p97基因治疗病毒感染的用途及其相关药物的制作方法
本发明涉及生物
技术领域:
,具体涉及p97基因在治疗病毒感染的用途及其相关药物。
背景技术:
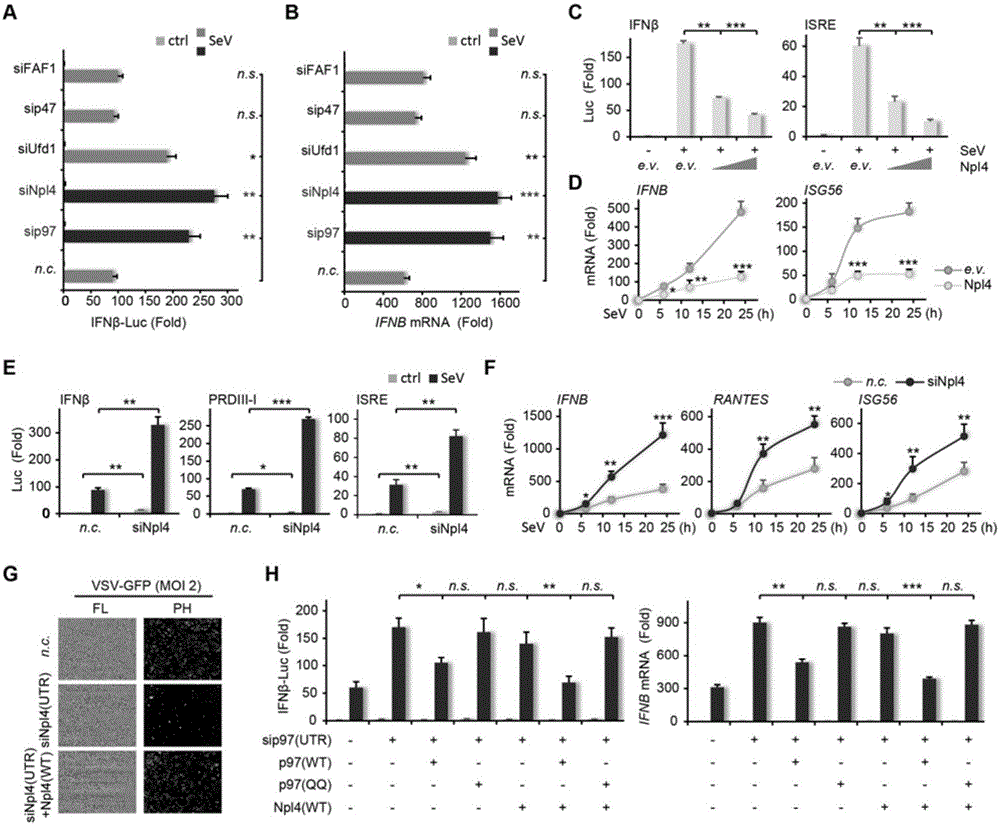
:病毒感染能够引起机体强烈的抗病毒免疫反应,天然免疫是机体抵抗病毒感染的第一道屏障,通过机体内的病原识别受体,包括toll样受体(TLR)、RIG-I样受体(RLRs)可以识别病毒核酸。被激活的受体可以启动下游转录因子NF-κB和干扰素调节因子IRF的信号转导途径,产生一系列促炎症细胞因子和I型干扰素(typeIinterferons,typeIIFN)。I型干扰素的释放能够促进下游基因的转录从而有效的抑制病毒的进一步复制。同时,I型干扰素的分泌还能够激活包括树突状细胞,NK细胞,单核细胞和巨噬细胞在内的参与固有免疫应答的细胞。此外,I型干扰素的分泌也能够直接激活T细胞和B细胞,进而产生适应性免疫应答反应。天然免疫的精确调控对于抗病毒感染以及防止自身免疫疾病的感染或机体产生免疫炎症都至关重要。泛素化是RLR受体介导信号通路的主要调控方式,以其代表性受体RIG-I为例,结合病毒RNA激活RIG-I产生构象变化,招募泛素连接酶,使RIG-I发生K63链泛素化,激活下游NF-κB以及IRF3的转录,促进促炎因子及I型干扰素的表达,增强其抗病毒活性。另一方面,泛素连接酶c-Cbl及RNF125使RIG-I发生K48链泛素化,终止该通路的免疫反应,抑制其抗病毒活性。p97属于II型ATP酶家族,它参与包括内质网及线粒体参与的蛋白降解,自噬,细胞膜重组,DNA修复,细胞周期以及性别决定等一系列的生理过程。p97与其辅助因子Npl4,Ufd1,p47,UBXD7以及FAF1结合,并利用其分离酶的活性将底物从其细胞环境中剥离,最终多数进入蛋白酶体降解。已有的研究表明p97与其辅因子Npl4组成复合物参与内质网及线粒体降解途径等一系列生理过程,p97-Npl4复合物能够通过介导IκBα的降解来促进TNFα诱导的NF-κB信号通路。近来也有研究发现p97参与TRIM21介导的病毒中和作用。但是现有技术中并未见p97在病毒感染免疫中的作用。技术实现要素:本发明的目的在于公开p97基因在治疗病毒感染中的用途及其相关药物。本发明通过广泛而深入的研究发现,p97与辅助因子Npl4结合形成的p97-Npl4复合物能够通过促进RNF125介导的RIG-I的K48链泛素化来促进RIG-I降解,从而抑制RIG-I介导的IFNβ信号通路,最终起到降低抗病毒感染能力的作用。为实现本发明的目的及其他相关目的,本发明采用以下技术方案:本发明的第一方面,提供了分离的p97基因在筛选病毒感染治疗药物中的用途。优选的,所述病毒感染为RNA病毒感染。更优选的,所述病毒感染为VSV病毒感染或SeV病毒感染。p97基因的核苷酸序列在Genebank中序列号为NM_007126.3的序列。所述p97基因筛选病毒感染治疗药物中的用途,是指以p97基因为作用靶点,一方面筛选能敲除或抑制该基因表达的物质作为病毒感染治疗候选药物;另一方面筛选能阻断或抑制p97与Npl4相互作用的物质作为病毒感染治疗候选药物。所述通过分离的p97基因筛选获得的病毒感染治疗药物包括但不限于:核酸分子、碳水化合物、脂类、小分子化学药、抗体药、多肽、蛋白或干扰慢病毒。所述病毒感染治疗药物的施用量为足够改变人p97基因的转录或翻译,或者足够改变人p97蛋白的表达或活性的剂量;或者有效阻断或抑制p97与Npl4的相互作用的剂量。采用前述病毒感染治疗药物治疗病毒感染的方法,主要是通过降低人p97基因或者有效阻断或抑制p97与Npl4的相互作用,从而抑制RIG-I降解,促进RIG-I介导的IFNβ信号通路,最终达到提高抗病毒感染能力、治疗病毒感染的目的。具体的,治疗时,将能抑制p97基因表达的物质或有效阻断或抑制p97与Npl4的相互作用的抑制剂者给药于患者。进一步的,本发明的第二方面,提供了一种筛选病毒感染治疗候选药物的方法,所述方法将p97基因作为作用靶标,筛选能敲除或抑制p97基因表达的物质作为病毒感染治疗候选药物。优选的,所述方法包括步骤:将测试物质施用于常规细胞,并与未施用测试物质的常规细胞进行比较,其中施用后导致p97基因表达水平降低的测试物质作为病毒感染候选药物。优选的,所述常规细胞选自293细胞,293T细胞或MEF细胞。优选的,所述p97基因的表达至少被降低50%、80%、90%、95%或99%。本发明的第三方面,还提供了另外一种筛选病毒感染治疗候选药物的方法,所述方法以p97基因为作用对象,筛选能阻断或抑制p97与Npl4相互作用的物质作为病毒感染治疗候选药物。优选的,所述方法包括步骤:采用AlphaScreen实验,筛选对p97与Npl4相互作用的抑制率>30%的测试物质作为病毒感染治疗候选药物。更优选的,所述方法包括步骤:(1)将带His标签的Npl4和生物素标记的p97混合,加入反应缓冲液,获得将反应体系,室温孵育;(2)将测试物质溶解于DMSO中,并采用稀释缓冲液稀释,施加于步骤(1)的反应体系中,室温避光孵育;(3)将镍螯合acceptorbeads加入步骤(2)的反应体系中,室温避光孵育,再加入等量的streptavidin交联的donorbeads,室温避光孵育;(4)于激光激发下测得步骤(3)所得反应体系中acceptorbeads所发出的荧光信号值记为A;并以未添加测试物质的反应体系作为阳性对照,所测荧光信号值记为B;(5)计算测试物质对p97与Npl4相互作用的抑制率(%)=B-A/B,将抑制率>30%的测试物质作为病毒感染治疗候选药物。优选的,步骤(1)中,所述反应缓冲液含有20mMHepespH7.5,100mMNaCl,0.1%BSA。优选的,步骤(1)中,带His标签的Npl4的终浓度为125nM,生物素标记的p97终浓度为31.25nM。优选的,步骤(2)中,所述稀释缓冲液含有20mMHepes,100mMNaCl,pH7.5。优选的,步骤(2)中,稀释液稀释后的测试物质的浓度为100μM,施加量为1μl。优选的,步骤(3)中,镍螯合acceptorbeads的终浓度为10μg/ml。本发明的第四方面,提供了一种治疗病毒感染的药物组合物,所述药物组合物包含治疗有效量的能够阻断或抑制p97与Npl4的相互作用的抑制剂。优选的,所述抑制剂通过促进RIG-I介导的IFNβ信号通路从而提高抗病毒感染能力进而起到治疗病毒感染的作用。优选的,所述抑制剂通过抑制RIG-I的蛋白酶体降解从而起到促进RIG-I介导的IFNβ信号通路的作用。优选的,所述抑制剂通过抑制RNF-125介导RIG-I的K48链泛素化从而起到抑制RIG-I的蛋白酶体降解的作用。优选的,所述抑制剂通过抑制RNF125与RIG-I之间的相互作用从而起到抑制RNF-125介导RIG-I的K48链泛素化的作用。本发明的优选实施方式中,所述抑制剂为通佐溴胺(ThonzoniumBromide)或盐酸表柔比星(EpirubicinHydrochloride)。其中,通佐溴胺(ThonzoniumBromide)的CAS编号为553-08-2,盐酸表柔比星(EpirubicinHydrochloride)的CAS编号为56390-09-1。优选的,所述治疗病毒感染的药物组合物的药效成分选自通佐溴胺(ThonzoniumBromide)或盐酸表柔比星(EpirubicinHydrochloride)中的一种或多种的组合。更优选的,通佐溴胺(ThonzoniumBromide)或盐酸表柔比星(EpirubicinHydrochloride)中的一种或多种的组合为所述治疗病毒感染的药物组合物的唯一药效成分。进一步的,治疗病毒感染的药物组合物还包含药学上可接受的赋形剂。术语“包含”指“含有”和“由﹍构成”,例如“包含”X的组合物可以完全由X构成,或者可以含有X之外的物质,例如X-Y。本文所用的术语“治疗有效量”指治疗剂治疗、缓解或预防目标疾病或状况的量,或是表现出可检测的治疗或预防效果的量。该效果例如可通过化学标记或抗原水平来检测。治疗效果也包括生理性症状的减轻。对于某一对象的精确有效剂量取决于该对象的体型和健康状况,病症的性质或程度、以及选择给予的治疗剂和/或治疗剂的组合。因此,预先指定准确的有效量是没用的。