新的抗微生物化合物、包含该化合物的反应培养基、及其用途的制作方法
本发明涉及新的抗微生物化合物、包含该化合物的新反应培养基、以及他们的用途。具体而言,根据本发明的新化合物证明对于那些对抗微生物剂具有多抗药性的微生物是有效的,尤其是对于那些对常规抗菌药具有多抗药性的细菌是有效的。
背景技术:
对典型抗微生物剂(例如抗菌药)呈现出多抗药性的微生物的增加通常需要研制具有新的作用模式的新的抗微生物剂。特别是对于治疗多抗药性的革兰氏阴性菌,例如那些产生碳青霉烯酶(carbapenemase)的革兰氏阴性菌,新的抗微生物剂的缺乏是令人担忧的,并且证实是特别令人关注的。另外,如果在对于其它微生物而言是特异性的反应培养基中存在所述多抗药性的微生物,则这些多抗药性的微生物能够产生错误性的诊断,给出假阳性结果,并由此诱发检测特异性方面的重大问题。
微生物学诊断技术仍主要基于培养步骤,以分离目标微生物。如果这些技术用于寻找潜在性地存在于大量共生微生物菌丛中的小量病原体,则必须使用选择培养基。这些培养基主要基于特异性抑制剂的使用,这些抑制剂作用于主要共生菌种,但不抑制所要寻找的病原体的生长。
在20世纪70年代后期,“模拟肽”的研制是由于其抗菌性质。这些模拟肽通常由共价连接于一个或多个氨基酸上的“活性”抗菌部分组成,所述氨基酸用于促进这些“模拟肽”通过微生物肽转运系统的吸收。然后,细胞内水解断裂(由于氨肽酶活性)在目标细菌内释放出所述活性抗菌部分,用于与其目标相互作用。对于“模拟肽”,可以提到的有阿拉磷(L-丙氨酰-L-1-氨基乙基膦酸;CAS编号:60668-24-8),其是由Roche在20世纪70年代研发的。据该文献的描述,阿拉磷的细菌吸收是通过LL-二肽透酶进行的。一旦在目标细菌内部,则活性抗菌部分—此时为fosfalin(L-1-氨基乙基膦酸),被水解(更准确地说是通过水解将其与丙氨酸残基结合在一起的肽键而由丙氨酸残基中被“释放”),然后与丙氨酸消旋酶结合,由此阻止D-丙氨酸的合成,而D-丙氨酸对于生物合成肽多糖是必需的成分[1]。阿拉磷已被描述为具有广谱抗菌活性[2],无论是在活体外还是在活体内,都可以抵抗某些需氧革兰氏阳性微生物(金黄色葡萄球菌(Staphylococcus aureus),粪肠球菌(Enterococcus faecalis),但不包括A组链球菌(Group A streptococci)以及B组链球菌(Group B streptococci),如无乳链球菌(S.agalactiae),也不包括肺炎链球菌(Streptococcus pneumoniae))、厌氧菌(拟杆菌属(Bacteroides sp.)以及产气荚膜梭菌(Clostridium perfringens),但不包括艰难梭菌(Clostridium difficile))以及许多革兰氏阴性菌种,但不包括假单胞菌属(Pseudomonas)或不动杆菌属(Acinetobacter)。
在专利申请WO 02/22785以及专利EP-B-1325024中也描述了阿拉磷的应用,其是作为用于研发选择培养基的选择剂,所述选择培养基能够分离属于沙门氏菌属(Salmonella spp)家族的微生物。
然而,Gibson等人[3]在1984年披露了以下事实:由于许多抗药细菌菌株的高发生频率,证明阿拉磷对于这些菌株是无活性的。
在20世纪80年代研制的其他合成抗菌药使用了类似的给药方法和/或作用模式。例如,Cheung等人[4]研究了数种通过水解释放的卤代二肽的抗菌活性,包括L-β-氯丙氨酰-L-β-氯丙氨酸。该通过水解释放的β-氯丙氨酸似乎与某些酶系统(包括丙氨酸消旋酶)相互作用,而且还似乎表现出宽的抗菌活性谱[5]。在试图优化Cheung等人[4]的底物的工作框架中,β-Cl-L-丙氨酸(β-Cl-L-Ala)残基被结合入各种二肽和三肽中[5]。然而,不仅如此得到的肽中没有一个能够显著提高化合物L-β-氯丙氨酰-L-β-氯丙氨酸的抗菌活性,而且该文献的作者得出以下结论:包含L-丙氨酰残基的二肽和三肽尤其是对于革兰氏阴性微生物不具有抗菌活性。该文献的作者由此推断,由包含L-丙氨酰残基的二肽和三肽残基观察到的抗菌活性的缺失有可能与L-丙氨酸为革兰氏阴性菌中的丙氨酸消旋酶提供针对β-Cl-L-Ala残基的保护的性质有关。换言之,相对于丙氨酸消旋酶,在β-Cl-L-Ala残基和L-丙氨酰残基之间似乎发生竞争。
