利用二芳基碳酸酯制造脂肪族胺基二甲酸酯、脂肪族胺基多甲酸酯和聚脲的方法与流程
本发明是关于在不需任何催化剂存在下,由脂肪族二胺化合物、脂肪族多胺化合物,尤指脂肪族三胺化合物和脂肪族四胺化合物,或其混合物与二芳基碳酸酯制造脂肪族胺基二甲酸酯或脂肪族胺基多甲酸酯,并进一步制备聚脲的绿色合成方法。进一步研究发现,由一锅三步法可制得高性能的高分子聚脲(urea)弹性体或交联性聚脲热固型聚脲产物,所述方法更可在无溶剂下或水溶液中完成聚脲的制备,更加符合绿色化学的目的。
背景技术:
聚脲(Polyurea)和聚胺酯(Polyurethane;PU)皆为以异氰酸酯为原料进行逐步聚合反应(Step-growth polymerization)所合成的共聚物[1],常温下具有优异的机械性质[2],在日常生活中应用广泛,是现代社会中不可或缺的物质[3]。依照现行的PU制作方法,异氰酸酯(如MDI或HDI)为必要的聚合反应起始物[4]。然而,异氰酸酯毒性高,危害人体健康,Bhopal事件更是使异氰酸酯恶名昭彰,令人闻之色变;再则,在量产异氰酸盐的过程中[5],亦使用剧毒性的光气为主试剂,并产生大量腐蚀性的氯化氢气体副产物,造成环境安全、环境保护上的疑虑。因此,近四十年来,陆续有许多研究致力于非光气/非异氢酸盐(NPR/NIR)的全新聚胺酯-聚脲弹性体(Polyurethane-Polyurea Elastomer;PUaE)程序,若此绿色化学目标能成功实现,对于PUaE的永续发展更有正面意涵。
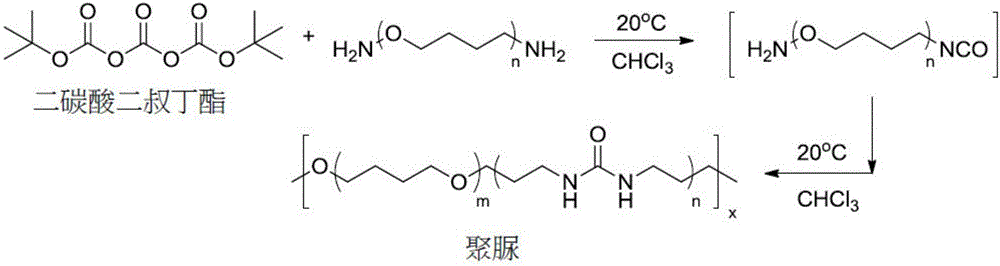
在非异氰酸酯法(NIR)合成PU的研究中,2005年和2006年Mejere[6]等人的研究最为突出,其使用二叔丁基三碳酸酯(Di-tert-butyl tricarbonate;DTBTC)取代异氰酸盐作为起始物,再与长链聚(四氢呋喃)-二胺(Poly(tertahydrofuran)-diamine)和短链二胺化合物,如1,2-乙烯二胺(1,2-ethylenediamine)反应合成聚脲,可在20℃的反应温度下的三氯甲烷溶液中进行,反应方程式如图1所示。Mejere在此研究中并举例,PUaE产物在热分析和机械性质分析中[7]所呈现的不同的弹性体特性可由DTBTC和长链与短链二胺化合物以不同莫耳比例进行调控而得。此论文亟符合NIR法合成的理想途径,但DTBTC价格昂贵,并非低廉的工业原料或试剂,无法普遍应用于工业化量产;且经NIR法合成的聚合物分子量不高(约68,000g/mol),因此绿色聚合物原料的选取,和聚合物合 成方法皆有改善深入研究的空间。
2011年C.E.Koning[8]等人的研究中使用天然原料(如1,4-丁二氨(1,4-Butanediamine,BDA))和催化剂,先合成中间体二甲酸酯,再进行聚合反应以合成聚脲,如图2所示,合成不同链段长的二甲酸酯,再接续以此不同链段长的二甲酸酯为原料、二甲基乙酰胺(DMAc)或N-甲基-吡咯啶酮(NMP)为溶剂,以聚丙二醇双(2-胺基丙基醚(Poly(propylene glycol)bis(2-aminopropyl ether))分子量400的PPGda-400或分子量2000的PPGda-2000为软链段,在130℃下反应4至24小时,合成聚脲。然而,所述聚合反应步骤繁琐又温度偏高。再以其所合成的二胺基甲酸酯(Biscarbamate)与PPGda-2000所合成的聚脲为例,分子量仅达35,700g/mol,拉伸率约42.4%,抗张强度为5.12MPa,物性偏低劣。但此文献以有机物作为催化剂,取代先前技术中使用的金属催化剂,并采用C4天然生质材料为原料,也部份符合绿色化学的原则。聚合时进行酯交换反应,并调整硬链段的长度,按所述硬炼段的长度决定PU的性质,在NIR的方法推进上有其贡献,但所述制法所获得的PUaE产率普遍偏低。
