确定供体和受体差异SNP的方法及应用与流程
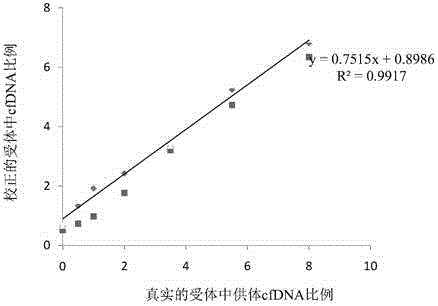
本发明涉及生物信息领域,具体的,本发明涉及确定供体和受体的差异SNP的方法及应用,更具体的,本发明涉及一种确定供体和受体差异SNP的方法、一种确定受体中供体来源cfDNA比例的方法、一种监测器官移植排斥的方法和装置。
背景技术:
:器官移植排斥临床常规检测方式通常是实验室和免疫学检查、影像学检查、细胞学与组织学检查。常规无创类检查敏感性及特异性不强,往往无法真实反映排斥程度。有创活检检查作为心脏移植等器官移植排斥检测的金标准,但有诸多限制,比如侵入性引起并发症及感染、不能准确定量、价格昂贵等。能否有一种便捷、早期、无创、准确移植排斥监测技术作为临床检测的辅助或补充呢?自血浆循环细胞游离DNA(cfDNA)发现以来,有文献指出cfDNA可能是血液中的潜在生物标志物,使得cfDNA的检测就如“液态组织活检(liquidbiopsy)”。cfDNA的检测已被广泛应用在孕期胎儿非整倍性染色体检测、肿瘤标记物检测、未知病原检测等领域。器官移植同时也是基因组移植,可通过检测受体血浆中供体细胞游离DNA(cell-freedonor-derivedDNA,cfdDNA),进而判断移植排斥的程度,这种方法为低创或无创检测,可最大程度化降低患者的机体损害。目前基于cfdDNA用于无创器官移植监测按性别区分主要有两种,分别是依赖于性别及不依赖于性别的检测。依赖于性别的检测,通常只适用于男性供体女性受体的情况,使用数字PCR技术(dPCR)进行对移植后受体尿液进行TSPY1(testisspecificprotein,Y-linked1)基因的扩增,通过区分男性供体Y染色体特异基因的表达情况,从而判断排斥程度。不依赖于性别的检测,大多是通过检测移植后受体中的cfdDNA的含量来判断。现有的确定受体中供体细胞游离DNA(cfdDNA)含量的方法,以及器官移植排斥检测手段,有待改进或补充。技术实现要素:本发明旨在至少解决上述问题至少之一或者提供至少一种可选择的商业手段。依据本发明的一方面,本发明提供一种确定供体和受体差异SNP的方法,包括以下步 骤:获得第一测序数据和第二测序数据,所述第一测序数据为所述供体的核酸序列测序结果,包括多个第一读段,所述第二测序数据为所述受体的核酸序列的测序结果,包括多个第二读段;分别将所述第一测序数据和所述第二测序数据比对到参考序列上,获得第一比对结果和第二比对结果;分别基于所述第一比对结果和所述第二比对结果进行SNP检测,获得第一分型结果和第二分型结果,所述第一分型结果包括多个所述供体的SNP的基因型,所述第二分型结果包括多个所述受体的SNP的基因型,其中包括,对于只有一类第一读段并且有多类第二读段比对上的位点、以及只有一类第二读段并且有多类第一读段比对上的位点,分别基于各类第二读段所占的比例和各类第一读段所占的比例进行基因分型,所述各类第一读段之间的区别在于其共同比对上的位点的对应位置上的碱基不同,所述各类第二读段之间的区别在于其共同比对上的位点的对应位置上的碱基不同;比较所述第一分型结果和所述第二分型结果,确定所述差异SNP,所述差异SNP为在所述供体和所述受体中基因型不相同的位点。依据本发明的第二方面,提供一种确定受体中供体来源cfDNA比例的方法,该方法包括:(1)获取来自所述受体的第三核酸样本,所述第三核酸样本包含cfDNA;(2)对(1)中的第三核酸样本中的至少一部分cfDNA进行序列测定,获得第三测序数据,所述第三测序数据包括多个第三读段;(3)将(2)中的第三读段与参考序列比对,获得第三比对结果;(4)基于(3)中的第三比对结果中包含的比对到差异SNP的第三读段的数量,确定所述供体来源cfDNA的比例,所述差异SNP利用上述本发明一方面的确定供体和受体差异SNP的方法来确定。上述本发明一方面的确定供体和受体差异SNP的方法、和/或确定移植后的受体中的cfdDNA的比例的方法的全部或部分步骤,可以利用包含可拆分的相应单元功能模块的装置/系统来施行,或者将方法程序化存储于机器可读介质,利用机器运行该可读介质来实现。依据本发明的第三方面,本发明提供一种确定供体和受体差异SNP的装置,该装置用以实施本发明一方面的确定供体和受体差异SNP的方法的全部或部分步骤,该装置包括:输入单元,用于输入数据;输出单元,用于输出数据;处理器,用以执行可执行程序,执行所述可执行程序包括完成本发明一方面的确定供体和受体的差异SNP的方法;以及存储单元,与所述输入单元、所述输出单元和所述处理器相连,用以存储数据,其中包括所述可执行程序。本领域技术人员能够理解,所称的可执行程序可以保存在存储介质中,所称存储介质可以包括:只读存储器、随机存储器、磁盘或光盘等。依据本发明的第四方面,本发明提供一种确定受体中供体来源cfDNA(cfdDNA)比例 的系统,该系统用以实施上述本发明一方面的确定受体中cfdDNA比例的方法的全部或部分步骤,该系统包括:样本获取装置,用以获取来自移植后受体的第三核酸样本,所述第三核酸样本包含cfDNA;测序装置,与所述样本获取装置相连,用以对样本获取装置中的第三核酸样本中的至少一部分cfDNA进行序列测定,获得第三测序数据,所述第三测序数据包括多个第三读段;比对装置,与所述测序装置相连,用以将来自测序装置的第三读段与参考序列进行比对,获得第三比对结果;确定cfdDNA含量装置,与所述比对装置相连,用以基于来自比对装置的第三比对结果中包含的比对到差异SNP的第三读段的数量,确定所述供体来源cfDNA的比例,所述差异SNP利用上述本发明一方面的确定供体和受体差异SNP的方法或装置来确定。依据本发明的第五方面,本发明提供一种监测器官移植排斥的方法,该方法包括:分别于不同时间点对受体进行采血,获得多个血液样本;利用本发明一方面的确定移植后受体中cfdDNA的含量的方法确定每个所述血液样本中供体来源cfDNA的比例;基于确定的多个所述供体来源cfDNA的比例,进行所述监测。依据本发明的第六方面,本发明提供一种监测器官移植排斥的装置,该装置能够实施上述本发明一方面的监测器官移植排斥的方法的全部或部分步骤,该装置包括:样本获取单元,用以分别于不同时间点对受体进行采血,获得多个血液样本;供体cfDNA比例确定单元,与所述样本获取单元相连,用以利用上述本发明一方面提供的确定受体中cfdDNA比例的方法来确定每个所述血液样本中供体来源cfDNA的比例;监测单元,与所述供体cfDNA比例确定单元相连,用以基于确定的多个所述供体来源cfDNA的比例,进行所述监测。