然而,对于某给定的症状而言,可以用常规实验来确定该有效量,临床医师是能够判断出来的。所述药学上可接受的赋形剂(或运载体)指用于治疗剂给药的赋形剂,其本身及其用量不应诱导接受该组合物的个体产生有害的抗体,且给药后没有过分的毒性。药学上可接受的赋形剂通常包括无毒的固体、半固体或液体填充剂、稀释剂、包裹材料或任何常规类型的制剂辅助物。合适的赋形剂包括但不仅限于水、葡萄糖、甘油、盐水、乙醇或其组合。所述赋形剂还包含有其他试剂如湿润剂或乳化剂、pH缓冲剂或增强制剂效力的佐剂。其他材料如抗氧化剂、保湿剂、粘度稳定剂和类似试剂可根据需要添加。脂质体也包括在药学上可接受的赋形剂的定义中。通常,可将药物组合物制成可注射剂,例如液体溶液或悬浮液;还可制成在注射前适合配入溶液或悬液中、液体载体的固体形式。本发明的第四方面,提供了一种治疗病毒感染的方法,包括对患者施用前述治疗病毒感染的药物组合物。本发明的有益效果为:本发明首次发现,p97与辅助因子Npl4结合形成的p97-Npl4复合物能够通过促进RNF125介导的RIG-I的K48链泛素化来促进RIG-I降解,从而抑制RIG-I介导的IFNβ信号通路,最终起到降低抗病毒感染能力的作用。基于此,筛选出了能够有效阻断或抑制p97与Npl4的相互作用的抑制剂,所述抑制剂通过促进RIG-I介导的IFNβ信号通路从而提高抗病毒感染能力进而起到治疗病毒感染的作用,极具临床和市场价值。附图说明图1.p97通过其ATP酶活性抑制IFNβ介导的抗病毒反应。(A)HEK293T细胞中转染p97小干扰RNA或对照RNA,SeV刺激12h后,IFNβ(左),PRDIII-I(中)以及ISRE(右)报告基因的转录活性。(B)在p97敲除的细胞中,SeV刺激后,IFNB(左),RANTES(中)以及ISG56(右)的转录水平。(C-D)p97敲除的细胞中,poly(I:C),poly(dA:dT),SeV或VSV-GFP处理后报告基因(C)以及Q-PCR(D)的结果。(E-F)转染了p97小干扰RNA的MEF细胞感染VSV-GFP病毒后,荧光相差显微镜检测(E)以及细胞流式实验结果(F)。(G-H)p97敲除细胞感染SeV后,以p97或其突变体进行rescue实验,检测IFNβ转录激活(G)以及IFNB的转录水平(H)。n.c.,表示对照siRNA;scr,表示对照shRNA;WT,野生型;PH,相差显微检测;FL,荧光显微检测(下同)。数据分析采用:标准误(三次独立平行实验的标准差),方差分析以及t-test。n.s.,无显著变化;*代表P<0.05;**代表P<0.01;***代表P<0.001。图2.Npl4协同p97抑制IFNβ介导的抗病毒反应。(A-B)转染了不同siRNA的细胞感染SeV后IFNβ启动子的转录活性(A)IFNB的转录水平(B)。(C-D)转染了不同剂量Npl4的细胞感染SeV后IFNβ(左)以及ISRE(右)启动子的报告基因的活性(C)以及IFNB(左)以及ISG56(右)的mRNA转录水平。(E-F)Npl4敲除的细胞中,SeV刺激后,报告基因(E)以及Q-PCR(F)的检测。(G)Npl4敲除的MEF细胞,感染VSV-GFP后的rescue实验。(H)敲除p97的细胞中转染不同质粒后,IFNβ转录活性以及IFNB转录水平。图3.p97-Npl4复合物结构以及突变分析。(A)Npl4及p97的结构域示意图。(B)果蝇Ter94-Npl4复合物的结构。相互作用界面上的残基以球-棒模型突出显示,括号中表示与果蝇该位点相对应的人源的氨基酸。(C)p97(左)-Npl4(右)相互作用的关键位点。(D)带有Strep标签的Npl4pulldown不同的p97突变体。(E)带有His标签的p97pulldown不同的Npl4突变体。(F)Npl4与p97或p973A突变体的免疫共沉淀。(G)p97与Npl4或Npl44A突变体的免疫共沉淀。(H-I)转染不同质粒的细胞中IFNβ的转录活性(H)以及IFNB转录水平(I)。3A代表p97-F52A/D55A/Y110A突变体;4A代表Npl4-I6A/R8A/R17A/R50A突变体。图4.p97-Npl4复合物促进RIG-I通过蛋白酶体降解抑制RIG-I信号通路。(A)poly(I:C)刺激p97敲除细胞后不同时间,通过免疫印迹实验分析RIG-I、MAVS、IRF3以及IRF3磷酸化。(B)poly(I:C)刺激过表达Npl4的细胞不同时间,通过免疫印迹实验分析蛋白表达水平。(C)poly(I:C)刺激沉默Npl4表达的细胞不同时间,通过免疫印迹实验分析RIG-I,MAVS,p-TBK1,TBK1,IKKε,IRF3以及IRF3磷酸化水平。(D)MG132处理p97敲除的细胞中过表达p97后,RIG-I的蛋白水平。(E)MG132处理细胞后过表达Npl4后RIG-I的蛋白水平。(F-G)敲除p97(F)敲除Npl4(G)的细胞SeV刺激后,RIG-I的泛素化水平。(H)敲除p97的细胞中转染p97野生型以及3A突变体后,RIG-I的泛素化水平。(I)转染了Npl4野生型以及4A突变体的细胞中,RIG-I的泛素化水平。图5.RNF125对于p97-Npl4复合物抑制RIG-I信号通路是必须的。(A)HEK293T细胞中转染对照组,敲除RNF125,敲除c-Cbl,或不同剂量的Npl4,在SeV刺激后,IFNβ荧光素酶报告基因的活性。(B)细胞中转染不同的RIG-ICARD结构域以及对照,敲除RNF125或敲除c-Cbl以及不同剂量的Npl4后,IFNβ荧光素酶报告基因的活性。(C)敲除RNF125的细胞中,转染或不转染Npl4的情况下,RIG-I的蛋白水平。(D)共转染Npl4与RNF125的细胞,SeV刺激后,RIG-I的泛素化水平。(E)SeV感染的细胞转染Npl4、RNF125后。(F)共转染RNF125以及HA-K48Ub后RIG-I泛素化位点的质谱分析。(G)共转染或分别转染Npl4、RNF125细胞中,转染野生型或突变体RIG-I后,RIG-I的免疫印迹分析。(H)共转染Npl4与RNF125的细胞中,转染野生型或突变体RIG-I后,RIG-I的泛素化水平。(I)共转染Npl4与RNF125的细胞中,转染野生型或突变体RIG-I后,IFNβ转录活性。(J)敲除Npl4以及RNF125的细胞中,转染RIG-I或其突变体后,VSV-GFP病毒感染细胞的荧光显微镜观察结果。图6.p97-Npl4复合物与RIG-I及RNF125相互作用。(A)SeV刺激及未SeV刺激情况下,细胞内Npl4与RIG-I免疫共沉淀。(B)SeV刺激及未SeV刺激情况下,Npl4与RIG-I的共定位。(C)体外pulldown实验验证Npl4与RIG-I的相互作用。(D-E)免疫共沉淀实验检测Npl4与RIG-I相互作用的具体结构域。(F)体外pulldown实验检测Npl4与RIG-I的CARD结构域的相互作用。(G)SeV刺激及未SeV刺激情况下,细胞内p97与RNF-125的免疫共沉淀实验。(H)体外pulldown实验检测p97与RNF125的相互作用。(I)体外pulldown实验检测p97-Npl4复合物对RIG-I与RNF125相互作用的影响。图7.pulldown实验验证小分子化合物破坏p97-Npl4相互作用。具体实施方式在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。可从Genebank获得人p97基因的核苷酸序列(NM_007126.3)及其氨基酸序列(NP_009057.1)。本文中同源基因指种间同源基因,不同物种中起源于一个共同的祖先基因的一些同源基因,这些基因通常保持相同的或相似的功能。根据NCBI网站BLAST的结果,人p97的同源基因包括小鼠VCP基因(NM_009503.4)和果蝇TEAR94基因(NM_001103780.2)。或者,可以“同源性”来定义本申请的同源基因。“同源性”指两条多核苷酸或两条多肽部分之间的相似性百分数。两条DNA或两条多肽序列在确定的分子长度内,当序列显示有至少约50%,优选至少约为75%、更佳至少约为80-85%、尤其佳至少约为90%、最佳至少约为95-98%序列相似性时,彼此“基本上同源”。如本文所述,基本同源也指与特定的DNA或多肽序列完全相同的序列。