在较实用的含羰基原料的应用上,1979年Yamazaki[9]曾尝试以二苯基碳酸酯(Diphenyl Carbonate;DPC)为起始物与芳香族的伸甲基二苯胺(Methylenedianiline;MDA)进行反应聚合成聚脲,如图3所示,显示DPC取代光气有其优势和可能性,但在聚脲的合成产率(80%)和黏度上皆偏低(ηinh=0.23),而使聚脲的性能欠佳而缺乏市场竞争力,显示此酯交换的活性和合成条件有所不足,有待較优选条件的创新提升,此以二苯基碳酸酯合成的路线改进乃为本研究的主轴。
申请人多年来致力于非光气法的研究,已发展出一渐趋完善的非光气法/非异氰酸酯法制备异氰酸酯和聚脲的策略和化学程序,如图4所示。
申请人业已在2012年所发表的文献中[10]指出,以二苯基碳酸酯(DPC)为起始物与芳香族二胺化合物,如伸甲基二苯胺(Methylenedianiline;MDA)反应合成主要中间体二苯基芳香族二异氰酸酯,再经由热裂解或酯交换聚合反应合成高纯度的异氰酸酯和高分子量的聚脲。在聚脲合成的研究中得知,1997年B.Thavonekham[11]的研究显示二甲亚砜(DMSO)和环丁砜(TMS)为酯交换反应效率较高的溶剂,但是DMSO与副产物酚的沸点(180℃)接近,溶剂回收后不易与副产物分离,因此,申请人选用高沸点的TMS(289℃)做为进行酯交换聚合的溶剂[10],经由减压蒸馏(120~140℃,7x10-3mmHg)轻易与苯酚分离,以达溶剂和副产物均可分离回收再利用的绿色化学目标。
本发明更突破了以往的研究窠臼,直接以二芳基碳酸酯取代光气的官能基,尤其改进了Yamazaki直接以二芳基碳酸酯与二胺化合物进行酯交换聚合反应,成功地藉由溶剂 的改善和减压蒸馏的辅助,双管齐下促成具有高分子量的PUaE的生成。本发明亦完整研究不同脂肪族二胺、三胺或四胺化合物和其进料顺序对聚合物的结构和性质的影响,并出乎意料地发现合成性质优异而且软硬区块显著分离的聚脲的合成程序关键。
技术实现要素:
本发明提供一种一锅法的连续进料合成高分子量的聚脲产物的方法,所述方法在适当溶剂存在下,直接利用二芳基碳酸酯与一或多种脂肪族二胺、三胺、四胺化合物或其混合物反应。
为更符合绿色化学的目标,本发明亦提供一种熔融聚合法,其于无任何有机溶剂存在下制造聚脲,所述方法包含利用熔融的二芳基碳酸酯依序与不同的脂肪族二胺化合物、脂肪族三胺化合物、脂肪族四胺化合物或其混合物进行转脲化反应。
为拓展产物应用性,本发明更进一步提供一种制造水性聚脲的方法,所述方法包含在藉由上述方法获得聚脲预聚物,再加入选自由磺酸钠盐、3-氨基丙基三乙氧基硅烷,或其他含胺基的亲水分散剂,例如二羟甲基丙酸(DMPA)/三乙胺(TEA)离子基团)所组成的群组的界面活性剂,以提供做为水分散的官能基,并可配合再加入丁酮(methyl ethyl ketone;MEK)或丙酮(Acetone)降低产物溶液黏度后,成功将聚脲产物水性化。
本发明所揭示的本发明的每个态样和每个实施例意欲与所有其他所揭示的本发明态样和实施例个别地组合和组合成其所有可能的态样。
附图说明
图1为DTBTC与二胺反应聚合成聚脲的反应式。