也是出于优化的角度,Atherton等人[6]试图在相同的分子中一方面结合阿拉磷,并且另一方面结合β-氯-L-丙氨酸(β-Cl-L-ala)。然而,该企图以失败告终,因为该杂化分子没有表现出任何的抗菌活性,虽然已知β-氯丙氨酸具有抗菌性质[7]。Atherton等人得出以下结论:该结果是由于L,L二肽透酶不能有效地在细菌内转运所述化合物。
对于典型/常规抗微生物剂的抗药性越来越多,因此一个目的是研发涉及人或者动物治疗的新的抗微生物化合物,而且还涉及使用这些新的抗微生物剂作为选择剂的活体外诊断。如前所述(参考文献[5]和[6]),现存抗微生物化合物的改进以及其它组合都有可能导致其抗微生物活性的降低或者甚至完全丢失,因此该任务变得更为困难。
技术实现要素:
申请人已令人惊奇地发现上述目的能够用具有以下通式(I)的化合物及其盐、衍生物和类似物,优选其盐来实现:
其中R1代表:
-由1-5个氨基酸残基的线性序列组成的肽部分P1;所述肽部分P1包含至少一个β-氯
丙氨酸残基,优选β-氯-L-丙氨酸;或者
-肽部分P2,所述肽部分P2由以下通式(II)代表:
其中:
X代表氢或氯原子,以及
Y代表氢原子或1-4个氨基酸残基的线性序列,有利地包括至少一个β-氯丙氨酸残基,优选β-氯-L-丙氨酸,和/或至少一个正缬氨酸残基,优选L-正缬氨酸,和/或至少一个甲硫氨酸残基,优选L-甲硫氨酸;
以及其中如果X代表氢原子,则Y如前所定义,但不包括氢原子或丙氨酸残基,无论该丙氨酸残基是在N-端位置(“在链端”)或其通过肽键(在所述丙氨酸残基的α氨基官能团与另一个氨基酸残基的α羧酸官能团之间)而结合至该另一个氨基酸残基上。
当然,所用的术语表示,尤其是术语“肽部分”,由多个氨基酸残基组成的线性序列意味着这些多个氨基酸残基是通过肽键结合在一起的。如生物化学领域众所周知的,肽键是在氨基酸的α碳所携带的羧基官能团(“α羧基官能团”)与该肽链中下一个氨基酸的α碳所携带的氨基官能团(“α氨基官能团”)之间所形成的共价键。该肽键相应于酰胺官能团。
优选的是,R1由通式(III)代表:
其中R2是氢原子或者1-4个氨基酸残基的线性序列,有利地包含至少一个β-氯丙氨酸残基,优选β-氯-L-丙氨酸,和/或至少一个正缬氨酸残基,优选L-正缬氨酸,和/或至少一个甲硫氨酸残基,优选L-甲硫氨酸。
如所定义的,如果R2是氢原子,则R1是包含单个氨基酸残基的肽部分,如β-氯丙氨酸残基,优选β-氯-L-丙氨酸。
如前所述,并根据优选实施方案,如果R2是1-4个氨基酸残基的线性序列,则其包含至少一个β-氯丙氨酸残基(优选β-氯-L-丙氨酸),和/或至少一个正缬氨酸残基(优选L-正缬氨酸),和/或至少一个甲硫氨酸残基(优选L-甲硫氨酸)。有利地,上述β-氯丙氨酸残基(优选β-氯-L-丙氨酸)、正缬氨酸残基(优选L-正缬氨酸)或甲硫氨酸残基(优选L-甲硫氨酸)通过肽键直接结合至通式(III)所代表的β-氯-L-丙氨酸残基(所述肽键是在所述β-氯丙氨酸、正缬氨酸或甲硫氨酸残基的α羧酸官能团与通式(III)中所表示的氨基官能团之间形成的)。
根据一个具体的实施方案,R2由正缬氨酸残基,优选L-正缬氨酸或甲硫氨酸残基,优选L-甲硫氨酸组成。
根据优选实施方案,R1由通式(IV)代表:
其中R3是氢原子或1-3个氨基酸残基的线性序列,后者有利地包含至少一个β-氯丙氨酸残基,优选β-氯-L-丙氨酸。
根据化学领域中的标记规则(特别是对于拓扑式),上述式(II)、(III)和(IV)中表示的所谓的“波浪线”或者“锯齿线”符号是用于表示在R1基团与通式(I)化合物的其余部分之间的键(通过肽键的形成)。
如所定义的,如果R3是氢原子,则R1是仅包含两个相同氨基酸残基的肽部分,例如两个β-氯丙氨酸残基,优选β-氯-L-丙氨酸,他们通过肽键相互结合在一起。以此方式形成的单元称为“β-氯丙氨酰-β-氯丙氨酸”。
根据一个具体的实施方案,R3包括正缬氨酸残基,优选L-正缬氨酸,或者甲硫氨酸残基,优选L-甲硫氨酸。有利地,所述正缬氨酸残基(优选L-正缬氨酸)或甲硫氨酸残基(优选L-甲硫氨酸)通过肽键(在所述正缬氨酸或甲硫氨酸残基的α羧酸官能团与通式(IV)所表示的β-氯丙氨酰-β-氯丙氨酸残基的氨基官能团NH-R3之间)直接结合至通式(IV)所代表的β-氯丙氨酰-β-氯丙氨酸残基(优选结合至β-氯-L-丙氨酰-β-氯-L-丙氨酸残基)。如本领域技术人员众所周知的,该肽键的形成是通过β-氯丙氨酰-β-氯丙氨酸单元(优选β-氯-L-丙氨酰-β-氯-L-丙氨酸)的伯氨基官能团与正缬氨酸(优选L-正缬氨酸)或甲硫氨酸(优选L-甲硫氨酸)的羧酸官能团之间的缩合反应而得到的,由此在所述β-氯丙氨酰-β-氯丙氨酸单元与所述正缬氨酸(优选L-正缬氨酸)或甲硫氨酸(优选L-甲硫氨酸)之间形成肽键。根据一个具体的实施方案,R3由正缬氨酸残基,优选L-正缬氨酸,或甲硫氨酸残基,优选L-甲硫氨酸组成。
