一种合成DNA的方法与流程
本发明涉及一种DNA合成方法,尤其涉及一种能够低成本合成DNA的方法。
背景技术:
DNA合成技术的发明极大地推动了合成生物学领域的进展,而随着合成生物学的深入发展,对DNA合成技术也提出了更多的挑战和要求。目前,随着高通量DNA测序技术的飞速发展,DNA测序的成本已经急剧下降,极大地推动了科研、医疗和生物技术等领域的发展。相比之下,DNA合成的成本却一直居高不下。为了降低合成成本,人们做出大量的努力,但是成本下降程度依然有限。
目前,在DNA合成中的主要成本之一是引物(或者寡核苷酸)的成本,例如,目前每个核苷酸的市场价格约为0.9元人民币。同时,用化学法合成的寡核苷酸不可避免地存在错误率,例如,大约有1/100概率会丢失核苷酸和1/400概率会插入非目标核苷酸。寡核苷酸中的错误会直接导致合成的DNA(或者基因)片段中带有大量的错误,并需要进行各种除错和测序验证的步骤,进一步增加了合成的成本。以现有市场上流行的合成方法来计算,由于各个步骤的成本(包括寡核苷酸成本、除错成本、克隆成本、测序成本和人力等成本)难以降低,所以造成了DNA合成的费用一直高居不下,也阻碍了生物相关行业的发展。
近年来,芯片合成寡核苷酸的技术得到了快速的发展,大大降低了寡核苷酸合成的成本(例如美国CustomArray Inc公司提供的合成价格折算下来,每个核苷酸不到0.3分)。目前,科学家们已经成功地利用芯片制备的寡核苷酸进行大片段DNA的合成(Zhou X.,Cai S.,et al.,Nucleic Acids Res.,2004,32(18):5409-5417;Borovkov A.Y.,Loskutov A.V.,et al.,Nucleic Acids Res.,2010,38(19):e180;Matzas M.,Stahler P.F.,et al.,Nat.Biotechnol.,2010,28(12):1291-1294;Eroshenko N.,Kosuri S.,et al.,Curr.Protoc.Chem.,2012)。然而,由于芯片制备的 寡核苷酸的错误率通常更高,用芯片制备的寡核苷酸进行DNA合成时,需要花费更多的精力来进行后续的除错(Saaem I.,Ma S.,et al.,Nucleic Acids Res.,2012,40(3):e23;Wan W.,Li L.,et al.,Nucleic Acids Res.,2014,42(12):e102)、测序筛选等步骤。也有一些方法来降低芯片制备的寡核苷酸的错误率,例如用高通量测序的方法从制备的寡核苷酸池中挑取正确的目的寡核苷酸用于后续的DNA合成(Matzas M.,Stahler P.F.,et al.,Nat.Biotechnol.,2010,28(12):1291-1294;Schwartz J.J.,Lee C.,et al.,Nat.Methods,2012,9(9):913-915),但是该方法的成本往往更高。同时,由于芯片制备寡核苷酸的特点是通量高(例如,美国CustomArray Inc公司提供12k和90k两种规模),常规DNA(或基因)合成的方法无法使用芯片制备的寡核苷酸;因此,芯片制备的寡核苷酸一般被加上标签,分为不同的组,然后利用特定的标签用PCR方法将各组寡核苷酸提取出来,进行后续的DNA(基因)合成(Eroshenko N.,Kosuri S.,et al.,Curr.Protoc.Chem.,2012)。这些操作都无疑增加了DNA合成的成本。因此,综合计算,该方法并没有大幅度降低基因合成的成本,因此目前也没有被商业公司广泛采用。