基本实验方法:1.目的基因的克隆:针对所需的目的基因设计引物,用PCR的方法将该目的基因从包含此目的基因的质粒或Hela细胞的cDNA中克隆出来,目的基因片段两边带有两个不同的酶切位点。用琼脂糖凝胶电泳的方法检测PCR产物的大小是否与所需的目的基因大小一致,并利用琼脂糖凝胶DNA回收试剂盒对该片段进行回收。在胶回收产物中加入与PCR产物两边酶切位点相对应的酶以及合适的buffer进行双酶切,37℃孵育过夜。同时,用相同的酶双酶切该片段所要连接的载体。利用普通DNA产物纯化试剂盒对酶切后的片段进行回收,利用琼脂糖凝胶DNA回收试剂盒对酶切后的载体进行回收,然后用T4连接酶将片段与载体连接起来,并将该连接产物转化DH5α。利用抗性筛选,验证及测序获得目的基因的克隆。2.质粒的定点突变:在突变位点附近设计一对引物,这两条引物的5’端有15-20bp的反向互补区域,突变位点应位于此反向互补区域的中间。3’端的非互补区域的大小至少为10bp。利用这一对引物,通过PCR的方法将质粒进行反向PCR扩增。利用普通DNA产物纯化试剂盒对PCR的产物进行回收,在回收产物中加入DpnI,37℃孵育2h,将甲基化的质粒模板去除。然后,将DpnI处理之后的样品直接转化DH5α。利用抗性筛选及测序获得目的基因的克隆。3.蛋白的表达与纯化:取原核表达质粒转化BL21,37℃培养12h。先进行接种,即挑克隆到7.5ml含有抗生素的LB培养基中,于37℃,220rpm震荡培养10-12h。然后再扩大培养,即将上述7.5ml菌液全部加入750ml含抗生素的TB培养基中,37℃,220rpm震荡培养至OD600为0.6-0.8(约3-4h)。此时,将摇床温度调节至16℃,待菌液完全降温至16℃后,按照1:2000的比例加入IPTG(终浓度为0.4mM),继续震荡培养16-18h。最后用Beckman的高速冷冻离心机收集菌体,即4℃,6000rpm离心10min,弃尽上清,将菌体称重后进行蛋白质纯化或保存于-80℃。(1)Ni柱亲和层析:Lysisbuffer:20mMHEPESpH7.0,500mMNaCl,20mMimidazole,1mMDTT;Washbuffer:20mMHEPESpH7.0,500mMNaCl,40mMimidazole,1mMDTT;Elutionbuffer:20mMHEPESpH7.0,500mMNaCl,500mMimidazole,1mMDTT;取适量的beads灌入层析柱中,用大量的ddH2O冲洗beads,一般为10CV×5(CV代表柱体积,下同)。用Lysisbuffer进行平衡:10CV×5。将平衡好的beads移入菌体破碎后得到的上清中,在4℃冷库Mix1-2h。500g,4℃离心2min,收集上清和流穿液。加入50mlWashbuffer,500g,4℃离心2min,收集上清和wash1。重复两次,收集的上清分别为wash2和wash3。将beads灌入层析柱中,继续用Washbuffer洗一次。用Elutionbuffer洗脱:0.5CV×10,用EP管分别收集洗脱液。SDS-PAGE电泳鉴定。(2)MBP柱亲和层析:Lysisbuffer:20mMHEPESpH7.0,500mMNaCl,1mMDTT;Washbuffer:20mMHEPESpH7.0,500mMNaCl,1mMDTT;Elutionbuffer:20mMHEPESpH7.0,500mMNaCl,10mMamylose,1mMDTT;取适量的beads灌入层析柱中,用大量的ddH2O冲洗beads,一般为10CV×5(CV代表柱体积,下同)。用Lysisbuffer进行平衡:10CV×5。将平衡好的beads移入菌体破碎后得到的上清中,在4℃冷库Mix1-2h。500g,4℃离心2min,收集上清和流穿液。加入50mlWashbuffer,500g,4℃离心2min,重复两次,。将beads灌入层析柱中,继续用Washbuffer洗一次。用Elutionbuffer洗脱:0.5CV×10,用EP管分别收集洗脱液。SDS-PAGE电泳鉴定。(3)StrepTactin柱亲和层析:Lysisbuffer/Washbuffer:100mMTrispH8.0,150mMNaCl,10mMβ-ME;Elutionbuffer:100mMTrispH8.0,150mMNaCl,2.5mMdesthiobiotin,10mMβ-ME;取适量的beads灌入层析中。用Lysisbuffer进行平衡:10CV×5。将破碎后的上清灌入装有beads的层析柱中,控制流速为1滴/2s,收集流穿液,再重复上样一次。用Washbuffer冲洗:10CV×5,收集washes。用Elutionbuffer洗脱:0.5CV×6,用EP管分别收集洗脱液。SDS-PAGE电泳鉴定。(4)凝胶过滤层析(Superdex75/200):Buffer:20mMHEPESpH7.0,100mMNaCl,1mMDTT;ddH2O冲洗2CV:流速:1ml/min,压力上限:0.3MPa(下同)。Buffer平衡2CV。样品离心:12000rpm,4℃离心40min。冲洗上样环:ddH2O洗5个体积,Buffer洗5个体积。上样:用注射器上样,一定要排尽注射器里的气泡。Buffer洗脱约40min后,开始注意观察紫外吸收峰的出现,一旦出峰立即开始收集。样品收集完毕后,继续用Bindingbuffer冲洗直到紫外吸收基线水平。ddH2O冲洗2CV。20%乙醇冲洗2CV后暂停,关闭仪器。(5)亲和标签的去除(TEV酶切):将Ni柱亲和层析之后的样品脱盐到Lysisbuffer中。测定洗脱液中蛋白的浓度,并根据体积估算出目的蛋白的总量。以TEV与目的蛋白的质量比为1:15的比例在eluates中加入足量的TEV,室温酶切2小时。将酶切后的样品回挂Ni柱,收集流出的流穿液,除去被切下的His标签和加入的TEV酶。4.晶体结构的解析:采用单波长反常散射法(Singlewavelengthanomalousdispersion,SAD)对Ter94-N/Npl4-UBD复合物的结构进行解析。利用在硒的反常散射波长下收集得到的衍射数据,通过Phenix软件包中AutoSol程序的解析得到衍射相位。然后,采用COOT软件进行模型的建立,利用Phenix中的Refinement和CCP4中的Refmac5进行结构的修正。5.Pull-down实验:取所需量的亲和层析beads,用适量pull-downbuffer洗3次。按照摩尔比为1:1的比例混合两个用于pull-down的蛋白,并用pull-downbuffer将混合好的蛋白溶液的终体积定容到500μl。在此蛋白混合液中加入20μl预处理过的亲和层析beads,在4℃冷库的旋转混合仪上缓慢旋转孵育2h。在4℃以500g的速度离心2min,将亲和层析beads离心至EP管底,弃尽上清。加入1mlpull-downbuffer清洗beads,在4℃以500g的速度离心2min,弃尽上清。重复清洗两次。加入40μlelutionbuffer进行洗脱,将结合在beads上的蛋白洗脱下来。在eluates中加入10μl的5×SDS上样缓冲液,100℃变性10min。利用SDS-PAGE分析两个蛋白的结合情况。Pull-downbuffer:Hispull-downbuffer:100mMTrispH8.0,150mMNaCl,40mMimidazole,10mMβ-ME;Hiselutionbuffer:100mMTrispH8.0,150mMNaCl,500mMimidazole,10mMβ-ME;Streppull-downbuffer:100mMTrispH8.0,150mMNaCl,0.1%TritionX-100;Strepelutionbuffer:100mMTrispH8.0,150mMNaCl,2.5mMdesthiobiotin,0.1%TritionX-100。6.双荧光素酶报告基因实验:双荧光素酶报告基因实验是先以荧光素为底物来检测萤火虫荧光素酶的活性,后以腔肠荧光素为底物来检测海肾荧光素酶的活性,并且在后续加入海肾荧光素酶的底物同时加入了抑制萤火虫荧光素酶催化虫荧光素的物质,使后续检测仅仅测定到海肾荧光素酶的活性,实现双荧光素酶报告基因的检测。