优选的是,通式(I)化合物的肽部分R1由2-3个、优选2个氨基酸残基组成;如上所解释的,这些氨基酸残基通过肽键结合在一起。
通常而言,所述氨基酸残基选自甘氨酸、肌氨酸,以及L和D形式、优选L形式的β-氯丙氨酸、丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酸、γ-谷氨酸、谷氨酰胺、组氨酸、异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、正缬氨酸、苯丙氨酸、脯氨酸、焦谷氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸和缬氨酸的残基。有利地,所述氨基酸残基选自L和D形式、优选L形式的β-氯丙氨酸、丙氨酸、甲硫氨酸、正缬氨酸和缬氨酸的残基。
根据另一个实施方案,根据本发明的抗微生物化合物由上述通式(I)限定,其中R1代表所述肽部分P2,并且其中X代表氢原子。有利地,在该另一个实施方案中,Y是选自以下的氨基酸残基:甘氨酸、肌氨酸,L和D形式、优选L形式的β-氯丙氨酸、精氨酸、天冬酰胺、天冬氨酸、半胱氨酸、谷氨酸、γ-谷氨酸、谷氨酰胺、组氨酸、异亮氨酸、亮氨酸、赖氨酸、甲硫氨酸、正缬氨酸、苯丙氨酸、脯氨酸、焦谷氨酸、丝氨酸、苏氨酸、色氨酸、酪氨酸和缬氨酸的残基;有利地,Y是选自以下的氨基酸残基:L和D形式、优选L形式的甲硫氨酸和正缬氨酸的残基。
根据特别优选的实施方案,根据本发明的抗微生物化合物包括:
‐β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选β-氯-L-丙氨酰-L-1-氨基乙基膦酸,和/或
‐β-氯-L-丙氨酰-β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸,和/或
‐L-正缬氨酰-β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选L-正缬氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸,和/或
‐L-甲硫氨酰-β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选L-甲硫氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸,和/或
‐L-正缬氨酰-L-丙氨酰-D/L-1-氨基乙基膦酸,优选L-正缬氨酰-L-丙氨酰-L-1-氨基乙基膦酸,和/或
‐L-甲硫氨酰-L-丙氨酰-D/L-1-氨基乙基膦酸,优选L-甲硫氨酰-L-丙氨酰-L-1-氨基乙基膦酸;
有利地,所述抗微生物化合物包括β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选β-氯-L-丙氨酰-L-1-氨基乙基膦酸,和/或
β-氯-L-丙氨酰-β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸。
有利地,根据本发明的抗微生物化合物包括β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选β-氯-L-丙氨酰-L-1-氨基乙基膦酸,和/或β-氯-L-丙氨酰-β-氯-L-丙氨酰-D/L-1-氨基乙基膦酸,优选β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸,或这两者的混合物。
根据本发明的优选实施方案,根据本发明的抗微生物化合物的N-端氨基官能团用保护基进行保护,所述保护基例如是叔丁氧基羰基、9-芴基甲氧基羰基或苄基氧基羰基。