技术实现要素:
针对现有技术方案合成DNA成本较高的问题,本发明提供了一种新的合成DNA的方法。
本发明第一个方面是提供一种合成DNA的方法,所述合成DNA的方法的步骤包括:
提供寡核苷酸池,所述寡核苷酸池中含有至少两种寡核苷酸;
采用PCR方法,从寡核苷酸池中取出所需寡核苷酸序列组,并将所取出的寡核苷酸序列生成双链DNA;
将双链DNA进行酶切,在所述双链DNA末端形成互补的粘性末端(cohesive end);
将切割后的DNA片段进行连接;
高通量DNA测序,筛选出正确的DNA序列。
本发明第二个方面是提供另一种合成DNA的方法,所述合成DNA的方法的步骤包括:
提供寡核苷酸,所述寡核苷酸两端分别连接有标签DNA序列,得到寡核苷酸池;其中,所述寡核苷酸序列结构如下:
TagF-E1-S-E2-TagR
其中,TagF和TagR分别为两端的标签DNA序列,其特异性标记靶标DNA;E1和E2为酶切位点,S为靶标DNA序列;各个寡核苷酸序列中的E1和E2可以相同或不同,各个寡核苷酸序列的E1之间可以相同或不同,各个寡核苷酸序列的E2之间可以相同或不同;
利用TagF和TagR,通过PCR方法,从寡核苷酸池中取出所需寡核苷酸序列组,并将所取出的寡核苷酸序列生成双链DNA;
将双链DNA经过E1和E2进行酶切,在所述双链DNA末端形成互补的粘性末端;将切割后的DNA片段进行连接;
高通量DNA测序,筛选出正确的DNA序列。
本发明第三个方面是提供另一种合成DNA的方法,所述合成DNA的方法的步骤包括:
提供寡核苷酸,所述寡核苷酸两端分别连接有标签DNA序列,得到寡核苷酸池;其中,所述寡核苷酸序列结构如下:
TagF-E1-S-E2-TagR
其中,TagF和TagR分别为两端的标签DNA序列,其特异性标记靶标DNA;E1和E2为酶切位点,S为靶标DNA序列;各个寡核苷酸序列中的E1和E2可以相同或不同,各个寡核苷酸序列的E1之间可以相同或不同,各个寡核苷酸序列的E2之间可以相同或不同;
通过PCR方法,从寡核苷酸池中取出所需寡核苷酸序列组,并将所取出的寡核苷酸序列生成双链DNA;其中,每组寡核苷酸序列中,有至少两条序列结构如下:
TagF-E1x-S-E2x-TagR
TagF-E1y-S-E2y-TagR
其中,E1x、E2x、E1y、E1y之间可以相同或不同,并且可以与其他寡核苷酸序列的E1和E2相同或不同;并优选为E1x、E2x不同,E1y、E2y不同;更优选为E1x与其他寡核苷酸序列的E1不同,E2y与其他寡核苷酸序列的E2不同;更优选 为E1和E2y相同,但是与其他的E1和E2不同;
将双链DNA进行E1和E2酶切,在所述双链DNA末端形成互补的粘性末端;并且经过E2x、E1y酶切后,保留TagF-E1x-S和S-E2y-TagR;
将切割后的DNA片段进行连接;
并通过PCR方法在连接后的DNA片段两端引入一段末端核苷酸序列;
高通量DNA测序,筛选出正确的DNA序列。
本发明第四个方面是提供另一种合成DNA的方法,所述合成DNA的方法的步骤包括:
提供寡核苷酸,所述寡核苷酸两端分别连接有标签DNA序列,得到寡核苷酸池;其中,所述寡核苷酸序列结构如下:
TagF-E1-S-E2-TagR
其中,TagF和TagR分别为两端的标签DNA序列,其特异性标记靶标DNA;E1和E2为酶切位点,S为靶标DNA序列;
通过PCR方法,从寡核苷酸池中取出所需寡核苷酸序列组,并将所取出的寡核苷酸序列生成双链DNA;
将双链DNA进行E1和E2酶切,在所述双链DNA末端形成互补的粘性末端;将切割后的DNA片段进行连接;