萤火虫荧光素酶可以催化虫荧光素氧化成氧合虫荧光素,在虫荧光素氧化的过程中,会发出生物荧光。海肾荧光素酶可以催化腔肠荧光素氧化成coelenteramide,在腔肠荧光素氧化的过程中也会发出生物荧光。然后可以通过荧光测定仪也称化学发光仪或液闪测定仪测定上述荧光素酶催化的氧化过程中释放的生物荧光。萤火虫荧光素酶催化虫荧光素发光的最强发光波长为560nm。海肾荧光素酶催化腔肠荧光素发光的最强发光波长为465nm。通过荧光素酶和其底物这一生物发光体系,可以极其灵敏、高效的检测基因的表达。通常把感兴趣的基因转录的调控元件克隆在萤火虫荧光素酶的上游或其它适当的地方,构建成报告基因质粒,然后转染细胞,适当刺激或处理后裂解细胞,测定荧光素酶活性。通过荧光素酶活性的高低判断刺激前后或不同刺激对感兴趣的调控元件的影响。海肾荧光素酶更多的被用作转染效率的内参,以消除细胞数量和转染效率的差异。实验步骤:培养HEK293FT细胞(或其它目的细胞),并接种于24孔板中,生长12-24小时(80%汇合度)。用lipo2000转染表达萤火虫荧光素的荧光素酶报告基因质粒和海肾荧光素酶内参报告基因质粒,以及所需的目的基因表达质粒。24-48小时之后,加入细胞裂解液(5Xlysisbuffer用水稀释到1X,购自promega):24孔板每孔加120μl,48孔板每孔加60μl。将该板放-80℃冻1h。将板从-80℃取出,室温融化后,将细胞裂解液吸出,转移到EP管中。每管裂解液取5μl加入到384孔板中。加入10μl萤火虫荧光素酶的反应底物,测报告基因的荧光值。加入10μl的反应底物,测海肾荧光素酶的荧光值。报告基因与海肾荧光素荧光值的比值即为每个样品的相对荧光信号值。在检测化合物对细胞内报告基因表达影响的实验中,在HEK293FT细胞中转染Flag标签的RIG-I(residues1-238),以及IFNβ或ISRE的报告基因,荧光素酶报告基因。12小时后,消化细胞,以10000个细胞/每孔的密度接种至384孔板中,加入50μl的细胞培养基。7小时后,加入AlphaScreen实验筛选获得的化合物,化合物的终浓度为10μM。37℃培养15h后,检测荧光素酶活性。7.免疫印迹:根据实验要求制备蛋白样品,100℃变性10分钟,13200rpm离心2分钟,取等量的上清加到SDS-PAGE胶的上样孔中。蛋白样品在浓缩胶中时电压为80V,在分离胶中时电压为120V。电泳结束后,取下凝胶,按以下顺序安装转膜装置:(负极)、滤纸、凝胶、硝酸纤维素膜、滤纸、(正极)。将转膜装置放入4℃冷库,100V恒压转印1h。转膜完成后,取出硝酸纤维素膜,将膜浸泡在用TBST缓冲溶液配制的5%的脱脂牛奶中,室温下在摇床上孵育1h。用TBST缓冲溶液洗膜,5min×3次。加入按规定比例用5%BSA溶液稀释的一抗,在4℃冷库的摇床上孵育过夜。用TBST缓冲溶液洗膜,10min×3次。加入按规定比例用5%牛奶稀释的二抗,室温下在摇床上孵育40-60min。用TBST缓冲溶液洗膜,10min×3次。将显色底物覆盖在硝酸纤维素膜上,室温显色2分钟。用LAS4000冷光/生物发光影像分析仪进行拍摄。所需试剂的配制:5%BSA溶液:称取5gBSA粉末溶于100ml1×PBS溶液中,再加入0.02%的叠氮钠,贮存于4℃。10×Westernblot转膜缓冲液:称取30.3gTris碱和144g甘氨酸,加入300ml甲醇,再用去离子水定容到1L。8.RNA的抽提:将转染后的细胞弃去培养基,并用预冷的PBS将细胞清洗一遍。六孔板的每个孔加入500μlTrizolReagent,室温处理5分钟。将细胞裂解液转移到1.5mlEppendorf管中。每份样品加入100μl的氯仿,vortex低速混匀后静置5分钟。4℃,12000g离心15分钟,转移240μl的上层水相至新的Eppendorf管中。每份样品加入240μl的异丙醇,颠倒混匀,室温放置10分钟。4℃,12000g离心15分钟,弃去上清,留下白色的RNA沉淀。每份样品加入500μl的75%乙醇,颠倒混匀。4℃,12000g离心5分钟,弃尽上清。室温晾干。加入适量DEPC处理过的去离子水,充分溶解。用NanoDropND1000测定所提取的RNA的浓度,260nm的吸光值与280nm的吸光值的比值应维持在1.9-2.0之间。将所提取的RNA用于逆转录或直接冻到-80℃保存。所需试剂的配制:磷酸缓冲盐溶液(PBS):800ml蒸馏水溶解0.2gKCl,8gNaCl,0.24gKH2PO4和1.44gNa2HPO4。用HCl调节溶液的pH值至7.4,定容至1L。高压蒸汽灭菌或过滤除菌。9.免疫共沉淀:将转染后的细胞弃去培养基,并用预冷的PBS将细胞清洗一遍。加入适量的细胞裂解缓冲液RIPA(含蛋白酶抑制剂),冰上裂解30min,将细胞裂解液于4℃,最大转速离心30min后取上清。取所需量的proteinA/G琼脂糖珠,用适量裂解缓冲液洗3次。取少量裂解液以备Westernblot分析,剩余裂解液中加入20μl预处理过的proteinA/G琼脂糖珠和1μg相应的抗体,在4℃冷库的rotator上缓慢旋转孵育过夜。免疫沉淀反应完成后,在4℃以7000rpm的速度离心1min,将琼脂糖珠离心至EP管底。将上清小心吸去,琼脂糖珠用300μl裂解缓冲液洗2次,再用300μlPBS洗2次。最后加入20μl的2×SDS上样缓冲液,100℃煮10分钟。利用SDS-PAGE和Westernblot分析与抗体结合的蛋白所需试剂的配制:RIPAbuffer:150mMNaCl,100mMTrispH8.0,1%TritonX-100,5mMEDTA,10mMNaF.10.细胞培养:HEK293、HEK293T、MEF细胞培养于DMEM(Hyclone)培养液中,培养液中添加10%血清,100ug/ml盘尼西林,100μg/ml链霉素。细胞于37℃培养,二氧化碳浓度为5%。11.实时定量荧光PCR:实时定量荧光PCR采用AppliedBiosystems公司两步法实时PCR(Realtime-PCR)系统,检测相对CT值。使用定量荧光PCR预混液(Toyobo公司提供GreenRealtimePCRreagent)配置反应体系检测并定量目标基因的表达水平,GAPDH作为内参。12.数据分析:采用SAS数据软件分析包(9.1.3)对数据进行分析,统计数据的平均数±标准差。单因子变异数分析(ANOVA)以及Student’st-test用于分析连续变量。置信区间为P<0.05。实施例1p97负调控I型干扰素信号通路依赖于其ATP酶活1.实验目的:确定p97是否参与I型干扰素应答以及抗病毒免疫的信号通路。2.实验方法:(1)细胞感染实验:人HEK293,HEK293T,小鼠MEF细胞细胞购自中国科学院上海生命科学研究院细胞库。细胞培养方法如基本实验方法10所述。Sendaivirus仙台病毒(SeV)以及GFP-Vesicularstomatitisvirus带有绿色荧光的水疱性口炎病毒(GFP-VSV)由武汉大学舒红兵教授赠予,SeV与VSV-GFP同市售,VSV-GFP改造方法同ConstructionandexpressionofhightiterpseudotypedGFP-retrovirusesbycotransfectionofVSV-GgeneJournalofShanghaiJiaotongUniversity(MedicalScience)》2007-07。poly(I:C),poly(dA:dT)购自Sigma(St.Louis,MO),poly(I:C)采用转染方式处理细胞,poly(dA:dT)直接加入细胞培养基中处理细胞。(2)IFNβ,IFNβ调控结构域PRDIII以及PRDI(PRDIII-I),ISRE的报告基因由中国科学院上海生化与细胞所王琛教授赠予,上述报告基因载体由市售载体插入相应报告基因元件改造而得,载体相关信息详见TRIM21IsEssentialtoSustainIFNRegulatoryFactor3ActivationduringAntiviralResponse2009vol.182no.63782-3792。