本发明还涉及包含至少一种如上所定义的抗微生物化合物的反应培养基,所述化合物的最终存在浓度为0.002至1024.0mg/L,优选0.003至32.0mg/L,有利地为0.2至8.0mg/L,优选为0.2至2.0mg/L。
根据优选实施方案,所述反应培养基是一种能够检测和/或鉴定和/或计数样品中至少一种靶微生物、优选至少一种靶细菌的培养基,所述样品能够包含所述靶微生物,例如是工业来源或者临床来源的样品,其中所述至少一种化合物是至少一种能够抑制非靶微生物的存活和/或生长以有利于所述至少一种靶微生物的存活和/或生长的选择剂。
有利地,所述反应培养基是包含至少一种营养剂的培养基,所述营养剂能够使至少一种靶微生物生长,其中所述至少一种选择剂能够抑制非靶微生物的生长,由此有利于所述至少一种靶微生物的生长。
优选的是,所述反应培养基还包含对所述至少一种靶微生物的酶活性具有特异性的酶底物。
优选的是,所述反应培养基包含至少一种选自以下的选择剂:
‐β-氯-L-丙氨酰-L-1-氨基乙基膦酸,优选其最终浓度为0.002至1024.0mg/L,优选0.003至32.0mg/L,有利地为0.2至8.0mg/L,优选0.2至2.0mg/L,或
‐β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸,优选其最终浓度为0.002至1024.0mg/L,优选0.003至32.0mg/L,有利地为0.2至8.0mg/L,优选0.2至2.0mg/L,或者
‐这两者的混合物。
根据有利的实施方案,所述反应培养基能够检测和/或鉴定和/或计数至少一种靶微生物(目标微生物,microorganism of interest),所述至少一种靶微生物是:
‐至少一种革兰氏阴性靶微生物,其属于沙门氏菌属,例如肠沙门氏菌(Salmonella enterica);属于鼠伤寒沙门氏菌(Salmonella Typhimurium)血清型或肠沙门氏菌(Salmonella Enteridis)血清型;属于不动杆菌属,例如鲍氏不动杆菌(Acinetobacter baumannii);属于伯克霍尔德氏菌属(Burkholderia),洋葱伯克霍尔德氏菌(Burkholderia cepacia);属于假单胞菌属,例如铜绿假单胞菌(Pseudomonas aeruginosa);有利地,所述至少一种革兰氏阴性微生物属于沙门氏菌属;或者
‐至少一种革兰氏阳性靶微生物,其属于利斯特氏菌属(Listeria),例如属于单核细胞增生利斯特氏菌(Listeria monocytogenes),或属于链球菌属(Streptococcus),例如无乳链球菌(Streptococcus agalactiae)和/或肺炎链球菌和/或酿脓链球菌(Streptococcus pyogenes);有利地,所述至少一种革兰氏阳性靶微生物属于利斯特氏菌属。
根据有利的实施方案,所述反应培养基能够检测和/或鉴定和/或计数至少一种属于沙门氏菌属的靶微生物。
根据特别有利的实施方案,所述反应培养基能够检测和/或鉴定和/或计数至少一种属于沙门氏菌属的靶微生物,所述培养基包含:
‐至少一种营养剂,例如蛋白胨,如猪或牛来源的,其浓度为0.2至30.0g/L,
‐任选存在的缓冲剂,
‐至少一种显色标记物,例如酯酶酶底物和/或α-半乳糖苷酶酶底物,其浓度为0.05至15.0g/L,
‐琼脂,其浓度为9.0至28.0g/L,以及
‐至少一种如权利要求12-16之一所限定的选择剂,优选其浓度为0.002至1024.0mg/L,优选0.003至32.0mg/L,有利地为0.2至8.0mg/L,优选为0.2至2.0mg/L。
有利地,所述反应培养基包含至少一种酯酶酶底物和/或至少一种α-半乳糖苷酶酶底物的混合物。其还可包含葡糖苷酶酶底物(有利地为β-葡糖苷酶)和/或半乳糖苷酶酶底物。根据优选实施方案,所述反应培养基包含至少一种酯酶酶底物或至少一种α-半乳糖苷酶酶底物以及至少一种β-葡糖苷酶酶底物。根据特别优选的实施方案,所述反应培养基包含至少一种酯酶酶底物以及一种β-葡糖苷酶酶底物。
该类型的反应培养基的例子示于下表2中(参见以下实施例3.1)。
优选的是,所述一种或多种非靶微生物是在以下组内:
-肠杆菌科(Enterobacteriaceae)家系中的属,例如肠杆菌属(Enterobacter)(例如阴沟肠杆菌(Enterobacter cloacae))和/或埃希氏菌属(Escherichia)(例如大肠埃希氏菌(Escherichia Coli))和/或克雷伯氏菌属(Klebsiella)(例如肺炎克雷伯氏菌(Klebsiella pneumoniae))和/或沙雷氏菌属(Serratia)(例如粘质沙雷氏菌(Serratia marcescens))和/或耶尔森氏菌属(Yersinia)(例如小肠结肠炎耶尔森氏菌(Yersinia enterocolitica)),和/或
-肠球菌属(Enterococcus)(例如粪肠球菌和/或屎肠球菌(Enterococcusfaecium)),和/或
-葡萄球菌属(例如表皮葡萄球菌(Staphylococcus epidermidis)和/或金黄色葡萄球菌)
优选所述一种或多种非靶微生物对至少一种常规抗菌药是具有抗药性的。
肠杆菌科家系中的细菌(肠细菌),例如大肠埃希氏菌(E.coli),是生物样品中经常遇到的革兰氏阴性菌,所述样品包含与革兰氏阴性靶细菌,例如沙门氏菌属的细菌的混合物。这些肠细菌容易在培养基上生长,并由此遮盖所述革兰氏阴性靶细菌(也称为目标革兰氏阴性菌)。因为根据本发明的抗微生物化合物相对于革兰氏阴性靶细菌(例如沙门氏菌属的细菌)选择性地并且在低浓度下抑制某些肠细菌,根据本发明的所述抗微生物剂特别适合于制备用于检测和/或鉴定和/或计数革兰氏阴性靶细菌(例如沙门氏菌属的细菌)的反应培养基。由以上可以得到以下结果:根据本发明的抗微生物化合物特别推荐用于制备针对检测和/或鉴定和/或计数沙门氏菌属的细菌的反应培养基。
另外,肠道球菌以及特别是那些属于粪肠球菌种的肠道球菌是生物样品中经常遇到的革兰氏阳性细菌,所述样品包含与其他革兰氏阳性细菌的混合物。这些肠道球菌容易在培养基上/中生长,并由此有可能遮盖革兰氏阳性靶细菌(目标革兰氏阳性细菌),例如利斯特氏菌属的细菌(例如属于单核细胞增生利斯特氏菌种)、金黄色葡萄球菌种的细菌、链球菌属的细菌(包括无乳链球菌、肺炎链球菌或酿脓链球菌)。因为根据本发明的抗微生物化合物相对于革兰氏阳性靶细菌(例如利斯特氏菌属的细菌)选择性地并且在低浓度下抑制上述肠道球菌,根据本发明的所述抗微生物化合物特别适合于制备用于检测和/或鉴定和/或计数革兰氏阳性靶细菌(例如如上所述的那些)的反应培养基。
本发明的另一个目的涉及反应培养基在检测和/或鉴定和/或计数样品中的至少一种靶微生物、优选至少一种靶细菌的活体外应用,所述样品能够包含所述靶微生物,例如是工业来源或者临床来源的样品。
本发明的另一个目的涉及检测和/或鉴定和/或计数样品中的至少一种靶微生物、优选至少一种靶细菌的方法,所述样品能够包含所述靶微生物,例如是工业来源或者临床来源的样品,所述方法包括以下步骤:
a)用所述样品接种如上定义的反应培养基,
b)如果需要,温育该组件足够长的时间,以便能够检测和/或鉴定和/或计数至少一
种靶微生物,
c)鉴定由所述至少一种靶微生物形成的菌落。
本发明还涉及如上所定义的抗微生物化合物,其用作人或兽用药物。
本发明的另一个目的涉及如上定义的化合物作为治疗微生物感染、优选细菌感染的人用或兽用药物的应用。
根据优选的实施方案,在浓度为0.002至1024.0mg/L、优选0.003至32.0mg/L、优选0.2至8.0mg/L、有利地为0.2至2.0mg/L时,式(I)化合物及其盐、衍生物和类似物(特别是其盐)对于对常规抗生素(特别是经典抗生素)具有多重抗药性(也称为多抗药性)的细菌是特别有效的。
因此,本发明涉及新的抗微生物化合物,优选抗菌化合物(例如杀菌抗生素或抑菌抗生素),使得能够治疗难以用常规抗菌剂(例如经典抗生素,即、那些通常用于治疗微生物感染的抗菌剂)治疗的微生物感染,优选细菌感染。
在本发明中,“氨基酸残基”应理解为目标氨基酸在其与至少一个其它氨基酸建立至少一个肽键后的剩余部分。例如,如果检查上述通式(IV),则明显的是:通式(III)所代表的分子结合至β-氯丙氨酸残基,其本身则结合至取代基R3;所述β-氯丙氨酸残基本身因此具有以下式:
‐i)如果R3是氢原子,或
‐ii)如果R3是1-3个氨基酸残基的线性序列(链);在此情况
下,由于在β-氯丙氨酸残基的α氨基官能团与另一个氨基酸残基的α羧酸官能
团之间形成肽键,所述β-氯丙氨酸残基结合至所述另一个氨基酸残基。
当然,相同的原理也类似地适用于丙氨酸残基之外的氨基酸残基。