图2为不同链段长的Biscarbamate的合成反应式。
图3为Yamazaki以DPC和MDA直接合成聚脲的反应式。
图4为本发明的非光气聚脲的制造方法。
图5为以TMS为溶剂合成聚脲弹性体的三段式合成法。
图6为聚脲弹性体水性化流程图。
图7为合成聚脲弹性体的FT-IR穿透率-波数变化图。
具体实施方式
在本说明书和申请专利范围中,除非上下文另外明确规定,否则单数形式「一」和「所述」包括复数。除非另外主张,否则使用本文所提供的任何和所有实施例或例示性 语言(例如「诸如」)仅欲更好地说明本发明,而不对本发明的范畴形成限制。本说明书中的语言不应解释为指示任何未主张的要素为实施本发明所必需。
<二芳基碳酸酯>
本发明方法中所使用的二芳基碳酸酯为由下式(1)表示的化合物:
其中R1和R2各自独立地为C6-C20芳基,且优选为C6-C12芳基。R1和R2可为未经取代或经一或多个脂族或芳香族取代基取代。在芳基具有两个或两个以上取代基的情况下,此等取代基可彼此相同或彼此不同。
R1和R2的取代基选自C1-C8烷基,例如甲基、乙基、丙基和丁基;C3-C12环烷基;C7-C15芳烷基,例如苯甲基和苯乙基;C6-C14芳基,例如苯基和甲苯基;C1-C12烷氧基(醚基),例如甲氧基、乙氧基、丙氧基、丁氧基和三氟甲氧基;C1-C12硫烷氧基,例如硫甲氧基和硫乙氧基;C6-C14芳氧基,例如苯氧基;卤素,例如氟、氯和溴;硝基;羟基;氰基;和二烷基胺基,例如二甲基胺基。
R1和R2可为例如苯基、萘基、蒽基、甲苯基、二甲苯基、乙基苯基、丙基苯基、辛基苯基、壬基苯基、十二烷基苯基、联苯、甲氧基苯基、氯苯基、二氯苯基、三氯苯基、五氯苯基、溴苯基、二溴苯基、三溴苯基、五溴苯基、硝基苯基、二硝基苯基、羟基苯基、氰基苯基和二甲基胺基苯基,上述基团可为未经取代的或经取代的。
此外,上述芳基包括其邻、间和对异构体,且连接至芳基的取代基包括正、异、第二和第三异构体。
特定言之,具有彼此相同且未经取代的芳基的二芳基碳酸酯可选自,但不限于二苯基碳酸酯、二-1-萘基碳酸酯、二-2-萘基碳酸酯和二-9-蒽基碳酸酯。
具有彼此相同且各自经至少一个烷基取代的芳基的二芳基碳酸酯可选自,但不限于双(2-甲苯基)碳酸酯和双[4-{第三丁基}苯基]碳酸酯。
具有彼此相同且分别经至少一个芳基取代的芳基的二芳基碳酸酯可为,但不限于双(4-联苯苯基)碳酸酯。
具有彼此相同且各自经至少一个烷氧基取代的芳基的二芳基碳酸酯可选自,但不限于双(2-甲氧基苯基)碳酸酯和双(3-丁氧基苯基)碳酸酯。
具有彼此相同且各自经至少一个卤素原子取代的芳基的二芳基碳酸酯可选自,但不限于双(2-氯苯基)碳酸酯、双(2,4-二氯苯基)碳酸酯和双(2,4,6-三氯苯基)碳酸酯。
具有彼此相同且各自经至少一个硝基取代的芳基的二芳基碳酸酯可选自,但不限 于双(2-硝基苯基)碳酸酯和双(2,4-二硝基苯基)碳酸酯。
具有未经取代的芳基和经至少一个烷基取代的芳基的二芳基碳酸酯可选自,但不限于3-甲苯基苯基碳酸酯和4-甲苯基苯基碳酸酯。
具有未经取代的芳基和经至少一个芳烷基取代的芳基的二芳基碳酸酯可为,但不限于4-苯甲基苯基(苯基)碳酸酯。
具有未经取代的芳基和经至少一个烷氧基取代的芳基的二芳基碳酸酯可选自,但不限于4-甲氧基苯基苯基碳酸酯和4-乙氧基-1-萘基苯基碳酸酯。
具有未经取代的芳基和经至少一个硫烷氧基取代的芳基的二芳基碳酸酯可选自,但不限于4-甲基硫苯基苯基碳酸酯。