利用上述本发明的方法和/或装置系统,能够确定出供体和受体的差异SNP,以作为区分混合cfDNA中供体和受体来源cfDNA的标记;而通过这些标记位点得到的测序读段的支持情况,利用本发明的方法和/或装置,可准确确定移植后的受体cfDNA样本中的cfdDNA的含量;而将其应用于器官移植排斥检测,由于其为低创或无创的检测,且具有可接受的成本、直观的数字化结果展示,能够作为一种便捷、早期、无创和准确的移植排斥监测辅助技术,为临床判断移植排斥程度提供建议,或者作为临床检测移植排斥的辅助或补充手段。附图说明本发明的上述和/或附加的方面和优点从结合下面附图对实施方式的描述中将变得明显和容易理解,其中:图1是本发明的一个实施例中的基于确定供体和受体差异SNP的器官移植排斥监测实验的流程图。图2是本发明的一个实施例中的基于高通量测序平台的基因分型实验的流程图。图3是本发明的一个实施例中的基于高通量测序平台的血浆cfDNA检测实验的流程图。图4是本发明的一个实施例中的捕获范围模似结果的示意图。图5是本发明的一个实施例中的校正供体cfDNA比例与真实供体cfDNA比例关系的示意图。图6是本发明的一个实施例中的数据分析方法的流程图。具体实施方式下面结合附图和具体实施方式对本发明进行详细说明。所述实施方式在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。在本文中,所使用的术语“第一”、“第二”等仅用于描述目的,而不能理解为指示或暗示相对重要性、隐含指明所指示的技术特征的数量或者具有顺序关系。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括一个或者多个该特征。在本发明的描述中,除非另有说明,“多个”的含义是两个或两个以上。在本文中,除非另有明确的规定和限定,术语“顺序连接”、“相连”、“连接”等术语应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通。对于本领域的普通技术人员而言,可以根据具体情况理解上述术语在本发明中的具体含义。在本文中,所称的供体和受体,是相对个体,是基于移植,例如器官移植时的器官供给一方和接受一方来说的。供体和受体可以是相同物种,也可以是亲缘关系近的能够或者可能能够进行器官或组织移植的不同物种。在本文中,所称的差异SNP,是指供体和受体基因组上或染色体上的具有不同基因型的相同位点,其中包括方便基于测序数据用于确定移植后受体中cfdDNA含量、和/或监测移植排斥的位点,即诊断SNP。下文的公开了不同的具体实施方式或实施例用来实现本发明的不同方法步骤或装置结构。为了简化本发明的公开,下文中对特定例子的步骤和设置进行描述。当然,它们仅仅 为示例,并且目的不在于限制本发明。此外,本发明可以在不同例子中重复参考数字和/或参考字母,这种重复是为了简化和清楚的目的,其本身不指示所讨论各种实施方式和/或设置之间的关系。根据本发明的一个具体实施方式提供的确定供体和受体差异SNP的方法,包括以下步骤:S10获得第一测序数据和第二测序数据。所称第一测序数据为供体的核酸序列测序结果,包括多个第一读段,所述第二测序数据为移植前受体的核酸序列的测序结果,包括多个第二读段。所称的测序数据通过对核酸序列进行测序得来,测序依据所选的测序平台的不同,可选择但不限于Illumina公司的Hisq2000/2500测序平台、LifeTechnologies公司的IonTorrent平台和单分子测序平台,测序方式可以选择单端测序,也可以选择双末端测序,获得的下机数据是测读出来的片段,称为读段(reads)。根据本发明的一个实施例,所称第一或者第二测序数据中的读段的长度不相同,测序数据利用华大基因的BGISEQ-100测序平台或者LifeTechnologies公司的IonTorrent系列中的Proton测序平台对基因组核酸序列进行测序获得。所测核酸序列通常是将来个体的基因组DNA样本经过打断获得的,接着根据所选用的测序方法或测序平台进行相应的测序文库(library)制备,进而将测序文库上机测序,获得下机数据即测序数据。根据本发明的实施例,S10获得第一测序数据和第二测序数据包括:S11获取第一核酸样本和第二核酸样本,所称第一核酸样本和第二核酸样本分别来自供体和移植前的受体,第一核酸样本和第二核酸样本均包含基因组DNA;S13分别提取第一核酸样本和第二核酸样本中的基因组DNA;S15分别对来自S13的基因组DNA进行捕获,对应获得第一目的片段和第二目的片段;S17对第一目的片段和第二目的片段进行序列测定,以获得所称的第一测序数据和第二测序数据。其中,S15分别对所述基因组DNA进行捕获包括基因组文库构建以及从基因组文库中获得目标文库:S150分别对来自S13的基因组DNA进行片段化,获得第一DNA片段和第二DNA片段,较佳的,片段化时使获得的第一DNA片段和第二DNA片段的大小均为150-250bp;S151分别对来自S150的第一DNA片段和第二DNA片段进行末端修复,获得第一修复片段和第二修复片段;S152分别对来自S151的第一修复片段和第二修复片段进行测序接头连接,获得第一连接产物和第二连接产物;S153分别对来自S152的第一连接产物和所述第二连接产物进行大小选择,获得预定大小的第一连接产物和预定大小的第二 连接产物,基于S150片段化时获得的第一DNA片段和第二DNA片段的大小均为150-250bp以及S152所连接的测序接头的长度一般不大于100bp,较佳的,所选择的预定大小的第一连接产物和预定大小的第二连接产物的大小均为210-270bp;S154对来自S153的预定大小的第一连接产物和预定大小的第二连接产物进行扩增,获得第一扩增产物和第二扩增产物;以及S155分别对来自S154的第一扩增产物和第二扩增产物进行捕获。捕获可以利用固相芯片进行,也可以利用液相芯片进行,本实施例对捕获方式不作限制。根据本发明的实施例,利用液相芯片进行所述捕获,设计探针使能够捕获的区域,即目标区域包括基因组每条染色体上每Mb的20个SNP位点,所称的每Mb的20个SNP位点为该Mb上次等位基因频率最接近0.5的20个SNP。基于测序数据包含这些捕获的SNP,足以获得足够数目的差异SNP来区分供受体的cfDNA。根据本发明的一个较佳实施例,为了后续能够准确确定移植后受体中的cfdDNA的含量,需确保捕获足够的SNP位点以能够从其中确定出足够数目的差异SNP作为区分供体和受体的cfDNA的标记,基于此,发明人评估了差异SNP个数对计算得的移植后的受体中的cfdDNA比例的影响,发明人将从上述捕获的SNP位点确定得的差异SNP个数按1至0.1的比例依次递减0.1,发现计算得的cfdDNA比例基本保持不变,由此确定更节约数据、捕获成本以及测序成本的一个较佳实施例为,使捕获的区域包括基因组每条染色体上每Mb的2个或者至少2个SNP位点,所称每Mb的2个或者至少2个SNP位点为该Mb上次等位基因频率最接近0.5的SNP。