并通过DNA连接方法在连接后的DNA片段两端引入一段接头序列,接头序列含有一段末端核苷酸序列和酶切位点E;其中两端的酶切位点E可以相同或不同;高通量DNA测序,筛选出正确的DNA序列。
本发明第五个方面是提供另一种优选合成DNA的方法,在上述任意方法的基础上,所述合成DNA的方法的步骤还包括:
筛选出正确的DNA序列后,采用根据所述末端核苷酸序列设计的引物,对所筛选出的正确的DNA序列进行PCR扩增,并将扩增产物进行E2x和E1y酶切或E酶切,得到目标序列。
其中,所述扩增产物中目标序列长度优选为至少50bp,更优选为至少75bp,更优选为至少80bp,更优选为至少90bp,更优选为至少95bp,更优选为至少100bp,更优选为至少110bp,如至少150bp,至少200bp,至少250bp,至少300bp,如300-500bp。
本发明第六个方面是提供上述任意一种合成DNA的方法的应用。
其中,所述应用可以是制备自然界中已经存在DNA序列,也包括人工设计或人工预测的、自然界中不存在的DNA序列。
其中,所述应用包括在基因修饰中的应用,如优化密码子、基因修正等。
其中,所述应用包括在制备基因突变株中的应用,所述基因突变株也包括SNPs和其它突变株。
其中,所述应用包括在制备克隆抗体(如克隆人-鼠抗体)和/或重组抗体中的应用。
其中,所述应用包括在制备cDNA中的应用;更优选为在制备微芯片中的应用。
其中,所述应用包括在制备核酸疫苗中的应用。
其中,所述应用包括在制备核酸药物中的应用。
其中,所述应用包括在制备基因改造生物中的应用。其中,所述生物可以是微生物、植物、动物中的任意一种或几种,也可以是上述生物的细胞、器官或组织中的任意一种或几种。
其中,所述应用包括在制备蛋白和/或重组蛋白中的应用。
其中,所述应用包括在制备抗体药物中的应用。
本发明上述内容中,所述寡核苷酸序列长度优选为≤500nt,更优选为至少≤400nt,更优选为≤300nt,更优选为≤250nt,更优选为≤200nt,更优选为≤1500nt,更优选为≤100nt,更优选为≤50nt,更优选为≤20nt。
本发明上述内容中,E1、E2、E1x、E2x、E1y、E2y和E可以分别独立地选自Type I、Type II、Type III型酶切位点中的任意一种或几种,比如AatII、Acc65I、AccI、AciI、AclⅠ、AcuI、AfeI、AflII、AflIII、Agel、AhdI、AleI、AluI、AlwI、AlwNI、ApaI、ApaLI、ApeKI、ApoI、AscI、AseI、AsiSI、AvaI、AvaII、AvrII、BeaI、BamHI、BanI、BanII、BbsI、BbvCI、BbvI、BccI、BceAI、BcgI、BciVI、BclI、BfaI、BfuAI、BglI、BglII、BlpI、Bme1580I、BmgBI、BmrI、BmtI、BpmI、Bpu10I、BpuEI、BsaAI、BsaBI、BsaHI、BsaI、BsaJI、BsaWI、BsaXI、BseRI、BseYI、BsgI、BsiEI、BsiHKAI、BsiWI、BslI、BsmAI、BsmBI、BsmFI、BsmI、BsoBI、Bsp1268I、BspCNI、BspDI、BspEI、BspHI、BspMI、BspQI、BsrBI、 