(3)IFNB,RANTES以及ISG56在细胞中转录水平的检测:细胞抽提总RNA方法如基本实验方法8所述,AMV逆转录酶合成第一链,并以其为模板,在DNAtaq酶的作用下用对应的引物进行PCR扩增;逆转录体系见表1,逆转录反应程序见表2,细胞样品扩增PCR引物见表3,Realtime-PCR反应体系见表4,Realtime-PCR扩增程序见表5。表1逆转录反应体系表2逆转录程序37℃15min50℃5min95℃5min4℃20min表3引物序列表4Realtime-PCR反应体系表5PCR反应体系程序设定(4)siRNA敲除细胞内p97:针对p97编码区及非编码区的siRNA以及对照siRNA由上海吉玛制药技术有限公司合成,siRNA序列如表6所示。表6siRNA序列(5)Rescue实验验证p97功能:p97野生型及p97QQ突变体由中科院上海生化与细胞所赵允教授赠予,上述载体由市售载体插入p97野生型或p97QQ突变体片段而得,具体载体信息详见Ter94ATPasecomplextargetsk11-linkedubiquitinatedcitoproteasomesforpartialdegradation.DevCell.2013Jun24;25(6):636-44。3.实验结果及分析:在HEK293细胞中转染IFNβ,IFNβ的调节结构域PRDIII,IFNβ的调节结构域PRDI,ISRE的报告基因,以及针对p97编码区的siRNA。之后用SeV感染细胞,诱导I型干扰素信号通路。siRNA敲除p97后,IFNβ,PRDIII-I以及ISRE启动子的活性明显上调(图1A)。同样,IFNB,RANTES以及ISG56的mRNA水平随着SeV感染时间的增加,不断上调(图1B)。在人293T细胞以及小鼠MEF细胞中,以poly(I:C)、poly(dA:dT)、VSV-GFP诱导I型干扰素信号通路,获得了类似的结果(图1C-1D)。以上结果说明,敲除p97可以增强I型干扰素的信号通路。以感染复数为2的VSV-GFP感染MEF细胞,敲除p97的细胞抗病毒感染的能力更强,GFP阳性,即病毒感染的细胞比对照组少(图1E)。通过细胞流式检测,在p97敲除的细胞中,GFP阳性,即病毒感染的细胞比例仅占9%,而对照组的细胞中,病毒感染的细胞比例高达93%(图1F)。在转染了针对p97非编码区siRNA的细胞中,进行rescue实验验证p97的功能。过表达野生型的p97抑制Sev诱导的IFNβ报告基因的活性以及IFNB的转录,而使其缺失ATP酶活性的p97QQ突变体没有上述功能(图1G-1H)。这表明,p97负调控抗病毒免疫,该功能依赖于其ATP酶活性。实施例2Npl4协同p97调控抗病毒免疫应答1.实验目的:p97与不同的辅助因子结合以参与不同的信号通路的调控,本实施例目的在于确定在抗病毒免疫应答的调控中,p97结合的辅助因子。2.实验方法:(1)siRNA敲除细胞内p97,Npl4,Ufd1,p47,FAF1:上述siRNA由上海吉玛制药技术有限公司合成,p97siRNA序列如表6所示,Npl4,Ufd1,p47,FAF1如表7所示。表7siRNA序列(2)报告基因的活性以及基因的转录水平的检测同实施例1。(3)在细胞中过表达的Npl4载体构建以实施例1中的方法及体系抽提细胞总RNA,并逆转录为cDNA,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表8,PCR反应体系见表9,PCR扩增程序见表10。表8引物序列表9PCR反应体系表10PCR扩增程序扩增获得的片段,通过1%琼脂糖凝胶电泳,确认大小与目的条带相同,切下胶,使用琼脂糖凝胶回收试剂盒回收(天根生化科技(北京)有限公司,货号DP209)。Npl4片段使用限制性内切酶NcoI和SalI(thermoscientific)进行双酶切,酶切反应体系如表11所示,反应在37度进行2小时。表11片段酶切反应体系酶切后,通过普通产物纯化试剂盒(天根生化科技(北京)有限公司,货号DP204),纯化获得酶切后的片段。片段(HindIII/NheI双酶切)与载体pCDNA3(HindIII/NheI双酶切),进行连接,连接体系表12所示,连接反应在室温(25℃)进行2小时。载体双酶切反应体系如表13所示,反应在37℃进行4小时。反应完成后使用天根普通产物纯化试剂盒纯化获得酶切后的载体。连接后的产物转化DH5α感受态细胞,通过抗性筛选,获得的阳性克隆,并测序验证。表12连接反应体系表13载体酶切反应体系3.实验结果及分析:在HEK293T细胞中敲除不同的p97辅助因子,包括Npl4,Ufd1,p47以及FAF1后,检测SeV感染后IFNβ报告基因的活性以及IFNB的转录水平。敲除Npl4后,IFNβ的转录活性以及IFNB的转录水平明显上调,与Npl4形成复合物的Ufd1也能上调SeV感染介导的I型干扰素的信号通路。因而,在抗病毒免疫信号通路中,Npl4-Ufd1是p97的辅助因子(图2A-2B)。我们在感染了SeV的细胞中转染不同剂量的Npl4,结果表明Npl4可以抑制SeV诱导的IFNβ的激活以及ISRE报告基因的活性,并且其抑制作用呈现计量依赖效应(图2C)。同样,过表达Npl4后,SeV刺激细胞后IFNB以及ISG56的mRNA水平明显下调(图2D)。以siRNA敲除内源的Npl4,可以大大激活IFNβ,PRDIII-I以及ISRE的报告基因(图2E)。同样,在Npl4敲除的细胞中,IFNB,RANTES以及ISG56的转录水平随着SeV刺激时间不断上调(图2F)。Rescue实验表明,以针对Npl4非编码区的siRNA敲除内源Npl4可以降低细胞感染VSV的比例,而后过表达Npl4,可使细胞更易感染VSV(图2G)。将细胞中的p97敲除后,共转染Npl4与野生型的p97或Npl4与突变型p97QQ,只有共转染Npl4与p97野生型的细胞,SeV诱导的IFNβ报告基因斤的活性以及IFNB的转录受到抑制,单独的Npl4或Npl4与p97QQ突变体的共转均不能抑制IFNβ报告基因的活性以及IFNB的转录(图2H)。以上结果表明Npl4作为p97的辅助因子参与I型干扰素信号通路和抗病毒免疫反应的负调控。实施例3p97-Npl4复合物的结构解析1.实验目的:通过结构生物学的方法,以体外纯化获得的p97、Npl4蛋白组装复合物,解析其结构,研究蛋白相互作用方式。2.实验方法:(1)复合物结构解析的表达载体构建:HT-代表pET28a-HisTEV载体,为实验室自行构建的载体,以商品化载体pET28a(Merck)改造而得。质粒信息参见(Shi,Z.,etal.StructureoftheMST4incomplexwithMO25providesinsightsintoitsactivationmechanism.Structure21,449-461,2013)果蝇HT-Ter94-N(20-186),果蝇HT-Npl4-UBD(1-77)分别以果蝇Ter94全长,细胞逆转录的cDNA为模板,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表14,载体构建方法及体系同实施例2。连接的载体为上述的pET28a-HisTEV,酶切位点为NcoI/XholI。果蝇Ter94全长由中国科学院上海生化与细胞所赵允教授赠予,上述载体由市售载体插入Ter94片段而得,具体载体信息详见Ter94ATPasecomplextargetsk11-linkedubiquitinatedcitoproteasomesforpartialdegradation.DevCell.201324;25(6):636-44。细胞逆转录的cDNA的制备方法及体系同实施例1。表14引物序列(2)Ter94-N/Npl4-UBD复合物蛋白的纯化:按基本实验方法3进行纯化,得到了纯度高于95%的目的蛋白,再通过一步凝胶过滤层析均一蛋白。将纯化的HT-Ter94-N和HT-Npl4-UBD以摩尔比为1:1的比例混合,4℃孵育2h,再用凝胶过滤层析的方法进行纯化,出现了单一的紫外吸收峰,该峰的位置与单独纯化蛋白的紫外吸收峰位置相比,向左发生了明显的偏移,说明Ter94和Npl4能够以化学计量比为1:1的方式结合,形成稳定的二元复合物。(3)His-Ter94-N/His-Npl4-UBD复合物的结构解析:将His-Ter94-N/His-Npl4-UBD复合物蛋白浓缩到25mg/ml,采用坐滴法进行晶体生长条件的初步筛选。