由此,可以使用更小的捕获芯片、更低的数据量,也即更低的成本来检测cfdDNA,达到移植排斥监测的目的。通常所说的SNP都是二态性的,基因型是指同源染色体上一对等位位点的类型的组合。所称的SNP的次等位基因频率,也叫最小等位基因频率(minorallelefrequency,MAF)是指该SNP的频率较低的等位基因在给定人群中的频率。SNP的MAF可以依据数据库公开的信息,在该实施例中,挑选的MAF符合要求的SNP是通过查找NCBI中的dbSNP数据库信息来确定的。MAF越高,即越接近0.5,说明该SNP在群体中杂合频率越高,最后被确定为供体和受体的差异SNP的可能性越大。S20分别将第一测序数据和第二测序数据比对到参考序列上,获得第一比对结果和第二比对结果。将测序数据中的读段比对到参考序列上(readsmapping或者readsalignment),是指将测序得到的DNA片段(也就是reads)定位在基因组上。通过读段定位,在克服测序产生的Reads过短导致的技术困难的同时,也方便利用基因组位置作为桥梁,来将测序得到的数据与前期研究产生的注释结果进行整合。读段比对定位往往被作为测序数据分析的第一 步,其质量的好坏以及速度的快慢,都会直接对后续的分析工作产生影响。在比对过程中,根据比对参数的设置,reads最多允许有n个碱基错配(mismatch),n优选为1或2,若reads中有超过n个碱基发生错配,则视为该对reads无法比对到参考序列。具体比对时,可使用各种比对软件,例如SOAP(ShortOligonucleotideAnalysisPackage),bwa,Tmap等,本实施方式对此不作限定。所说的参考序列是已知序列,可以是预先获得的目标个体所属生物类别中的任意的参考模板,例如,同一生物类别的已公开的基因组组装序列,若混合核酸样本为来自人类,其基因组参考序列(也称为参考基因组)可选择NCBI数据库提供的HG19。比对结果包含各条读段与参考序列的比对情况,包括读段是否能够比对上参考序列、读段比对到参考序列的位置、比对到参考序列的唯一位置还是多个位置、某一位点多少读段比对上、比对上某位点的读段的相应位置的碱基类型等。S30分别基于第一比对结果和第二比对结果进行SNP检测,获得第一分型结果和第二分型结果。所称第一分型结果包括多个供体的SNP的基因型,第二分型结果包括多个受体的SNP的基因型,其中包括,对于只有一类第一读段并且有多类第二读段比对上的位点、以及只有一类第二读段并且有多类第一读段比对上的位点,分别基于各类第二读段所占的比例和各类第一读段所占的比例进行基因分型,所称各类或者说不同类的第一读段之间的区别在于其共同比对上的位点的对应位置上的碱基不同,所称各类或者不同类别第二读段之间的区别在于其共同比对上的位点的对应位置上的碱基不同。SNP检测或者SNP识别可以利用各种SNP识别软件,包括但不限于SOAPsnp、SomaticSniper、CaVEMan、SAMtools、MuTect和TVC。将比对上同一位点的读段分成不同类别,是基于比对上的读段中的对应位置的碱基不同来进行的,例如比对到参考序列碱基为A的位点的读段中,一部分读段的该位置上的碱基为A,另一部分读段的该位置上的碱基为G,则比对到该位点的读段分为两类。对于与参考序列碱基一致的或者不一致的纯合子位点,常规的SNP识别分型软件不能对其进行分型。根据本发明的一个实施例,利用TVC软件进行SNP识别及分型,对于与参考序列碱基一致的纯合位点,TVC软件没办法对其分型。为最大化位点分型,在本发明的实施例中,利用SNP的各类读段的支持情况来分型。所称的对于只有一类第一读段并且有多类第二读段比对上的位点、以及只有一类第二读段并且有多类第一读段比对上的位点,分别基于各类第二读段所占的比例和各类第一读段所占的比例进行基因分型,包括进行以 下a和/或b:a.依据所占的比例大于95%的那一类第一读段或者那一类第二读段,确定该位点的基因型;b.依据所占的比例大于等于25%且小于等于95%的多类第一段读段或者多类第二读段中的所占比例最大的前两类第一读段或者前两类第二读段,确定该位点的基因型。所称的只有一类第一读段并且有多类第二读段比对上的位点代表该位点在供体中的基因型为纯合子、在受体中的基因型可能为杂合子;类似的,所称的只有一类第二读段并且有多类第一读段比对上的位点,表示该位点在供体中的基因型可能为杂合子、在受体中的基因型为纯合子。这两类利用常规分型软件不能对其进行基因分型。而对于规则a,即比对上一位点的读段中的某一类读段的比例大于95%,则认为该位点为纯合子,组成碱基为比例大于95%的这类比对上的读段的相应位置的碱基。对于规则b,即对比上一位点的读段中有两类或者多于两类的读段的比例介于25%到95%,则认为该位点是杂合子,碱基组成为其中的比例最大,即相对最接近95%的两类读段相应位置上的碱基。为使上述依据读段的支持比例进行分型的分型结果准确,对后续分析有意义,在进行a和/或b之前,分别对第一读段和第二读段进行去重,去掉由于文库构建过程的扩增带来的重复。S40比较第一分型结果和第二分型结果,确定差异SNP。所述差异SNP为在所述供体和所述受体基因组中基因型不相同的位点。由于移植后的受体的血浆cfDNA样本中,受体来源的cfDNA的比例远高于cfdDNA,为方便依据确定的差异SNP的读段比对或支持数目进行移植后受体中的cfdDNA含量的确定、辅助或补充用于移植排斥检测,根据本发明的实施例,将所称差异SNP中包括的,在受体基因组中为纯合子、在供体基因组中为杂合子或者为不同纯合子的位点,定义为诊断SNP,可将诊断SNP相应表示为AAab或者AAbb,其中,AA表示所述差异SNP在受体中的基因型,ab或bb表示相同所述差异SNP在供体中的基因型。需要说明的是,用4个字母以及用大小写字母来表示诊断SNP的基因型,只是为方便表示以及区分其在供体或受体中的基因型,非指特定或真实碱基。根据本发明的另一个具体实施方式提供的一种确定受体中供体来源cfDNA比例的方法,该方法包括:(1)获取来自所述受体的第三核酸样本,所述第三核酸样本包含cfDNA;(2)对(1)中的第三核酸样本中的至少一部分cfDNA进行序列测定,获得第三测序数据,所述第三测序数据包括多个第三读段;(3)将(2)中的第三读段与参考序列比对,获得第三比对结果;(4)基于(3)中的第三比对结果中包含的比对到差异SNP的第三读段的数量,确定所述供体来源cfDNA的比例,所述差异SNP利用上述本发明一方面或者任一实 施例中的确定供体和受体差异SNP的方法来确定。根据本发明的一个实施例,第三核酸样本来自移植后的受体的外周血。外周血血液或血浆中包含细胞游离DNA。移植后的受体的外周血样本一般是混合核酸样本,包含目前技术能够检测区分出或者不能检测区分出的供体来源的cfDNA。