BsrBI、BsrDI、BsrFI、BsrGI、BsrI、BssHII、BssKI、BssSI、BstAPI、BstBI、BstEII、BstNI、BstUI、BstXI、BstYI、BstZ17I、Bsu36I、BtgI、BtgZI、BtsCI、BtsI、Cac8I、ClaI、ScpCI、CviAII、CviKI-1、CviQI、DdeI、DpnI、DpnII、DraI、DraIII、EaeI、EagI、EarI、EciI、EcoNI、EcoO109I、EcoP15I、EcoRI、EcoRV、FatI、FauI、Fnu4HI、FoKI、FseI、FspI、HaeII、HaeIII、HgaI、HhaI、HincII、HindIII、HinfI、HinP1I、HpaI、HpaII、HphI、Hpy188I、Hpy188III、Hpy99I、HpyAV、HpyCH4III、HpyCH4IV、HpycCH4V、KasI、KpnI、MboI、MboII、MfeI、MluI、MlyI、MmeI、MnlI、MscI、MseI、MslI、MspA1I、MspI、MwoI、NaeI、NarI、NciI、NcoI、NdeI、NgoMIV、NheI、NlaIII、NlaIV、NmeAIII、NotI、NruI、NsiI、NspI、PacI、PaeR7I、PciI、PflFI、PflMI、PhoI、PleI、PmeI、PmlI、PpuMI、PshAI、PsiI、PspGI、PspOMI、PspXI、PstI、PvuI、PvuII、RsaI、RsrII、ScaI、SacII、SalI、SapI、Sau3AI、Sau96I、SbfI、ScaI、ScrFI、SexAI、SfaNI、SfcI、SfiI、SfoI、SgrAI、SmaI、SmlI、SnaBI、SpeI、SphI、SspI、StuI、StyD4I、StyI、SwaI、TaqaI、TfiI、TliI、TseI、Tsp45I、Tsp509I、TspMI、TspRI、Tth111I、XbaI、XcmI、XhoI、XmaI、XmnI、ZarI和酶切位点中的任意一种,更优选为Type II酶切位点中的任意一种或几种,如AlwI、ApaI、BamHI、BbsI、BglII、BsaI、BsmBI、BspQI、EcoRI、FokI、HindIII、KpnI、MmeI、NcoI、NdeI、NheI、NotI、SacI、SalI、SapI、SphI、XbaI和XhoO中的任意一种或几种,更优选为Type IIS酶切位点中的任意一种或几种,如AlwI、BbsI、BbvI、BfuAI、BsaI、BsmBI、BspMI、BspQI、FokI、HgaI、MmeI、SapI、SfaNI中的任意一种或几种。
本发明上述内容中,所述酶切优选为使用双链DNA限制性内切酶进行酶切,更优选为所述双链DNA限制性内切酶的识别位点在切割位点之外,并产生非回文性粘性末端(cohesive end)。其中,所述双链DNA限制性内切酶最优选为IIS型限制性内切酶(Type IIS限制性内切酶)。
本发明上述内容中,所述连接在DNA连接酶的存在下进行,其中,所述DNA连接酶优选为T4DNA连接酶、E.coli DNA连接酶中的任意一种或几种,并更优选为T4DNA连接酶。
本发明上述内容中,所述粘性末端长度优选为1-6nt,如3-5nt。
本发明上述内容中,TagF和TagR可以相同或不同。TagF和TagR长度优选 为5-50nt、更优选为5-45nt、更优选为8-40nt、更优选为10-35nt、更优选为15-30nt、更优选为15-25nt、更优选为15-20nt。
本发明上述内容中,所述末端核苷酸序列(DNA条形码,barcode)可以是随机序列,长度优选为5-50nt、更优选为5-45nt、更优选为8-40nt、更优选为10-35nt、更优选为15-30nt、更优选为15-25nt、更优选为15-20nt。