“抗微生物化合物”应理解为对微生物有活性的化合物,即、用于对抗后者。根据优选实施方案,该抗微生物化合物是抗菌化合物,即、对细菌具有活性(并且用于对抗后者)。对于本发明,所述抗菌化合物可以是杀菌抗生素,即、其破坏细菌,或者是抑菌抗生素,即、其抑制细菌生长;换言之,其阻止细菌增殖但不必杀死它们。应注意的是,抗生素在低剂量时可以是抑菌的,而在高剂量时为杀菌的。
根据本发明的另一个实施方案,“抗微生物化合物”可包括“抗真菌”化合物。“抗真菌化合物”应理解为任何能够阻止或者减慢酵母或者霉菌生长的化合物。例如,可以特别提及的有两性霉素B、氟康唑、伊曲康唑、伏立康唑和放线菌酮。优选的是,如果使用至少一种抗真菌剂,则其使用浓度是本领域技术人员已知能够得到上述效果的浓度。
“反应培养基”应理解为包含对于表达微生物的代谢和/或存活和/或生长而言所必需的所有成分的培养基。该反应培养基既可以仅作为警戒培养基(a revealingmedium),或者也可以作为培养及警戒培养基(a culture and revealing medium)。在第一种情况下,微生物可在接种之前培养,而在第二种情况下,该反应培养基也可构成所述培养用培养基。该反应培养基可以是固体、半固体或液体的。“半固体培养基”应理解为例如凝胶化培养基。琼脂是微生物学中培养微生物所用的常规胶凝剂,但是也可以使用其他的胶凝剂,例如脱乙酰吉兰糖胶、明胶、琼脂糖,以及其他的天然或人工胶凝剂。许多制剂是可以市售得到的,例如Columbia琼脂、Trypcase-soy琼脂、MacConkey琼脂、Mueller Hinton琼脂,或者那些在微生物学培养基手册(Handbook ofMicrobiological Media(CRC Press))中描述的更为常规的。根据本发明的反应培养基还可包含可能的添加剂,例如氨基酸、蛋白胨(本领域技术人员已知的合适浓度,避免消除作为本发明目的之抗微生物化合物的抑制作用)、一种或更多种生长因子、糖、氨基酸、核苷酸、矿物质、维生素、一种或更多种选择剂、缓冲剂、一种或更多种胶凝剂等。所述反应培养基还可包含染料。作为指示,可能的染料可以是Evans蓝、中性红、绵羊血、马血、例如二氧化钛的遮光剂、硝基苯胺、孔雀绿、亮绿、一种或更多种代谢指示剂、一种或更多种代谢调节剂等。该反应培养基可例如为液体形式,即、随时可用的凝胶,如用于接种在试管中、烧瓶中或者在Petri皿上。
本领域技术人员也可以使用双平板,这使得可以容易地比较两个包含不同底物或者不同的选择性混合物的培养基,在该双平板上已沉积相同的生物样品。该反应培养基可包含一种或更多种选择剂。
“选择剂”应理解为能够阻止或者减缓所谓“非靶”微生物(即、靶微生物之外者)之生长的任何化合物。没有任何限制,对于本发明而言特别合适的是,其浓度为0.002至1024.0mg/L,优选0.003至32.0mg/L,有利地为0.2至8.0mg/L,优选0.2至2.0mg/L。通常而言,我们推荐选择剂的浓度为0.01mg/l至5.0g/l。
“检测”应理解为用裸眼或者使用光学仪器检测是否存在靶微生物(优选靶细菌)的生长。如果希望由其检测靶微生物的反应培养基包含显色或者荧光底物,则该检测对于荧光底物而言可以使用光学仪器,而对于显色底物而言可以用裸眼或者使用光学仪器来进行。
“计数至少一种靶微生物”应理解为记录/定量靶微生物的数量,例如为细菌菌落的数量,如果靶微生物是细菌。
“样品”应理解为用于分析的小份或者分离的少量实体。该样品可以是工业来源的,或者根据非穷尽的名单,还可包括空气样品、水样品、取自表面、部件或制成品、或食品的样品。在食物来源的样品中,可以非穷尽地提及以下食品的样品:奶制品(酸奶、奶酪…)、肉、鱼、蛋、水果、蔬菜、水、饮料(牛奶、果汁、苏打水等)。这些食物样品也可来自调味汁或准备好的餐食。最后,食物样品可来自于动物饲料,例如动物或植物餐。该样品可以是生物来源的,动物、植物或人来源的。在此情况下,其相应于生物流体(全血、血清、血浆、尿、脑脊液、器官分泌物、粪便…)的样品、外部样品(皮肤、鼻子、喉咙、会阴、直肠、阴道…)或组织样品或分离的细胞。该样品可原样使用,或者在分析之前根据本领域技术人员已知的方法,通过富集、提取、浓缩或纯化进行制备。
微生物学对照相应于分析其目的是检测和/或计数怀疑/能够存在于样品中的微生物的样品。
作为能够使至少一种靶微生物在根据本发明的反应培养基上或在该反应培养基内生长的营养剂,可以特别提及的有氨基酸、蛋白胨(本领域技术人员已知的合适浓度,避免消除作为本发明目的之抗微生物化合物的抑制作用)、糖、核苷酸、矿物质和维生素。