前述对测序、读段、比对、参考序列、比对结果以及差异SNP的定义、包含的情况、可采取的方式以及参数或设置的影响,在该方法(1)-(3)步骤中同样适用,在此不再赘述。比对结果中,将比对上某位点的读段中的相应位置与该位点包含的任一碱基类型的相同的读段称为支持该位点的读段,例如,某一杂合SNP为C/T,在DNA上本来碱基为C(参考序列上为C),比对上该杂合位点的读段中,若读段上的该位置的碱基为C或T,则为支持该位点的读段;类似的,若读段上的该位置的碱基为C,则称该类读段为支持该位点的这一等位位点的读段。根据本发明的实施例,所述差异SNP包括在所述受体中为纯合子、在所述供体中为杂合子或者为不同纯合子的位点,表示这些差异SNP为AAab或AAbb,其中,AA表示所述差异SNP在受体中的基因型,ab或bb表示相同所述差异SNP在供体中的基因型;(4)包括:利用以下公式计算所述供体来源cfDNA的比例,供体来源其中,N表示第三读段的数量,NAB(B)表示支持AAab位点的等位位点b的第三读段的数量,NBB(B)表示支持AAbb位点的等位位点b的第三读段的数量,NAB(A)表示支持AAab位点的等位位点A或a的第三读段的数量,NAB(A)表示支持AAbb位点的等位位点A的第三读段的数量。为消除或减少测序文库构建、测序和/或比对错误带来的影响,根据本发明的一个较佳实施例,(4)还包括:利用比对到在所述供体和所述受体中为相同纯合基因型的位点的第三读段的数量,对所述供体来源cfDNA的比例进行错误校正,以获得校正后的供体来源cfDNA的比例,表示所述相同纯合基因型的位点为CC。利用以下公式确定所述校正后的供体来源cfDNA的比例,校正后的供体来源其中,NCC(error)表示比对到CC位点的该位置非C的第三读段的数量,即比对到CC位点但不支持C的第三读段的数量;NCC(C)表示比对到CC位点的该位置为C的第三读段的数量,即比对到该位点且支持该等位位点的第三读段的数量。发明人进行试验模拟,模拟出多个cfdDNA比例确定的cfDNA样本,通过上述校正后的公式计算出的cfdDNA比例值,与真实的cfdDNA 比例呈高度线性相关(r>0.99),说明利用该方法能够准确确定移植后的受体cfDNA中的cfdDNA含量。根据本发明的再一个实施方式提供的一种确定供体和受体差异SNP的装置,该装置用以实施上述本发明任一实施方式中的确定供体和受体差异SNP的方法的全部或部分步骤,该装置包括:输入单元,用于输入数据;输出单元,用于输出数据;处理器,用以执行可执行程序,执行所述可执行程序包括完成本发明任一实施方式中的确定供体和受体的差异SNP的方法;以及存储单元,与所述输入单元、所述输出单元和所述处理器相连,用以存储数据,其中包括所述可执行程序。本领域技术人员能够理解,所称的可执行程序可以保存在存储介质中,所称存储介质可以包括:只读存储器、随机存储器、磁盘或光盘等。上述对本发明的任一实施方式中的确定供体和受体差异SNP的方法的技术特征和优点的描述,也适用于本装置,在此不再赘述。根据本发明的又一个实施方式提供的一种确定受体中供体来源cfDNA(cfdDNA)比例的系统,该系统用以实施上述本发明任一实施方式中的确定受体中cfdDNA比例的方法的全部或部分步骤,该系统包括:样本获取装置,用以获取来自移植后受体的第三核酸样本,所述第三核酸样本包含cfDNA;测序装置,与所述样本获取装置相连,用以对样本获取装置中的第三核酸样本中的至少一部分cfDNA进行序列测定,获得第三测序数据,所述第三测序数据包括多个第三读段;确定cfdDNA含量装置,与所述测序装置相连,用以基于来自测序装置的第三测序数据中支持差异SNP的第三读段的数量,确定所述供体来源cfDNA的比例,所述差异SNP利用上述本发明任一实施方式的确定供体和受体差异SNP的方法或装置来确定。上述对本发明的任一实施方式中的确定受体中cfdDNA比例的方法的技术特征和优点的描述,也适用于本系统,在此不再赘述。本领域技术人员可以理解,为实施上述具体实施例中的确定受体中cfdDNA比例的方法中的具体步骤,可以进一步利用包含可拆分的相应单元功能模块的装置来施行。根据本发明的一个实施方式提供的一种监测器官移植排斥的方法,该方法包括:分别于不同时间点对受体进行采血,获得多个血液样本;利用上述本发明任一实施方式中的确定移植后受体中cfdDNA的含量的方法确定每个所述血液样本中供体来源cfDNA的比例;基于确定的多个所述供体来源cfDNA的比例,进行所述监测。上述对本发明的任一实施方式中的确定受体中cfdDNA比例的方法的技术特征和优点的描述,也适用于本方法,在此不再赘述。基于准确定量多个时间点的移植后受体cfDNA样本中的cfdDNA含量,进行移植排斥监测,由于其为低创或无创的检测,且具有可接受的成本、直观的数字化结果展示, 能够作为一种便捷、早期、无创和准确的移植排斥监测辅助手段,为临床判断移植排斥程度提供建议,或者作为临床检测移植排斥的辅助或补充手段。根据本发明的又一个实施方式提供的一种监测器官移植排斥的装置,该装置能够实施上述本发明任一实施方式中的监测器官移植排斥的方法的全部或部分步骤,该装置包括:样本获取单元,用以分别于不同时间点对受体进行采血,获得多个血液样本;供体cfDNA比例确定单元,与所述样本获取单元相连,用以利用上述本发明一方面提供的确定受体中cfdDNA比例的方法来确定每个所述血液样本中供体来源cfDNA的比例;监测单元,与所述供体cfDNA比例确定单元相连,用以基于确定的多个所述供体来源cfDNA的比例,进行所述监测。上述对本发明的任一实施方式中的确定受体中cfdDNA比例的方法以及对任一实施方式中的监测器官移植排斥的方法的技术特征和优点的描述,也适用于本装置,在此不再赘述。利用上述本发明的方法和/或装置系统,能够确定出供体和受体的差异SNP,作为区分混合cfDNA中供体和受体来源cfDNA的标记;而通过这些标记位点得到的测序读段的比对或支持情况,利用本发明的方法和/或装置,可准确确定移植后的受体cfDNA样本中的cfdDNA的含量;而将其应用于器官移植排斥检测,由于其为低创或无创的检测,且具有可接受的成本、直观的数字化结果展示,能够作为一种便捷、早期、无创和准确的移植排斥监测辅助技术,为临床判断移植排斥程度提供建议,或者作为临床检测移植排斥的辅助或补充手段。以下结合附图和具体实施例对本发明的方法、装置和/或系统进行详细的描述。下面示例,仅用于解释本发明,而不能理解为对本发明的限制。除另有交待,以下实施例中涉及的未特别交待的试剂、序列(接头、标签和引物)、软件及仪器,都是常规市售产品或者开源的,比如购自LifeTechnologies等。