其中,所述末端核苷酸序列(DNA条形码,barcode)可以是至少包括PCR引物序列,并优选为随机引物序列,如优选为5-50nt随机序列、更优选为5-45nt随机序列、更优选为8-40nt随机序列、更优选为10-35nt随机序列、更优选为15-30nt随机序列、更优选为15-25nt随机序列、更优选为15-20nt随机序列。
应当理解的是:
本发明上、下文内容中,“寡核苷酸序列池”可以是含有利用现有化学方法合成的多条DNA寡核苷酸序列,一般为单链DNA序列;如:可以是利用芯片合成技术制备的寡核苷酸序列,并且可以是未经纯化的粗产物。
本发明上、下文内容中,“合成DNA”(或“合成基因”)是指利用化学法合成的寡核苷酸序列来合成一段较长的DNA序列,一般为双链DNA序列。
本发明上、下文内容中,“引物”可以是单链RNA或单链DNA,并优选为单链DNA。
本发明上、下文内容中,“高通量测序”(High-Throughput sequencing、Next Generation Sequencing,NGS)是相对于传统的桑格测序(Sanger Sequencing)而言的。目前高通量测序的主要平台代表为Illumina、Life Technologies(Thermo Fisher Scientific)、罗氏公司(Roche)和Pacific Biosciences的测序平台。
本发明中,在利用芯片等高通量制备的寡核苷酸进行DNA合成之后,引入高通量测序的方法对合成得到的每一条序列进行测序验证;并在每一条序列的两端加上末端核苷酸序列(DNA条形码),方便后续调取正确的DNA序列。本发明方法极大地结合了“芯片等高通量制备寡核苷酸的低成本”和“高通量测序解决高错误率”的优势,极大地降低了合成DNA的成本和费用。
附图说明
图1为本发明一种优选实施例中合成DNA方法流程示意图;
图2为本发明另一种优选实施例中合成DNA方法流程示意图。
具体实施方式
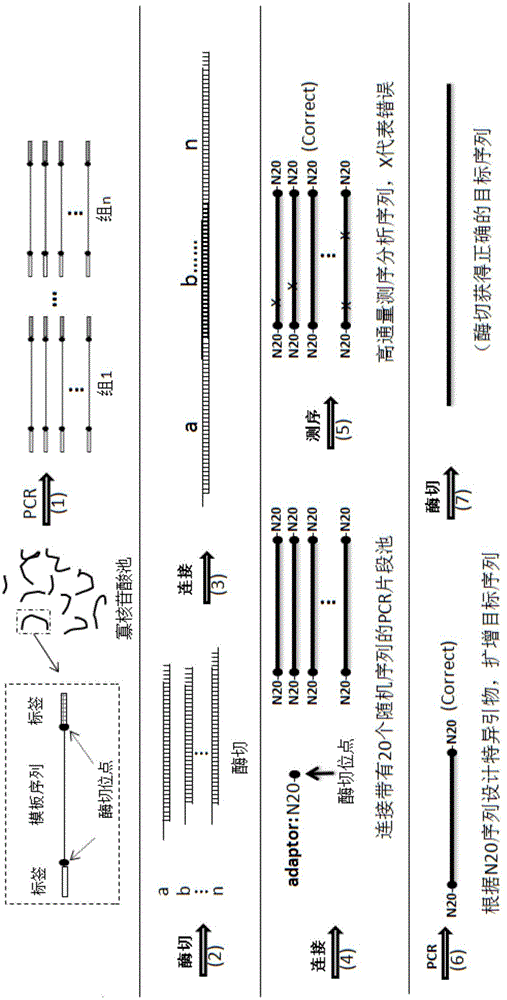
本发明人通过广泛而深入的研究,建立了一套DNA合成的技术方案:首先用PCR的方法从寡核苷酸池中调出每一组寡核苷酸(含2条、3条、4条、5条或以上寡核苷酸),并生成双链。然后利用IIS型限制性内切酶切割以产生互补的粘性末端,并利用连接酶把一组的DNA片段连接起来。在此基础上,利用高通量DNA测序方法,从连接好的DNA序列中筛选出正确的序列,并利用每条序列两端的DNA条形码(DNA barcode)将正确的序列扩增出来,以便于后续的进一步组装。实验结果表明,采用上述技术方案成功地进行了多个DNA片段的合成
下面结合附图、并参照具体实施例对本发明合成DNA的方法进行详细说明。
参照图1,首先,根据所需DNA序列结构,芯片合成不同的寡核苷酸序列,得到寡核苷酸序列池,每一条寡核苷酸序列两端含有一对标签序列(Tag),和一组酶切位点,结构如下:
TagF-E1-S-E2-TagR
其中,一般情况下,S长度在200nt以下。由于芯片合成寡核苷酸序列错误率较高,因此,寡核苷酸序列池中,含有一些错误的S序列。
通过PCR方法将寡核苷酸分组,并分别将各组取出。以第一组为例,该组中有两条核苷酸序列的一端带有第一酶切位点(不标有*的位点),另一端带有第二酶切位点(标有*的位点),其他寡核苷酸序列两端均为第一酶切位点。本实施例中,上述的酶切位点均为Type IIS酶切位点。将该组内的寡核苷酸合成双链DNA。