具有未经取代的芳基和经至少一个芳氧基取代的芳基的二芳基碳酸酯可为,但不限于4-苯氧基苯基苯基碳酸酯。
具有未经取代的芳基和经至少一个卤素原子取代的芳基的二芳基碳酸酯可选自,但不限于2-氯苯基苯基碳酸酯和4-氯苯基苯基碳酸酯。
具有未经取代的芳基和经至少一个羟基取代的芳基的二芳基碳酸酯可选自,但不限于3-羟基苯基苯基碳酸酯和4-羟基苯基苯基碳酸酯。
适用于本发明方法的其他二芳基碳酸酯包括例如4-甲氧基苯基-4'-硝基苯基碳酸酯、4-氰基苯基-4'-硝基苯基碳酸酯、4-硫甲氧基苯基-4'-硝基苯基碳酸酯、2-氯苯基-4'-硝基苯基碳酸酯、2-二甲基胺基苯基苯基碳酸酯、2-溴-4-氰基-6-硝基苯基苯基碳酸酯,和五溴苯基-2',4',6'-三溴苯基碳酸酯。
在上述二芳基碳酸酯中,优选使用二苯基碳酸酯、双(2-甲苯基)碳酸酯、双(4-氯苯基)碳酸酯、双(4-硝基苯基)碳酸酯和双(3,5-二甲氧基苯基)碳酸酯,且較优选使用二苯基碳酸酯。
适用于制备本发明的脂肪族胺基二甲酸酯和聚脲的脂肪族二胺化合物是选自由脂肪族C1-C6烷基二胺、芳族C1-C6烷基二胺、分子量为400至3000的脂肪族聚醚二胺、分子量为400至3000的芳族聚醚二胺、二甲苯二胺和其组合所组成的群组,其包括醚二胺,诸如1,8-二胺基-3,6-二氧杂辛烷;长链聚醚二胺,诸如聚乙氧基化或聚丙氧基化二胺(D-2000);脂族二胺,诸如1,6-己二胺(1,6-HDA);环状脂族二胺,诸如异佛尔酮二胺(IPDA)或氢化MDA(H12MDA)。
适用于制备本发明的脂肪族胺基三甲酸酯和聚脲的脂肪族三胺化合物是选自由脂肪族C1-C6烷基三胺、芳族C1-C6烷基三胺、分子量为400至3000的脂肪族聚醚三胺、分子量为400至3000的芳族聚醚三胺和其组合所组成的群组。
适用于制备本发明的脂肪族胺基四甲酸酯和聚脲的脂肪族四胺化合物是选自由脂肪族C1-C6烷基四胺、芳族C1-C6烷基四胺、分子量为400至3000的脂肪族聚醚四胺、分子量为400至3000的芳族聚醚四胺和其组合所组成的群组。
<制造聚脲>
根据本发明,可由二芳基碳酸酯与脂肪族二胺化合物、脂肪族三胺化合物或脂肪族四胺化合物或其混合物直接采一锅法制备聚脲。在极性溶剂的存在下,或为熔融二芳基碳酸酯的状态下,藉由上述方法获得的反应中间体脂肪族胺基二甲酸酯、脂肪族胺基多元甲酸酯和脂肪族胺基四甲酸酯等脂肪族胺基多甲酸酯与脂肪族二胺化合物、脂肪族三胺化合物或脂肪族四胺化合物或其混合物反应来制造最终产物聚脲。
当以二芳基碳酸酯与脂肪族二胺化合物进行反应时,所获致的最终产物聚脲为热塑性聚合物;当以二芳基碳酸酯与脂肪族三胺化合物或脂肪族四胺化合物等多胺化合物进行反应时,所获致的最终产物为热固性聚合物。然当以二芳基碳酸酯与脂肪族二胺化合物和脂肪族三胺化合物和/或脂肪族四胺化合物的混合物进行反应,且其中脂肪族三胺化合物和/或脂肪族四胺化合物所占比例以全部脂肪族二/多胺化合物重量计约2至5%时,交联情况不明显,仅改变最终产物的物性,而不会改变其大体上呈现热塑性聚合物的性质;而当脂肪族三胺化合物和/或脂肪族四胺化合物所占比例以全部脂肪族二/多胺化合物重量计超过10%~50%时,交联情况趋明显,最终产物为热固性聚合物。
适用于根据本发明制造聚脲的转脲化方法的优选极性溶剂包括二甲基乙酰胺(DMAc)、N-甲基-吡咯啶酮(NMP)、二甲亚砜(DMSO)或环丁砜(TMS;沸点=289℃);优选使用DMSO或TMS;較优选为TMS。