实施例一将本发明的确定差异SNP的方法和/或装置用于器官移植免疫排斥监测,实验部分的思路如图1所示,可分为两步:(1)对器官移植的供体和受体进行目标区域捕获测序,用于基因分型,使能够从基因层面区分供体和受体;(2)对各个采血点的血浆cfDNA进行全基因组测序,分析评估各采血点血浆中供体cfDNA占总cfDNA的百分比。1、基因组目标区域捕获进行SNP分型该部分实验流程如图2所示,先将基因组DNA打断成主带为150-250bp小片段DNA,打断后DNA片段进行末端补平,加接头后,取1ug构建完成的文库,通过自主设计地NimblegenSeqCapEZChoiceLibrary将目标区域进行富集,然后通过PCR扩增后产物即可用于测序分析,其具体步骤如下:1.1外周血样本基因组DNA的提取;1.2取1μg基因组DNA,超声波打断至主带位于150-250bp左右为止;1.3把打断成小片段的DNA修复成平末端;1.4加上接头后,用琼脂糖电泳切胶法选择210-230bp、230bp-250bp或者250-270bp大小的DNA片段;1.5PCR扩增目地片段,用SeqCapEZChoiceLibrary进行SNP位点捕获;1.6PCR扩增捕获的片段,Agilent2100检测文库质量合格后,通过BGISEQ-100测序平台进行高通量测序。2、各采血点的血浆cfDNA检测血浆cfDNA检测实验流程如图3所示,各采血点血浆分离后,提取cfDNA,末端修复后加接头,最后进行一个PCR扩增,具体步骤如下:2.1两步离心法分离各采血点血浆,避免基因组污染,提取血浆cfDNA;2.2浆cfDNA修复成平末端;2.3加上接头并进行PCR扩增,用Agilent2100进行文库质量检测合格后,通过BGISEQ-100测序平台进行高通量测序。上述步骤1.5中所用的SeqCapEZChoiceLibrary为华大基因自主设计,罗氏合成的SNP捕获芯片。本液态芯片的目标区域的确定包括:从HapMap所有三个阶段以及千人基因组计划(1kGP)里面选取杂合的位点为原始目标区域;在每条染色体上每Mb长度内选择20个次等位基因频率最接近0.5的SNP位点,确定共有56049个SNP点可进行探针设计,委托罗氏公司进行捕获探针的合成。获得最终芯片大小5.7M,包含56049个目标SNP位点,针对目标区域7.7M,一次可以捕获15-20个样本。对于器官移植患者,无论哪种排斥反应(急性、慢性),控制不及时都可能导致病人死亡。因此在临床工作中,早期发现和诊断排斥反应,对于及时采取防治措施具有重要指导意义。目前已有的高通量测序检测方法成本过高,难以推广。利用本发明的方法和/或装置能够成功解决成本问题,而且技术上的主要有益效果还体现在以下两个方面:一、在一定程度上缩小捕获范围不会引起供体比例的显著变化;对器官移植患者的供体和受体进行目标区域捕获测序,会降低测序的物理覆盖度,为研究目标区域捕获测序对供受体cfDNA含量百分比的影响,我们进行了一个模似分析。如图4所示,取市售的全基因组SNP芯片(180M)对移植前供受体样本进行基因分型,对全基因组测序所得的可用SNP数目,按梯度10%减少其数据量,计算各个梯度的供受体cfDNA百分比。从图4中可以看出,可用SNP位点个数随机去除90%后,供受体百分比从3.1%下降为2.95%,没有发生显著的变化。因而我们采取目标区域捕获测序代替芯片分型技术是可行的。二、利用目标区域捕获芯片,大幅降低成本。cell-freeDNA的检测首先依赖于对供体及受体进行基因分型,此过程涉及对上万个SNP位点的多态性进行分析,这一技术我们选择的是高通量芯片捕获测序平台,从而相对于已有的基于芯片分型的技术成本大幅降低;但是目前市面上高通量芯片的设计和合成几乎均依靠国外公司,因此在实验成本和实验周期上都带来很大的负担。发明人基于信息分析平台,开发了探针芯片设计流程,能够根据项目需求快速的对捕获芯片进行设计和调整,同时依赖于芯片合成平台,能够自行合成国产芯片。因此在降低芯片成本和缩短实验周期上都带来了很大的提升。实施例二为证实实验方法可行,发明人设计了本实施例。设计思路如下:取2个正常人血样(取自志愿者),一个作为供体,另一个作为受体进行模拟。采取的血样分离血细胞与血浆,血细胞提取基因组DNA后,打断DNA并进行目标区域捕获测序,用于基因分型;血浆提取cfDNA后,Agelint2100测定其准确浓度,供受体的cfDNA按0%、0.5%、1.5%、3.5%、5.5%、8%比例混合,然后将混合的cfDNA建库测序,用以检测本实验方法的可靠性。按照上述实验步骤,将该实施例步骤分为两步,1、基因组目标区域捕获测序;2、各混合cfDNA建库测序。具体如下。实施例中接头、PCR扩增引物由Invitrogen公司合成,所使用的C0T1DNA购买于Invitrogen公司。所用试剂信息如下表所示:1、基因组目标区域捕获测序1)分离血浆和血细胞①抗凝管取血(5ml)后,颠倒混匀5-6次充分混匀;②水平离心机,1600g,4℃离心10min;③将上清(约1.5ml)分装到2ml管中,下层即为血细胞;④16000g,4℃离心10min去除残余细胞,将上清转放新的1.5ml管中,标记后-80℃保存。2)基因组DNA的提取取200μl分离的血细胞进行基因组DNA提取,具体步骤参见试剂盒说明书。3)样品打断(Fragmentation)①在Bioruptor仪器制冷水槽中加Milli-Q的水,水面介于MAX线与MIX线之间;②开启制冷开关并浆制冷仪的温度设置为4℃;③当制冷水槽的水温达到4℃时开启Bioruptor仪器,制冷水槽中的水会被运送到打断运行槽中,并开始循环流动;④用Nuclease-freewater或(1×TE)将1μg的gDNA稀释到100μL,混匀后用移液器小心的转入打断小管中;点击“set”,按下表设置参数:ON30s,OFF30s,5个Cycle;将打断管放入打断转盘装置,并放进打断槽中。点击“run”按钮,盖上仪器盖子,样本开始 打断;仪器停止后将样品取出,涡旋振荡10s混匀瞬离后冰浴3min,重复步骤④共6次;⑤取出2μL的样品用于电泳检测打断效果,主带位于150-250bp左右视为合格。4)打断后DNA的纯化(AgencourtAMPurebeads)①使用前将磁珠置于室温下平衡30min;②将100μL打断后的DNA转入1.5mL的EP管中,加入1.8倍体积的磁珠(180μL),用移液器吹打10次混匀;③室温下静置10min使磁珠与DNA充分结合,然后瞬时离心3秒;④将EP管放到磁力架上至液体澄清,用移液器小心的去除上清;⑤保持EP管在磁力架上,加入500μL70%的乙醇洗涤磁珠表面,以除去盐离子以及未吸附的DNA等,除去乙醇,重复一次;⑥瞬时离心,尽量完全去除乙醇,并将磁珠置于开盖置于磁力架上,至磁珠表面没有光泽(约10分钟);⑦加入25μLElutionBuffer,轻轻将磁珠从管壁冲洗下来并吹打10次混匀;室温静置10min以使DNA完全从磁珠上洗脱下来;⑧将EP管放到磁力架上至液体澄清。