条件为:聚苯乙烯聚合物研磨珠粒径为0.2-0.4mm,搅拌轴转速为1000rpm。60分钟后取样,测粒径为238.5nm,电位为-5.9mv。
实施例3:一种蟾毒配基纳米晶体的制备
称取大豆卵磷脂0.1g,聚维酮k300.05g,海藻糖0.02g,加入50ml蒸馏水中,使其充分溶解溶胀,在磁力搅拌下加入0.05g蟾毒配基原料药,超声30分钟,得粗混悬液。然后将此粗混悬液转移至纳米研磨机中进行研磨。研磨条件为:玻璃研磨珠粒径为0.3-0.5mm,搅拌轴转速为6000rpm。15分钟后取样,测粒径为973.8nm,电位为-20.9mv。
实施例4:一种蟾毒配基纳米晶体的制备
称取壳聚糖0.08g,交联羧甲基纤维素钠0.15g,加入50ml蒸馏水中,使其充分溶解溶胀,在磁力搅拌下加入0.5g蟾毒配基原料药,超声混悬20分钟,得粗混悬液。然后将此粗混悬液转移至高压均质机中进行均质。均质条件为:压力为200mpa,温度为60℃,循环2次,测粒径为720.8nm,电位为-20.9mv。
实施例5:一种蟾毒配基纳米晶体的制备
称取泊洛沙姆4070.1g,羟丙基纤维素0.05g,交联羧甲淀粉钠0.1g,加入50ml蒸馏水中,使其充分溶解溶胀,在磁力搅拌下加入0.6g蟾毒配基原料药,搅拌20分钟,得粗混悬液。然后将此粗混悬液转移至高压均质机中进行均质。均质条件为:压力位50mpa,温度为5℃,循环20次,测粒径为608.8nm,电位为-2.9mv。
实施例6:蟾毒配基纳米晶体的表面修饰:
称取0.1g的羟丙基三甲基氯化铵壳聚糖(hacc),将其溶解于100ml的蒸馏水中得0.1%的hacc溶液。将此hacc溶液于探头超声作用下缓慢加入到实施例1所制得的蟾毒配基纳米晶体中(纳米晶体与修饰液的体积比为1:4),然后磁力搅拌3小时,测定经修饰的纳米晶体的粒径及电位。
实施例7:蟾毒配基纳米晶体冷冻干燥粉末的制备:
取制备的蟾毒配基纳米晶体新鲜样品,加入蔗糖作为冻干保护剂,其浓度为1%(m/v),然后在-70℃预冻12小时,然后转移至冷冻干燥机中进行冻干,冷阱温度为-60℃,冷冻干燥时间为24小时,得其冻干粉。
实施例8:溶出度测定:
对蟾毒配基原料药和实施例2制得的蟾毒配基纳米晶体冷冻干燥粉末进行溶出度测定,方法如下:
采用《中国药典》2015版四部溶出度与释放度测定法(第二法),分别以900ml的纯化水、ph6.8磷酸盐缓冲液、ph4.5醋酸缓冲液作为漏槽条件下的溶出介质。溶出条件和实验操作如下:量取经过脱气处理的溶出介质900ml,注入1000ml溶出杯中,桨法,转速为50rpm,温度为(37±0.5)℃。待温度稳定后,分别向各溶出杯中投入样品,分别于0,5,10,15,30,45,60,90,120min取样5ml,用0.22μm的滤膜过滤,同时补加5ml新介质,取续滤液作为供试品溶液。另取蟾毒灵、华蟾毒配基对照品各3mg,脂蟾毒配基对照品约5mg,精密称定,溶于10ml甲醇后,取其中1ml以溶出介质定量稀释至10ml,作为对照品溶液。采用高效液相色谱法进行含量测定,计算蟾毒配基原料药和蟾毒配基纳米晶体冻干粉末中蟾毒灵、华蟾毒配基和脂蟾毒配基的溶出度,结果见图3。
- 还没有人留言评论。精彩留言会获得点赞!