如果根据本发明的反应培养基还包含对所述至少一种靶微生物的酶活性具有特异性的酶底物,则优选使用显色和/或荧光底物。
“显色和/或荧光底物”应理解为能够通过直接或间接可检测的信号来检测靶/搜寻微生物之酶或代谢活性的底物。对于直接检测,该底物可结合至起荧光或着色标记物作用的部件上[8]。对于间接检测,根据本发明的反应培养基也可包括pH指示剂,其对由底物消耗所诱发的pH变化敏感并展现靶微生物的代谢。所述pH指示剂可以是发色团或荧光团。发色团的例子可以是溴甲酚紫、溴麝香草酚蓝、中性红、苯胺蓝和溴甲酚蓝。
可在本发明中使用的显色酶底物可以是不同性质的。
首先应该提及的是基于吲哚酚及其衍生物的底物,其在水解之后且有氧存在时产生由蓝至粉红变化的沉淀物。在本发明中,这些基于吲哚酚及其衍生物的底物是特别优选的,这是因为它们相对容易使用并且在检测和/或计数细菌的工作框架内具有良好的敏感性。它们的应用基本上涉及osidase型、酯酶型、脂酶型和磷酸酶型酶活性(磷酸酶是磷酸的酯酶活性)。由于非常适合用于固体或半固体载体(过滤器、琼脂、电泳凝胶等)上,因此他们很少用于液体培养基(形成沉淀物)。
某些型吲哚酚衍生物代表本发明感兴趣的酶底物,因为着色沉淀物的出现不需要任何添加(氧、金属盐等)。因此,此等酶底物的使用已证明在倾注接种细菌的工作框架内是特别有利的。这些型吲哚酚衍生物是具体的吲哚酚衍生物(1H-吲哚-3-基),即、连接在环状胺上的基于吲哚酚的底物(N-芳基化),如PCT申请公布WO 2010/128120(申请人为AG[CH])中所公开的。这些酶底物可得自于AG,尤其是可通过AG网站http://www.biosynth.com进行订购。
其次可以提及的是基于羟基喹啉、二羟基蒽醌、儿茶酚、二羟基黄酮或七叶亭及其衍生物的酶底物,其在有铁盐存在时产生着色沉淀物。它们的应用也涉及osidase型、酯酶型和磷酸酶型的酶活性。
第三可以提及的有基于硝基酚和硝基苯胺及其衍生物的酶底物,其导致黄色化合物的形成。对于基于硝基酚的底物,它们可以检测osidase、酯酶和磷酸酶活性,而对于基于硝基苯胺的底物,它们可以检测肽酶活性。然而,在检测肽酶活性时,所释放的硝基苯胺对于期望鉴定或表征的细菌是毒性的,这证明对于正在进行或者随后进行的分析是有害的。另一方面,它们不太适合用在固体载体上,而更适合用于液体培养基中。另外,颜色(黄色)在生物培养基中具有低对比度(这影响相应微生物学测试的灵敏度)。
第四可以提及的是基于萘酚和萘胺及其衍生物的酶底物。在此情况下,酶-底物反应按照两个步骤进行,由酶活性释放的萘酚或萘胺在有重氮盐存在时经历“偶氮偶联”,该重氮盐是在显色时添加的,导致不溶性有色化合物的形成。它们使得能够通过萘酚检测osidase和酯酶活性,并通过萘胺检测肽酶活性。“偶氮偶联”反应是在对于细菌而言通常为化学侵略性、毒性的介质中,并且该样品不适合其他分析,另外萘胺化合物是致癌的。
在适合于本发明使用的荧光酶底物中,可以特别提及的是香豆素、荧光素、若丹宁、吩噁嗪和羟基黄酮的衍生物,或者是酶底物97或其衍生物。
根据优选的实施方案,如果根据本发明的反应培养基于其中使用根据本发明的抗微生物化合物作为选择剂以特异性地靶向革兰氏阴性(Gram-)菌,例如属于沙门氏菌属的细菌,则所述反应培养基还可包含至少一种抗革兰氏阳性(anti-Gram+)选择系统和/或抗真菌选择系统。该抗革兰氏阳性和/或抗真菌选择系统对于本领域技术人员是已知的。例如可以提及表面活性剂、糖肽、两性霉素、azoles、结晶紫(非穷尽列表),其浓度为得到所预期效果而对于本领域技术人员已知的,即、消除革兰氏阳性细菌。类似地,如果根据本发明的反应培养基使用至少一种新的抗微生物化合物作为至少一种革兰氏阳性(Gram+)细菌(例如金黄色葡萄球菌种的细菌)的选择剂,则可以使用至少一种对本领域技术人员已知的抗革兰氏阴性和/或抗真菌选择系统。例如,可以提及的是氨曲南、polymixins、萘啶酸。
如果需要温育以使靶/搜寻微生物生长,则温育接种有待测试的生物样品的根据本发明的反应培养基。“温育”应理解为升高并保持适当的温度共1至48小时、优选4至24小时、更优选16至24小时,该温度通常为20℃至50℃、优选30至45℃。
“至少一种靶微生物”在本发明中应理解为至少一种期待检测和/或鉴定和/或计数的微生物。