将25μL洗脱下来的DNA转入一个新的EP管中。5)末端修复在1.5ml的离心管中配制末端修复反应体系:上述100μL反应混合物轻微振荡混合均匀,瞬时离心,在Thermomixe或水浴锅中20℃温浴30min。6)末端修复产物的纯化(AgencourtAMPurebeads)加入1.8倍体积的磁珠(180μL)纯化,用22μLElutionBuffer洗脱7)Adapter的连接(AdapterLigation)在1.5ml的离心管中配制Adapter连接反应体系,体系如下表。上述100μL反应混合物轻微振荡混合均匀,瞬时离心后置于Thermomixer中20℃温浴15min。8)AgencourtAMPurebeads纯化连接产物加入1.5倍体积的磁珠(150μL)纯化,用32μLElutionBuffer洗脱9)片段选择①每个样品称取1.3g的琼脂糖于65ml的1×TAE中;②点样之前加入1μl上样缓冲液再次检查胶孔是否漏液;③使用NEB50bpDNALadder,须取出1μl与2μl上样缓冲液充分混匀后点样;④将来自步骤5.8的样品分别至少与10μl上样缓冲液充分混合;⑤先130使样品跑入胶中,再100V电压下电泳120min;⑥100ml的电泳缓冲液1×TAE,加入10μl核酸染料EB充分混匀待用;⑦电泳结束后取出凝胶,放到染胶盘中染色10min;⑧在凝胶系统中拍照存档;⑨以Marker为参照,切230bp-250bp回收,再分别切210bp-230bp和250bp-270bp作为备份;⑩完成切胶后将剩下的胶块放在保鲜膜或PE手套上,用凝胶成像系统中拍照并存档。确认一切没有问题后可将所剩凝胶丢弃至垃圾桶中;10)片段凝胶回收(QIAquickGelExtractionKit)①往需回收的凝胶中加入6倍体积(600μl)缓冲液QG。②50℃孵育10min,期间颠倒混匀3~5次,以帮助凝胶溶解。③往步骤5.10.2的溶液中加入1倍体积(100μl)预冷的异丙醇,充分混匀。④将步骤5.10.3的溶液加入到核酸吸附柱(MinEluteSpinColumn)中,室温静置2min,17900g离心1min。⑤将步骤5.10.4的滤液重新加入到吸附柱中,室温静置2min,17900g离心1min,弃滤液。⑥往吸附柱中加入500μl缓冲液QG,17900g离心1min,弃滤液。⑦往吸附柱中加入750μl缓冲液PE,室温静置2~5min,17900g离心1min,弃滤液,重新17900g离心1min。⑧将吸附柱转移到新的1.5ml离心管中,环吸后室温静置数分钟以晾干吸附柱中残留的液体。⑨往核酸吸附柱的膜中间悬空加入35μl缓冲液EB,室温静置4min,17900g离心1.5min。11)片段浓度测定(Qubit)12)Non-Captured样品LM-PCR在0.2mL管中配制PCR反应体系:置于PCR仪中按照下列程序反应:72℃20min,95℃5min,8个循环的95℃30s/58℃30s/70℃1min/72℃5min,4℃Hold。13)PCR产物的纯化加入1.5倍体积的AgencourtAMPurebeads(150μL)纯化,用32μLElutionBuffer洗脱。14)Pooling将各文库等比例Pooling成1μg。15)杂交杂交前准备①将heatblock调到95℃②将分装好的4.5μLExomeLibrary从-20℃冰箱中拿出,放在冰上化冻。样品的杂交①在一个1.5mL的PE管中加入:②盖好管盖,用干净的50ml注射器针在分装的EP管盖上戳一个孔,将上述样品文库和block的混合物置于浓缩仪中蒸干,温度设置为60℃;③使用新的离心管管盖替换戳孔的管盖,标记,并分别加入以下两种试剂:④将样品震荡混匀后置于离心机上全速离心10秒。将离心后样品转移至95℃heatblock中10分钟使DNA变性;⑤将样品取出,震荡混匀后室温条件下全速离心10秒;⑥将上述杂交混合物转入分装好的4.5μLExomeLibrary中;⑦震荡混匀后置于离心机上全速离心10秒;⑧放在PCR仪上57℃杂交64h,PCR仪热盖应设置保持在105℃;16)捕获序列的洗涤和洗脱准备链霉素磁珠①提前从冰箱中拿出链霉素磁珠,vortex磁珠1min,使其充分混匀;②在1.5mL的EP管中加入100μL磁珠(1个样品);③将EP管置于磁力架上至液体澄清,用移液器小心的去除上清;④保持EP管在磁力架上,加入200μL(2倍体积)的StreptavidinDynabeadBindingandWashBuffer;⑤从磁力架上取下EP管,vortex10s混匀,将EP管重新放回磁力架至液体澄清,用移液器小心的去除上清,用移液器小心的去除上清,重复洗两次;⑥用100μL的StreptavidinDynabeadBindingandWashBuffer悬浮磁珠,并将其转入0.2mL的小管中;⑦用磁力架结合磁珠,直到液体澄清,用移液器小心的去除上清,现在磁珠可以用来结合捕获的DNA了。将捕获到的DNA结合到链霉素磁珠上;①将杂交混合物吸出来(记录杂交后剩余体积)加到5.2准备好的磁珠中;②用移液器吹打10次混匀。③将小管放在PCR仪上57℃孵育45min(PCR仪热盖应设置保持在105℃,每隔10min拿出来vortex3s以防止磁珠沉淀。结合了捕获DNA的链霉素磁珠的洗涤①孵育45min后,将混合物从0.2mL的小管中转入1.5mL的EP管中,将EP管置于磁力架上至液体澄清,用移液器小心的去除上清。②加100μL预热到57℃的1XWashBufferI,vortex10s混匀,将EP管置于磁力架上至液体澄清,用移液器小心的去除上清。③从磁力架上取下EP管,加入200μL预热到47℃的1XStringentWashBuffer,用移液器吹打10次混匀。57℃孵育5min,将EP管置于磁力架上至液体澄清,用移液器小心的去除上清。重复本次操作两次,即总共用1XStringentWashBuffer洗三次;④加200μL室温下放置的1XWashBufferI(不用47℃预热的),vortex2min混匀,如果液体溅到管盖上,用手指轻弹EP管使其集中到管低。将EP管置于磁力架上至液体澄清,用移液器小心的去除上清;⑤加200μL室温下放置的1XWashBufferII,vortex1min混匀。将EP管置于磁力架上至液体澄清,用移液器小心的去除上清。⑥加200μL室温下放置的1XWashBufferIII,vortex30s混匀。将EP管置于磁力架上至液体澄清,用移液器小心的去除上清。⑦从磁力架上取下EP管,加入30μLUltraPureWater。