三天后,在Indexkit(HamptonResearch)第69号条件中有不规则的片状晶体产生,组成成分为0.1MTrispH8.5,25%PEG3350,0.2Mammoniumsulfate,将晶体带到上海同步辐射光源生物大分子晶体学光束线站BL17U-MX进行衍射数据的收集。(4)复合物结构解析:为了解析Ter94-N/Npl4-UBD复合物的相位,我们在含有硒代甲硫氨酸的M9培养基中诱导表达HT-Ter94-N和HT-Npl4-UBD硒代蛋白,然后按照与母体蛋白相同的纯化方法进行硒代蛋白的纯化。利用硒代甲硫氨酸标记的Ter94-N/Npl4-UBD复合物蛋白重复该复合物母体晶体的生长条件Index-69和Index-75(HamptonResearch)。通过改变沉淀剂PEG的种类和浓度分别对这两个条件中长出的硒代晶体进行优化,得到了晶体。将这些硒代晶体带到北京同步辐射光源(BSRF)进行衍射数据的收集,在三个不同的波长下(peakinflection和remote)分别收集到了分辨率为和的衍射数据,空间群均为P212121。利用peak的衍射数据,通过单波长反常散射(SAD)的方法对Ter94-N/Npl4-UBD复合物进行了相位的解析和模型的建立,最终得到了该复合物的晶体结构。复合物的结构显示:Npl4的UBD结构域结合在了Ter94N端的两个亚结构域形成的疏水沟槽中,在不对称单元中的结合比例为1:1。(5)IFNβ激活以及IFNB转录的检测同实施例1。3.实验结果与分析Npl4有四个结构域:N端的UBD结构域(1-80)与p97结合,zf-Npl4结构域(104-246),Npl4结构域(248-557),以及C端的NZF结构域(580-608)与泛素结合(图3A)。缺失UBD结构域的Npl4不能抑制SeV诱导的IFNβ激活以及IFNB转录,这进一步证明Npl4协同p97抑制抗病毒免疫反应。我们将解析的果蝇Ter94-N/Npl4-UBD复合物的晶体结构分别与人源p97N端结构域的晶体结构(PDB编号为3QQ7)和鼠源Npl4UBD结构域的核磁结构(PDB编号为2PJH)进行比较,我们发现人源p97-N单独的晶体结构能够很好的与果蝇复合物中的Ter94-N叠加在一起,叠加之后Cα原子的均方根偏差(rmsd)为这与它们高度的序列同源性(78%)相一致。另一方面果蝇Npl4-UBD与鼠源Npl4-UBD这两个结构之间还是存在明显差异的。通过对不同物种的Cdc48/Ter94/p97-N和Npl4-UBD进行以结构为导向的序列比对,我们发现Ter94-N上与果蝇Npl4-UBD相互作用的关键残基Q47,F49,R50,D52,K106,Y107和Y140在不同物种间都是十分保守的。我们所解析的果蝇Ter94-N/Npl4-UBD复合物的晶体结构可能更精确的描述了p97与Npl4相互作用的普遍方式和特点。我们解析的Ter94/Npl4复合物结构与单独的Ter94,Npl4相似,Npl4的UBD结构域结合在了Ter94N端的两个亚结构域形成的疏水沟槽中,在不对称单元中的结合比例为1:1(图3B)。Ter94/Npl4复合物形成的异源二聚体有两个相互作用的位点。第一个相互作用的位点Npl4的第一以及第二个β片层上的L7,R9以及R18与Ter94N端的F49形成疏水相互作用,同时Npl4的R9,R18以及H69与Ter94的R50,D52andQ47形成氢键及离子键。第二个相互作用位点Npl4的A13以及R49与Ter94N端的Y107以及Y140通过疏水相互作用联结,Npl4的A13,Q11以及R49与Ter94的Y107通过氢键及离子键相互作用。此外,Npl4的R49与Ter94的Y140形成cation-π相互作用。上述这些相互作用的残基在果蝇和人中都是保守的,因而以果蝇的Ter94-Npl4解析的复合物结构,同样适用与人p97-Npl4复合物的结构分析(图3C)。实施例4p97-Npl4复合物的突变研究1.实验目的:通过突变分析研究p97-Npl4复合物的相互作用对于其在抗病毒信号通路中作用的影响。2.实验方法:(1)相互作用检测载体Strep-Npl4、His-Npl4、Strep-p97、p97-His的载体构建:Strep-Npl4、His-Npl4以HA-Npl4载体为模板,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表15,载体构建方法及体系同实施例2。Strep-Npl4采用表15中SEQIDNO:31,32扩增,连接的载体为市售载体pET51b-Strep,His-Npl4采用表15中SEQIDNO:31,33扩增,连接的载体为自行构建的pET28a-HisTev,酶切位点均为NcoI/SalI。表15引物序列p97-His、Strep-p97以V5-p97载体为模板,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表16,载体构建方法及体系同实施例2。p97-His采用表16中SEQIDNO:34,35扩增,连接的载体为市售载体pET21a,酶切位点均为NdeI/SalI。Strep-p97采用表16中SEQIDNO:36,37扩增,连接的载体为市售载体pET51b-Strep,酶切位点均为BamHI/SalI。表16引物序列(2)p97及Npl4的突变载体的构建:p97突变体F52A/D55A、Y110A,以p97-His为模板,采用表17中SEQIDNO:38-41所示引物进行PCR扩增后,使用DpnI去甲基化酶去除未突变的野生型p97模板,扩增获得带有特定位点突变的带有p97基因的载体片段,将PCR产物转化DH5α感受态细胞,通过抗性筛选,获得的阳性克隆,并测序验证。p97突变体3A,以p97-HisF52A/D55A为模板,采用表17中SEQIDNO:40,41所示引物进行PCR扩增后,使用DpnI去甲基化酶去除未突变的p97-HisF52A/D55A模板,扩增获得带有特定位点突变的带有p97基因的载体片段,将PCR产物转化DH5α感受态细胞,通过抗性筛选,获得的阳性克隆,并测序验证。表17引物序列Npl4突变体I6A/R8A、R50A,以Npl4-His为模板,采用表18中SEQIDNO:42,43,46,47所示引物进行PCR扩增后,使用DpnI去甲基化酶去除未突变的野生型Npl4模板,扩增获得带有特定位点突变的带有Npl4基因的载体片段,将PCR产物转化DH5α感受态细胞,通过抗性筛选,获得的阳性克隆,并测序验证。Npl4突变体I6A/R8A/R17A以Npl4-HisI6A/R8A为模板,采用表18中SEQIDNO:44,45所示引物进行PCR扩增后,使用DpnI去甲基化酶去除未突变的Npl4-HisI6A/R8A模板,扩增获得带有特定位点突变的带有Npl4基因的载体片段,将PCR产物转化DH5α感受态细胞,通过抗性筛选,获得的阳性克隆,并测序验证。Npl4突变体4A以Npl4-HisI6A/R8A/R17A为模板,采用表18中SEQIDNO:46,47所示引物进行PCR扩增后,使用DpnI去甲基化酶去除未突变的Npl4-HisI6A/R8A/R17A模板,扩增获得带有特定位点突变的带有Npl4基因的载体片段,将PCR产物转化DH5α感受态细胞,通过抗性筛选,获得的阳性克隆,并测序验证。表18引物序列3.实验结果与分析:我们根据结构信息构建了一系列人p97以及Npl4的突变体,通过pulldown实验,我们发现p97的(F52,Y110)(D55,Y110)对于Npl4的结合具有决定性的作用。Npl4的R50以及I6,R8,R17,R50的多点突变,可以打破Npl4与p97的相互作用(图3C-3E),免疫共沉淀实验也验证了这一结果(图3F-3G)。因而,F52A/D55A/Y110p97(3A)突变以及I6A/R8A/R17A/R50A的Npl4(4A)突变是影响p97与Npl4相互作用的关键位点。在敲除内源p97的HEK293T细胞中,SeV的刺激下,转染野生型的p97可以抑制IFNβ的转录激活以及IFNB的转录,转染p973A突变体不能(图3H)。同样,转染野生型的Npl4可以抑制IFNβ启动子的激活以及IFNB的转录,而Npl44A突变体不能(图3I)。