TMS(沸点=289℃)在此制程中的实用性表现最佳,此是因为可在较高温度(140℃)下进行转脲化聚合反应,且同时向反应混合物应用减压。此外,发现自混合物中移除苯酚极利于加速聚合反应速率,从而在短时间内形成高分子量的聚合物。例如,在80℃下由于高分子聚合不足而不能在DMSO中产生聚脲弹性体膜的芳族二胺(诸如MDA)和环二胺(诸如H12MDA)藉由本发明方法以TMS为溶剂,能改善所欲合成的聚脲膜。
反应温度一般在约20℃至约200℃范围内,优选在约20℃至约160℃范围内,且較优选在约20℃至约100℃范围内。反应压力可为减压、常压或高压,其中常压為优选。
对反应时间不存在特别限制。反应时间一般为0.001小时至100小时,优选为0.005小时至50小时,且較优选为0.1小时至10小时。
已大体上描述本发明,可藉由参考某些特定实施例获得进一步了解,所述等实施例在本文中仅出于说明的目的而提供且除非另外规定,否则不欲具有限制性。
实施例
实施例1
以TMS为溶剂合成聚脲弹性体,硬链段比例为30%
将二苯基碳酸酯(17.14g,0.08mole)与约一半当量的HDA(4.65g,0.04mole)先加入三颈反应瓶中,以TMS(186g,固含量20%)为溶剂,使用机械搅拌使其混合均匀,于室温下进行反应1小时,先形成类似二苯基异氰酸酯的化合物,再加入聚丙二醇双(2-胺基丙基醚(Poly(propylene glycol)bis(2-aminopropyl ether)的长链聚醚二胺(26g,0.013mole),升温至90℃反应1小时,最后再加入IPDA(4.6g,0.027mole),使其在相同温度下反应1至2小时,可在FT-IR中观察到特征峰1781cm-1完全消失而后生成特征峰于1640cm-1(如图7),接续减压蒸馏下将反应温度缓慢升至140℃,历时约1至2小时并同时蒸出酯交换所生成的苯酚,直至无蒸馏物(苯酚和TMS的混合物)为止,以促进酯交换的完成。待蒸馏反应步骤结束后,将产物倒入超纯水中析出,抽气过滤后,滤液进行减压浓缩,回收溶剂TMS,固体产物(SPUaE)则于真空烘箱120℃,6~12小时烘干移除其溶剂,如图5所示,其后再将产物(SPUaE)溶于NMP中,再倒入铁氟龙(Teflon)皿中,循环烘箱60℃,2~3天制成膜,并进行TGA、DSC、GPC、固有黏度和机械性质等测试,如表1所示。
表1-以非异氰酸盐三步法合成聚尿素高分子的各项性质比较
实施例2
利用熔融法合成聚脲弹性体
进行与实施例1相同的程序、试剂量和分析,除了不添加溶剂(TMS)和起始物二苯基碳酸酯为熔融态流体以外在约90℃下进行。产物所得的各项性质比较是如表2所示。由表2的数据可知,无溶剂状态下所得的产物具有优异的热稳定性和机械性质表现。
表2-无溶剂的环境中以非异氰酸盐三步法合成聚尿素高分子的各项性质比较
实施例3
以磺酸钠盐(ESA)为界面活性剂合成水性化聚脲
进行与实施例1相同的程序和分析,除了熔融法合成预聚物并使用磺酸钠盐取代部分链延长剂以外,反应过程如图6所示,于合成反应末了加入丁酮降低黏度以将聚脲转为水性。由表3可知,当磺酸钠盐的硫含量占全体产物的重量比为0.6%时,水性化的聚脲产物有较好的机械性质和热性质表现,如表3所示。
表3-以磺酸钠盐(ESA)做为界面活性剂的水性化聚脲弹性体各项性质比较
实施例4
以3--胺基丙基三乙氧基硅烷(APTES)为界面活性剂合成水性化聚脲