17)Captured样品LM-PCRLM-PCR在1.5mL管中为每个样品按下表配制PCR反应体系:置于PCR仪中按照下列程序反应。95℃5min、12个循环的95℃15s/58℃15s/70℃1min、72℃2min、4℃Hold。PCR产物的纯化(AgencourtAMPurebeads)①将PCR混合物(100μL)转入1个1.5mL的EP管中,将EP管放到磁力架上至液体澄清,将上清转移到一个新的EP管中,弃链霉素磁珠。②上清中加入1.5倍体积的磁珠(150μL)进行纯化,用52μLElutionBuffer洗脱;PCR产物的再次纯化(AgencourtAMPurebeads)加入1.5倍体积的磁珠(75μL)进行纯化,用32μLElutionBuffer洗脱;18)文库检测使用Agilent2100Bioanalyzer检测文库产量。2、各混合cfDNA建库测序1)cfDNA提取取200μl血浆到2ml的离心管中,加缓冲液GA到100μl终体积。②加入20μlProteinaseK溶液,涡旋混匀。③加入200μl的缓冲液GB,轻轻颠倒混匀,56℃孵育10min,并不时摇动样品。简短离心以去除管盖内壁的液滴。④加入200μl的无水乙醇。如果室温超过25℃,请将乙醇置冰上预冷。轻轻颠倒混匀样品,室温放置5min,简短离心以去除管盖内壁的液滴。⑤将上一步所得溶液添加到一个吸附柱CR2中(吸附柱放入收集管中),12,000rpm离心30sec,弃废液,将吸附柱CR2放回收集管中。⑥向吸附柱CR2中加入500μl缓冲液GD,12,000rpm离心30sec,弃废液,将吸附柱CR2放回收集管中。⑦向吸附柱CR2中加入600μl漂洗液PW,12,000rpm离心30sec,弃废液,将吸附柱CR2放回收集管中。⑧重复操作步骤⑦。⑨12,000rpm离心2min,倒掉废液。将吸附柱CR2置于室温放置2-5min,以彻底晾干吸附材料中残余的漂洗液。⑩将吸附柱CR2转入一个干净的离心管中,向吸附膜中间位置悬空滴加20-50μl洗脱缓冲液TB,室温放置2-5min,12,000rpm(~13,400×g)离心2min,将溶液收集到离心管中。2)QubitHS测定核酸浓度(2100检测)3)末端修复与纯化①按照下列的配比准备反应混合物:在Thermomixer中20℃,反应30min。②磁珠纯化加入1.8倍体积的磁珠(90μL)进行纯化,用24μLElutionBuffer洗脱;4)DNAAdaptor连接与连接产物纯化①按照下列的配比准备反应混合物在Thermomixer中,20℃反应20min。④磁珠纯化加入1.2倍体积的磁珠(84μL)进行纯化,用32μLElutionBuffer洗脱;⑤Qubit测定核酸浓度5)PCR反应与纯化①PCR体系和反应条件,扩增体系:反应程序:72℃20min、95℃5min、15个循环的95℃30s/60℃30s/70℃30s/70℃5min、12℃∞。②PCR产物的磁珠纯化加入1倍体积的磁珠(100μL)进行纯化,用32μLElutionBuffer洗脱。③Qubit测定核酸浓度。3、结果评价和分析发明人抽取两个自愿者的血液样本,一个做为供体(15ml血液),另一个做为受体(25ml血液),分离血浆和血细胞后,供体得到6.6ml血浆,7.5ml血细胞,受体得到11.4ml血浆,12ml血细胞。得到的样本用于实验。基因组DNA的差异SNP位点确定的实验结果及分析1)基因组DNA提取取200μl分离的血细胞用于提取DNA,用Qubit进行核酸浓度检测,提取的结果如表1所示,结果显示提取正常中,可用于下步实验。表1两志愿者的基因组DNA提取结果2)打断、加接头及胶回收各取1μg基因组DNA超声波打断后,加接头并用琼脂糖电泳进行DNA片段大小选择,我们切取230-250bp和250-270bp大小的片段,其中一份作为备份,胶回收核酸浓度(Qubit检测)如表2所示,胶回收的核酸总量达到杂交捕获的要求,可进行下步实验。表2打断后片段选择结果3)PCR后进行液态芯片杂交捕获进行一个PCR扩增后,各取500ng(共1μg)进行杂交捕获:4)出库浓度目地序列杂交捕获下来,洗脱,进行PCR扩增后即可进行下一步的测序上机,出库浓度如下表3所示,出库浓度符合5.7M芯片杂交正常水平,2100结果正常,可用于测序分析。表3基因组DNA杂交出库结果血浆cfDNA全基因组检测结果发明人人工模拟了0%、0.5%、1.5%、3.5%、5.5%、8%供体比例的实验,即把两个正常人的血浆cfDNA样本按上述比例混合在一起,然后用高通量测序进行检测。1)血浆提取cfDNA的提取供体用6.6ml血浆进行提取,受体用11.4ml血浆进行提取,得到的结果为表4所示,正常人的血浆cfDNA浓度较低,结果显示提取正常,总量能够用于后续实验。表4血浆cfDNA提取结果2)按模拟浓度比例混合cfDNA按我们设计的模拟浓度0%、0.5%、1.5%、3.5%、5.5%、8%进行混合,具体操作如下:3)出库浓度混合好的血浆cfDNA,末端修复后,加上不同的接头,进行一个PCR扩增后,出库浓度如表5所示,出库结果正常,可用于下步测序分析。表5血浆建库结果结果分析与画图上述的文库用BGISEQ-100测序平台进行测序,得到的数据通过生物信息学分析,得了各个点的供受体cfDNA比值,画成线性图后,校正供体比例与真实供体比例关系如图5所示,横坐标表示真实供体比例,纵坐标表示校正供体比例,可以看出,其符合线性规律(R2=0.9917),证明我们的确定差异SNP的方法的灵敏度是足够应用于监测器官移植免疫 排斥的。实施例三获得测序数据后,数据分析方法流程如图6所示,一般包括以下步骤:1.与参考基因组比对。对BGISEQ-100有效测序数据使用tmap工具比对到参考基因组上,得到精确的比对结果。其中tmap工具源自:https://github.com/iontorrent/TS/tree/master/Analysis/TMAP。2.比对结果去除PCR重复片段。对tmap工具比对后的结果(bam格式)使用BamDuplicates工具去除PCR重复片段。其中,BamDuplicates工具来自IonTorrentSystems公司。3.统计及质量控制。统计目标区域数据量占总数据量的比例、目标区域的平均测序深度、目标区域的覆盖率等,生成一系列质控指标用于判断测序数据的质量情况。前3步对目标区域捕获测序及血浆样本均适用。对于血浆样本,在第2步去重后,还需去掉多比对的reads,只获取唯一比对的reads。4.