以上结果表明,p97-Npl4形成复合物,在抗病毒信号通路中发挥作用。实施例5p97-Npl4复合物负调控RIG-I的蛋白水平1.实验目的:p97具有AAAATP酶活,可以调控蛋白的稳定性。为研究p97-Npl4复合物是否通过调控RLR介导的抗病毒信号通路中的分子的稳定性,我们研究了p97-Npl4复合物对RIG-I蛋白水平的调控。2.实验方法:(1)慢病毒重组shRNA敲除细胞内p97,Npl4,Ufd1,p47设计一段21个碱基的靶序列(shRNA),该序列可以形成反向互补的发卡状结构,沉默相关基因表达。对应基因的shRNA以及对照shRNA(scramble)连接至慢病毒载体pLKO.1-puro。shRNA(scramble)为对照。shRNA序列如表19所示。表19shRNA序列0.01M的正向和反向DNA链混合,稀释至50ul,经过如下反应:95℃5min70℃10min55℃30min37℃30min退火连接为双链。双链DNA与pLKO.1-puro(EcoRI/AgeI双酶切)载体连接,转化DH5α,鉴定得到的阳性克隆经测序验证后提取质粒。将质粒转染293细胞,用2μg/mlpuromycin筛选基因组稳定整合shRNA序列的细胞,48-72小时后,使用Vivaspin20ml(SartoriusStedimBiotech,Aubagne,France)以3000g的离心力,离心50min,浓缩病毒液。感染病毒后,可获得敲除相应基因的细胞。pLKO.1-puro质粒从中国科学院上海生命科学研究院生物化学与细胞生物学研究所季红斌研究员处获得,为市售的构建RNAi的常用载体,可从Addgene公司购得。(3)Flag标签RIG-I,Flag标签RNF125的载体构建:以实施例1中的方法及体系抽提细胞总RNA,并逆转录为cDNA,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表20,载体构建方法及体系同实施例2。Flag-RIG-I采用表20中SEQIDNO:60,61扩增,Flag-RNF125采用表20中SEQIDNO:62,63扩增,连接的载体为市售载体pCDNA3.1-Flag,酶切位点均为HindIII/XholI。表20引物序列(4)RIG-IK181突变体构建:以Flag-RIG-I为模板,采用表21中所示引物进行PCR扩增后,使用DpnI去甲基化酶去除未突变的模板,扩增获得带有特定位点突变的带有Npl4基因的载体片段,将PCR产物转化DH5α感受态细胞,通过抗性筛选,获得的阳性克隆,并测序验证。表21引物序列(5)抗体检测蛋白表达水平:实验方法同基本实验方法7。检测Flag,Myc以及β-actin的抗体购自Sigma(St.Louis,MO)。检测p97,RIG-I,MAVS,p-TBK,TBK,IKKε,p-IRF3,IRF3以及K48Ub的抗体购自CellSignaling(Danvers,MA)。检测HA,Ubqutinin,RNF125的抗体购自SantaCruz(SantaCruz,CA)。检测Npl4(IF)的抗体购自NovusBiologicals(Littleton,CO)。检测Npl4(IP以及WB)购自Abcam(Cambridge,UK)。3.实验结果与分析:我们检测了转染了shp97的细胞中RIG-I,MAVS以及IRF3的蛋白水平(图4A)。敲除p97可以显著增加poly(I:C)诱导的RIG-I表达,同样MAVS的蛋白水平也增加,IRF3的总蛋白水平没有变化,但其磷酸化水平明显增加。反过来,过表达Npl4显著降低了poly(I:C)诱导的RIG-I以及MAVS的表达,IRF3的磷酸化水平也显著降低(图4B)。此外,敲除Npl4可以显著增加poly(I:C)诱导的的RIG-I,MAVS的表达,增强IRF3的磷酸化水平,而TBK1,IKKε以及IRF3的蛋白水平没有变化(图4C)。同样,敲除p47,p97的另一个辅助因子,对RIG-I抗病毒通路并没有调控作用。因而p97对于RIG-I抗病毒信号通路的调控作用依赖于Npl4。过表达Npl4,并不会引起poly(I:C)诱导的RIG-I的mRNA水平变化,而以蛋白酶体抑制剂MG132处理细胞后p97以及Npl4介导的RIG-I蛋白水平下降也被抑制(图4D-4E)。这说明p97-Npl4复合物通过蛋白酶体降解调控RIG-I的蛋白水平。在HEK293细胞内敲除p97或Npl4,再转染Flag标签RIG-I,RIG-I的K48链泛素化水平明显下降(图4F-4G)。相反,在敲除p97的HEK293细胞中同时转染Flag标签RIG-I以及野生型p97,RIG-I的K48链泛素化水平明显增加,而同时转染Flag-RIG-I以及不能与Npl4结合的p973A突变体,对RIG-I的泛素化水平没有影响(图4H)。同样,过表达野生型的Npl4可以增加RIG-I的K48泛素化水平,而过表达不能与p97结合的Npl44A突变体对RIG-I的泛素化水平没有影响(图4I)。以上结果说明,p97-Npl4复合物通过促进RIG-I的K48链泛素化,使其通过蛋白酶体降解,抑制抗病毒免疫信号通路。p97-Npl4复合物通过介导RIG-I的降解调控抗病毒信号通路,而之前的研究表明E3泛素连接酶c-Cbl以及RNF125参与RIG-I的K48链泛素化,我们研究了参与p97-Npl4复合物调控的RIG-I降解的泛素连接酶。我们分别设计了RNF-125以及c-Cbl的shRNA,通过Q-PCR验证了其敲除效率。双荧光酶素报告基因实验表明,敲除内源的RNF125,Npl4无法下调SeV诱导的IFNβ启动子的激活,而敲除c-Cbl没有作用(图5A)。同样,在敲除RNF-125的细胞中,Npl4不能抑制过表达RIG-I的card结构域介导的IFNβ的转录激活,在敲除c-Cbl的细胞中,Npl4的抑制作用不受影响(图5B)。在敲除内源RNF125的细胞中,Npl4也不能降低RIG-I的蛋白水平(图5C)。以上结果说明,RNF125是p97-Npl4复合物抑制RIG-I抗病毒信号通路中特异的E3泛素连接酶。我们检测了转染Npl4或RNF125的细胞中总RIG-I的泛素化水平,过表达Npl4显著增加了RIG-I的K48链泛素化水平,而同时过表达Npl4以及RNF125可以进一步增加其泛素化水平(图5D),这表明Nlp4与RNF125协同作用,促进RIG-I的K48链泛素化。QPCR实验也表明共转染Npl4与RNF125对于IFNβ的转录抑制作用远高于单独转染Npl4或RNF125(图5E)。我们在细胞中过表达RNF125,并以为未过表达RNF125的细胞为对照,通过质谱分析RNF125催化的RIG-I的泛素化位点,结果表明其泛素化发生与181位赖氨酸(图5F)。通过点突变手段构建181位不被泛素化的RIG-I突变体K181,在细胞中共转染野生型或突变体的RIG-I与Npl4或RNF125,SeV刺激后,通过免疫印迹检测RIG-I的蛋白水平,结果表明Npl4或RNF125都能显著降低野生型的RIG-I的蛋白水平,但对突变体K181R的蛋白水平没有影响(图5G),这表明181位赖氨酸是p97-Npl4复合物通过RNF125调控RIG-I相关的抗病毒通路的主要位点。此外,Npl4可以增加RIG-I的K48链泛素化水平,但对K181R突变体的K48链泛素化水平没有影响(图5H)。同样,RNF125以及Npl4都可以明显抑制野生型RIG-I介导的IFNβ启动子的活性,但对K181R突变体没有影响(图5I)。在细胞中敲除Npl4或RNF125后,转染野生型的RIG-I可以抑制VSV感染(感染复数MOI=20),转染K181R突变体不能产生抗病毒作用(图5J)。我们进一步分析了RIG-I第181位赖氨酸的结构特征,该位点在结构上接近172位赖氨酸,即RIG-I激活相关的关键K63链泛素化位点,以及负调控RIG-I信号通路的磷酸化位点第170位苏氨酸,因而,RIG-I的第181位赖氨酸是RNF125/p97-Npl4介导的RIG-I泛素化的关键位点。实施例6p97-Npl4复合物与RIG-I以及RNF125结合1.实验目的:检测p97-Npl4复合物是否直接结合RIG-I以及RNF125。2.