采用第一种Type IIS限制性内切酶(Enzyme 1)将合成的双链DNA进行酶切,第一种Type IIS限制性内切酶在上述序列第一酶切位点的外部进行酶切,去除第一酶切位点一端的标签序列,并在酶切位置产生非回文性、互补的粘性末端。酶切后,前一个序列的3’端和后一个序列的5’端有3-5个碱基的重复序列(粘性末端),便于酶切产物后续的连接。
将该组酶切后的序列进行连接,比如DNA连接酶可以采用T4DNA连接酶 或者其它类型的连接酶。
采用20nt随机引物(N20),对连接后的产物进行PCR扩增,生成的产物两端带有20nt的随机序列片段。因此,每一条DNA序列的5’端和3’端都含有长达20个碱基的随机序列,这些随机序列可以作为每一条DNA序列的特定DNA条形码(DNA barcode)。
PCR扩增产物纯化后,用高通量测序方法来确定每一条DNA的序列。根据测序分析的结果,利用正确序列的特定DNA条形码来设计一对特异的引物,并利用这些引物从PCR产物中扩增出正确的目标序列。利用第二种Type IIS型限制性内切酶(Enzyme 2)将两端带有的第二酶切位点和标签切除,得到正确的目标序列,所述目标序列还可以用于后续进一步拼接更长的序列。
在本发明的另一种实施例中,组内的每个寡核苷酸两端酶切位点可以相同,如图2所示,PCR筛选出的组内,进行酶切后,两端Tag序列均被去除。合成双链DNA后进行连接,与上述实施例不同的是,此时连接的DNA链的两端不存在Tag序列,接下来,合成含有20nt随机引物的接头,通过DNA连接方法在已经连接的DNA链两端添加末端随机核苷酸序列,并且在末端随机核苷酸序列与DNA链之间形成酶切位点E,高通量测序分析筛选出正确的DNA序列。根据需要,PCR扩增正确的DNA序列,并进行E酶切,得到目标DNA序列。
在本发明的另一种实施例中,组内每个寡核苷酸的E1和E2也可以各不相同,采用多种酶依次进行酶切。
根据上述实施例可以看出,本发明具有如下优点:
1、由于经典的寡核苷酸方法成本较高,导致DNA合成(基因合成)的价格高居不下。然而,芯片等高通量制备的寡核苷酸的错误率太高,导致后续除错、修复的成本也非常高。本发明中,在利用芯片等高通量制备的寡核苷酸进行DNA合成之后,引入高通量测序的方法对合成得到的每一条序列进行测序验证。并在每一条序列的两端加上DNA条形码,方便后续调取正确的DNA序列。该方法极大地结合了“芯片等高通量制备寡核苷酸的低成本”和“高通量测序解决高错误率”的优势,极大地降低了DNA合成的费用。
2、本发明中,寡核苷酸序列首先被PCR扩增生成双链DNA,然后通过酶切产生互补的粘性末端。互补的粘性末端可以进一步在连接酶的作用下进行连接,并 获得更长的DNA序列。相对于其他的方法,如聚合酶循环组装(polymerase cycling assembly,PCA)、Gibson assembly等,该方法的优点在于处理含有复杂DNA序列时,成功率高。
3、本发明利用了高通量DNA测序,从大量的DNA分子中调选出正确的DNA分子。由于DNA测序的通量非常大,可以保证挑选出正确的序列,因此,本发明的技术方案可以节省大量除错、修复突变的费用。在后续的测序验证环节,也可以减少目的克隆的验证数量,节约成本。相比于经典的DNA合成方法,本发明可节约成本60%以上,相比于芯片等高通量制备核苷酸合成DNA的方法,本发明可节约成本30%以上。
以上对本发明的具体实施例进行了详细描述,但其只是作为范例,本发明并不限制于以上描述的具体实施例。对于本领域技术人员而言,任何对本发明进行的等同修改和替代也都在本发明的范畴之中。因此,在不脱离本发明的精神和范围下所作的均等变换和修改,都应涵盖在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!