进行与实施例1相同的程序和分析,除了熔融法合成预聚物并使用3-胺基丙基三乙氧基硅烷取代部分链延长剂以外。由表4可发现,利用溶液凝胶法(sol-gel method)可成功降低产物的粒径。其中采用溶液凝胶法可使聚脲尾端的硅烷(silane)基进入水后,先发生水解反应生成硅醇基(silanol),移除水后,进而缩合形成硅氧烷(siloxane)。
表4-以APTES做为界面活性剂的水性化聚脲弹性体各项性质比较
根据本发明,藉由完全略去自二胺基甲酸酯制造异氰酸酯的步骤的新颖简化聚脲 合成方法实为可行的经济而有效的绿色化学合成方法,其不使用毒性异氰酸酯和金属催化剂,且可被广泛应用。
本发明所属技术领域中具有通常知识者应了解,可相当容易地利用上文揭示的概念与特定实施例可作为修改或设计其它结构或制程而实现与本发明相同的目的。本发明所属技术领域中具有通常知识者亦应了解,这类等效建构无法脱离后附的申请专利范围所界定的本发明的精神和范围。
参考文献:
[1]徐武军,高分子材料导论,五南图书出版股份有限公司(2004).
[2]Huang W.B.,Xiang,J.Y.,Lv P.and Li X.M.,”Study on Mechanical Properties Aging of Spray Pure Polyurea for Hydraulic Concrete Protection”,Advanced Materials Research,374-377,pp.1325-1329(2011).
[3]黄微波,喷涂聚脲弹性体技术,化学工业出版社(2005).
[4]Randall,D.and Lee,S.,The Polyurethanes Book,John Wiley:New York(2000)
[5]张丰志,应用高分子手册,五南图书(2003)
[6]Versteegen,Ron M.,Sijbesma,Rint P.,Meijer,E.W.,Macromolecules,38(8),pp.3176-3184(2005).
[7]Versteegen,Ron M.;Sijbesma,Rint P.;Meijer,E.W.,Macromolecules,39(2),pp.772-783(2006).
[8]Tang,D.,Mulder,D.J.,Noordover,B.A.J.,Koning,C.E.,”Well-defined Biobased Segmented Polyureas Synthesis via a TBD-catalyzed Isocyanate-free Route”,Macromol.Rapid Commun.,32,pp.1379-1385(2011).
[9]Yamazaki,N.and Iguchi,T.,“The reaction of diphenyl carbonate with amines and its application to polymer synthesis”,Polymer Science:Polymer Chemistry Edition.,17,pp.835-841(1979).
[10]Chen,H.Y.,Pan,W.C.,Lin,C.H.,Huang,C.Y.and Dai,S.A.,Journal of Polymer Research,19(2),pp.9754-9765(2012).
[11]Thavonekham,B.,”A Practical Synthesis of Ureas from Phenyl Carbamates”,Synthesis,pp.1189-1194(1997).
- 还没有人留言评论。精彩留言会获得点赞!