实验方法:(1)Flag标签RIG-I1-238,200-803,804-925的载体构建:以Flag-RIG-I为模板,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表20,22,载体构建方法及体系同实施例2。连接的载体为市售载体pCDNA3.1-Flag,酶切位点均为HindIII/XholI。(“Flag标签RIG-I1-238扩增引物为表20引物60,表22引物66;Flag标签RIG-I200-803扩增引物为表22引物67,68;Flag标签RIG-I804-925扩增引物为表22引物69,表20引物61”)表22引物序列(2)HA-Npl4全长,ΔNZF1-579,ΔUBD(81-608),以及单独的UBD载体构建:以HA-Npl4为模板,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表8,23,载体构建方法及体系同实施例2。连接的载体为市售载体pCDNA3,酶切位点均为HindIII/NheI。(“HA-Npl4ΔNZF1-579扩增引物为表8引物25,表23引物70;HA-Npl4ΔUBD(81-608)扩增引物为表23引物71,表8引物26;HA-Npl4UBD扩增引物为表8引物25,表23引物72”)表23引物序列(3)MBP-RIG-ICARD,MBP-RNF125的载体构建:MBP-RIG-I以Flag-RIG-I为模板,MBP-RNF125以Flag-RNF125为模板,在Takara高保真DNA聚合酶的作用下用对应的引物进行PCR扩增;PCR引物见表24,载体构建方法及体系同实施例2。连接的载体为自行构建的载体pET28aMBPTev(载体构建信息参见Shi,Z.,etal.StructureoftheMST4incomplexwithMO25providesinsightsintoitsactivationmechanism.Structure21,449-461,2013)),酶切位点均为NcoI/XholI。表24引物序列3.实验结果与分析:我们在SeV刺激的HEK293T细胞中,进行免疫共沉淀实验,发现内源的RIG-I可以与内源的Npl4结合,也可以与RNF125结合(图6A)。同时,激光共聚焦显微镜检测也证明了,在SeV刺激下,Npl4与RIG-I存在共定位(图6B)。体外纯化的Npl4与RIG-I重组蛋白在pulldown实验中进一步证明,这两个蛋白可以直接相互作用(图6C)。我们截取了不同的RIG-I以及Npl4的结构域,研究其相互作用的具体区域,不同RIG-I片段与全长的Npl4共转染HEK293T细胞,免疫共沉淀实验表明,RIG-I的CRAD结构域(氨基酸1-238)与Npl4相互作用,helicase结构域与CTD结构域不参与其与Npl4的相互作用(图6D)。同样,Npl4的缺失突变体与全长的RIG-I共转染细胞,免疫共沉淀实验表明,缺失Npl4的NZF结构域的突变体完全丧失与RIG-I结合的能力,而缺失UBD结构域的Npl4依旧能与RIG-I结合(图6E),即Npl4通过NZF结构域与RIG-I结合。体外pulldown实验进一步证明Npl4可以直接与RIG-I的CARD结构域结合(图6F)。内源免疫共沉淀实验表明RNF125不仅能与RIG-I结合,也能与p97结合(图6G)。利用体外纯化的蛋白,我们发现p97可以与RNF125结合,两者之间存在直接的相互作用(图6H)。此外,RNF125可以直接结合RIG-I以及p97-Npl4复合物,p97-Npl4复合物可以进一步促进RNF125与RIG-I之间的相互作用(图6I)。以上结果表明,在SeV的刺激下,p97-Npl4复合物可以与RIG-I以及RNF125结合,RNF-125介导RIG-I的K48链泛素化,进而促进RIG-I的蛋白酶体降解,抑制RIG-I的抗病毒信号通路。实施例7p97-Npl4复合物抑制剂的筛选及其在抗病毒信号通路中的作用1.实验目的:确定可以特异性阻断p97-Npl4相互作用的小分子化合物并检测化合物对p97-Npl4介导的抗病毒信号通路的影响。2.实验方法:(1)AlphaScreen实验:在384浅孔板(PerkinElmer)中,加入2.5μl带His标签的Npl4,2.5μl生物素标记的p97,再加入反应缓冲液(20mMHepespH7.5,100mMNaCl,0.1%BSA),每孔終体积为11μl,带His标签的Npl4的终浓度为125nM,生物素标记的p97终浓度为31.25nM,将反应体系于室温孵育60分钟。用稀释缓冲液(20mMHepes,100mMNaCl,pH7.5)将溶解于DMSO中的化合物(10mM)稀释至100μM,将稀释后的化合物加1μl至反应体系中,室温避光孵育60分钟。将2.5μl镍螯合acceptorbeads(PerkinElmer)加入反应体系中,终浓度为10μg/ml,室温避光孵育60分钟后,再加入等量的streptavidin交联的donorbeads(PerkinElmer),室温避光孵育60分钟。在仪器EnVisionMultilabelPlateReader(PerkinElmer)读取数值,每块384孔板上均有阳性对照及阴性对照。(2)细胞内报告基因实验:在HEK293FT细胞中转染Flag标签的RIG-ICARD结构域(1-238),以及IFNβ或ISRE的报告基因,荧光素酶报告基因。12小时后,消化细胞,以10000个细胞/每孔的密度接种至384孔板中,加入50μl的细胞培养基。7小时后,加入AlphaScreen实验筛选获得的化合物,化合物的终浓度为10μM。37℃培养15h后,检测荧光素酶活性。3.实验结果:p97与Npl4的相互作用对于IFNβ介导的抗病毒信号通路是必须的,我们通过AlphaScreen这一筛选手段,以8120个具有生物活性的化合物组成化合物库进行高通量筛选,寻找可以打破p97-Npl4相互作用的抑制剂。在预实验中通过一系列的实验流程优化,实验的Z因子达到0.7,表明实验条件或测量方法可以应用到接下来的高通量实验中。在对8120个化合物进行筛选后,获得抑制率>50%的化合物132个。通过生物素标记Npl4,筛选假阳性的条件,我们确定了40个抑制率>30%,可以抑制p97-Npl4复合物形成的化合物。通过细胞内报告基因实验,我们检测了这40个化合物在抗病毒信号通路中的作用,其中三个化合物对于报告基因IFNβ以及ISRE的激活效果>30%,即ThonzoniumBromide(TB),EpirubicinHydrochloride(EH)以及Purpurin。TB对于IFNβ的激活倍率为102.6%,对ISER的激活倍率为107.6%;EH对于IFNβ的激活倍率为89.5%,对ISER的激活倍率为207%,因而这两个化合物对抗病毒信号通路具有明显的激活作用。进一步通过pulldown实验验证TB、EH对于p97-Npl4复合物的影响,采用实施例4中p97、Npl4相互作用检测的pulldown体系,在体系中分别加入对照溶液和化合物,检测两者间的相互作用,对电泳结果采用灰度分析,表明这两种小分子化合物大大消弱了两者的相互作用(图7)。结果表明,上述筛选获得的小分子化合物可以通过破坏p97-Npl4复合物的形成,促进RIG-I相关的抗病毒信号通路。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 基于温敏阳离子聚合物的siRNA载体及其制备方法与流程
- 一种提高DC疫苗抗肝癌效果的基因靶标、抑制剂和DC肿瘤疫苗的制造方法与工艺
- 一种基因递送系统的制造方法与工艺
- LncRNA ENST00000424523.1及其基因在诊断和治疗药物中的应用的制造方法与工艺
- 核酸扩增反应方法、核酸扩增反应装置以及核酸扩增反应用试剂与流程
- 一种人胎盘绒毛膜间充质干细胞培养体系的制造方法与工艺
- GO‑PLL‑RGDS/VEGF‑siRNA靶向基因药物的制备、活性和应用的制造方法与工艺
- 纤维变性易感性IL22RA2基因及其用途的制造方法与工艺
- G6PD基因及其表达产物在治疗结直肠癌中的应用的制造方法与工艺
- 基于基因治疗的半导体微针组件及其制造模具的制造方法与工艺