供受体样本基因分型使用TVC工具(默认参数targetseq_germline_lowstringency_p1_parameters.json文件)(参考:http://ioncommunity.lifetechnologies.com/community/products/torrent-variant-caller)分别检测供受体的生殖系SNP(GermlineSNP),得到部分基因分型位点。对TVC工具无法分型的位点,通过频率(读段支持频率)来分型,最大化基因分型位点。频率分型的具体操作步骤为:(1)对供受体去重后的数据分别进行累积排序(pileup),可利用工具samtools进行[LiH,HandsakerB,WysokerA.,etal.1000GenomeProjectDataProcessingSubgroup(2009)TheSequencealignment/map(SAM)formatandSAMtools.Bioinformatics,25,2078-9.[PMID:19505943]。该工具官网地址:http://samtools.sourceforge.net/index.shtml,对pileup结果进行每个位点ACTG碱基的reads支持数统计。(2)统计每个位点的基因型。对读段支持频率>95%的位点定义为纯合子,频率在25%(包含25%)至95%(包含95%)间的位点定义为杂合子。对于有多种基因型的杂合子,取最大频率的碱基作为该SNP的组成碱基。5.获取诊断位点、控制位点获取可区分供受体的单核苷酸多态性(SNP)位点即受体为纯合子且供体为杂合子或与受体不同基因型的纯合子,作为诊断位点。诊断位点包括以下4种情况,见以下表6,其 中,B表示可区分供受体的供体SNP的组成碱基。表6受体基因型受体基因型标签供体基因型供体基因型标签0/0(与参考序列碱基一致的纯合子)AA1/1BB0/0(与参考序列碱基一致纯合子)AA0/1AB1/1(与参考序列碱基不同的纯合子)BB0/0AA1/1(与参考序列碱基不同的纯合子)BB0/1AB获取控制位点即供受体均为基因型相同的纯合子,用来评估比对错误及校正供体比例。包括以下2种情况,见下表,表7。表7受体基因型受体基因型标签供体基因型供体基因型标签0/0AA0/0AA1/1BB1/1BB6.计算供体比例对去重且唯一比对(比对到参考序列唯一位置)的血浆数据,统计诊断位点中来自供体或受体的reads(读段),通过加权公式,计算供体比例,模拟实验发现,加权公式计算的供体比例值与真实供体含量呈准确线性关系(=0.9917),通过线性关系进一步评估真实供体含量。主要包含两部分:(1)校正供体比例(供体cfDNA比例)计算校正供体比例是指已经评估过比对和/或测序错误的供体比例。先计算供体cfDNA比例及比对错误。以下将受体中cfdDNA比例简称为供体比例。A.供体比例=供体来源的reads数/(供体+受体)来源reads数,具体加权公式如下:其中,N表示reads数,NAB(B)表示受体纯合供体杂合的诊断位点中来自供体的reads,NBB(B)表示受体纯合供体纯合且与受体不同基因型诊断位点中来自供体的reads,同理,NAB(A)和NBB(A)及表示诊断位点中来自受体的reads。B.对无法区分供受体的位点即受体纯合供体纯合且基因型相同的位点,将其作为控制位点,可以评估比对和/或测序错误,公式如下:其中,N表示reads数,NCC(C)表示控制位点中正确reads数,NCC(error)表示控制位点中错误reads数。C.校正供体比例为供体比例与比对和/或测序错误的差,即校正供体比例=供体比例- 比对和/或测序错误。(2)与真实cfdDNA的线性拟合我们模拟了0%、0.5%、1.5%、3.5%、5.5%、8%供体比例的实验,计算出真实cfdDNA含量与校正供体比例间的线性拟合关系(r=0.9917)。真实cfdDNA含量=(校正供体比例-0.8986)/0.7515。使用不同大小的捕获芯片,具体的线性系数有所不同。利用程序语言,将上述该示例提供的基于BGISEQ-100测序平台的器官移植无创排斥监测方法编码成一软件包,具有以下优点:1.该软件包程序化本发明的全面、有效的基因分型算法。包括使用TVC等一般工具对供受体进行基因分型,对一般工具无法分型的位点,通过读段支持频率(即各碱基的频率)来分型,最大化基因分型位点。对于目标区域捕获测序来说,为使测序成本在合理范围内,捕获的SNP位点数较少,但使用此基因分型算法,可以获取最多的诊断位点。2.检测值准确反映真实供体含量。模拟实验发现,经加权公式计算所得校正供体比值与真实供体含量呈准确线性关系(=0.9917),通过线性关系细化检测供体比例,使其尽可能接近真实供体含量。3.灵活、一体化的软件包实现,可移植性强,可独立部署、高效运行。4.低创或无创的检测、可接受的成本、直观的数字化结果展示,可作为一种便捷、早期、无创、准确的移植排斥监测技术,可作为临床免疫排斥检测的辅助或补充手段。实施例四选择正常供体(样品名D)及受体(样品名R)的血细胞样品进行目标区域捕获测序,对混合了供体DNA的受体血浆进行全基因组测序,混合比例分别为0%、0.5%、1.5%、3.5%、5.5%、8%,对测序有效数据通过tmap比对、BamDuplicates去重、质量控制(QC)、供受体基因分型、获取诊断位点及控制位点、供体比例计算,最终获得模拟实验的供体含量检测报告,以评估器官移植排斥程度。本检测系统各部流程方法都已整合到软件Donor_Fraction_Calculation_main中,本软件的运行环境为Unix/Linux操作系统,通过Unix/Linux命令行运行。1、具体操作步骤如下:在LINUX操作系统计算机终端中输入以下命令:perlDonor_Fraction_Calculation_main.pl-d-llist-oresultDonor_Fraction_Calculation_main命令行参数见表8参数说明。表8参数说明一个完整的list表举例如下:>RDreceptor1.bamdonor2.bam03.bam0.54.bam1.55.bam3.56.bam5.57.bam88.bam该list表示名为RD(受体供体)的模拟实验,需检测混合了供体DNA比例分别为0%、0.5%、1.5%、3.5%、5.5%、8%采样点文库的供体比例。2、分析结果表9显示统计及质量控制分析的部分节选结果。表9以下表10显示供体cfDNA比例的部分结果节选。表10续:续:续:续:从模拟实验发现,加权公式计算的校正供体比例值与真实供体含量呈准确线性关系(=0.9917),具体线性关系见图5。在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发 明的范围由权利要求及其等同物限定。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1