相反地,“非靶微生物”应理解为不期望检测和/或鉴定和/或计数的微生物,而且因为不期望检测和/或鉴定和/或计数它们,对这些非靶微生物不具有任何兴趣。避免该一种或多种非靶微生物是关键的,因为它们能够诱导假阳性结果,并因此影响检测和/或鉴定和/或计数所述至少一种靶微生物的特异性。
附图说明
图1表示6个抗微生物化合物A-F的结构,其抑制主要细菌组的抑制能力在以下实施例2中进行评估(这些根据本发明的新的抗微生物化合物如结构C和E所示,结构A、B、D和F涉及现有技术的化合物)。
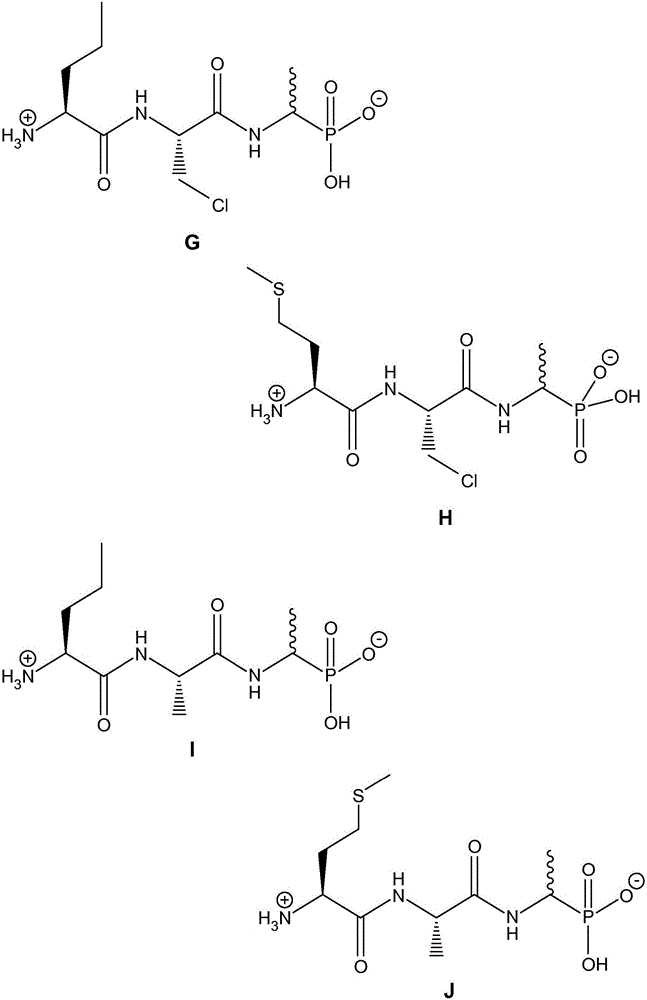
图2显示的是根据本发明的4种其他抗微生物化合物(即化合物G-J)的结构。
图3显示的是用于制备β-氯-L-丙氨酰-L-1-氨基乙基膦酸18(图1中结构C所代表的)和β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸24(图1中结构E所代表的结构)的合成方法的各个步骤。
图4示意性地表示根据本发明的另一个抗微生物化合物的合成方法,即、化合物L-正缬氨酰-L-β-氯丙氨酰-D/L-fosfalin 37(图2中的化合物G)。
图5显示的是能够得到化合物L-正缬氨酰-L-β-氯丙氨酰-D/L-fosfalin 37的反应中间体,即β-氯-L-丙氨酸O-苄基酯32(由tBoc-L-丝氨酸 41起始)的各个合成步骤。
图6代表能够得到化合物L-正缬氨酰-L-β-氯丙氨酰-D/L-fosfalin 37的另一个反应中间体,即D/L-fosfalin二乙基酯35的各个合成步骤。
具体实施方式
以下实施例能够使本发明更容易理解。然而,这些实施例仅用于说明,绝非是对本发明范围的限制。
实施例1:根据本发明的抗微生物化合物C和E的合成方法(图1)
1.1总体方案
合成图1中式A、B、D和F所代表的现有技术中的化合物的方法对于本领域技术人员是已知的。根据本发明的新的抗微生物化合物的合成方法如以下所示,其是用于图1中式C和E所代表的化合物。该合成方法另外概括在图3中。该合成方法框架中所用的试剂和条件如下:
i)苄基溴,DBU,苯;ii)Cl3CCN,PPh3,DCM,N2,t.a.;iii)H2,10%Pd/C,MeOH;iv)PFP,DCC,EtOAc;v)DCC,DCM,DMF;vi)HBr,AcOH,然后氧化丙烯
NMR光谱是在Bruker Ultrashield 300光谱仪(对于1H光谱是在300MHz,而对于13C光谱是在75MHz)上得到的。化学位移是由四甲基硅烷低磁场的ppm表示的,其中使用残留氯仿(1H NMR)或CDCl3碳三重峰的中间峰13C NMR)作为内标。熔点是通过Reichart-Kofler加热板显微镜得到的,但未进行校正。红外光谱是用PerkinElmer Spectrum BX FT-IR仪器记录的。低分辨率质谱是在使用正离子模式电蒸发源(positive ion mode electrovaporisation source)的Bruker Esquire 3000plus分析仪上记录的。高分辨率质谱是在纳米雾化电离模式(nanospray ionisation mode)的LTQ Orbitrap XL设备上得到的。元素分析是是用Exeter Analytical CE-440元素分析仪进行的。所有市售可得的试剂和溶剂都得自于Sigma-Aldrich、Alfa-Aesar、Fisher Scientific和Fluka,而且在使用时没有任何附加的纯化。薄层色谱是在Merck硅胶板(60F-254)上进行的。
1.2制备tBoc-L-丝氨酸苄基酯12
将tBoc-丝氨酸11(16.60g,81.0mmol)溶解在苯(250mL)中,然后添加DBU(18.90g,21.6mL,124.0mmol)。接着将苄基溴(21.25g,15mL,124.0mmol)滴加至经搅拌的反应混合物中。在室温下搅拌过夜后,该反应用1M HCl溶液(150mL)进行中和。在低压下除去所述苯,而残留物放入乙酸乙酯中处理。该溶液用盐水洗涤,有机层在Na2SO4上干燥,然后真空除去溶剂,得到粗产物。柱色谱(50%石油醚,50%乙酸乙酯)后,得到白色固体形式的tBoc-丝氨酸苄基酯12(16.90g,57.0mmol,71%);熔点61-63℃(文献中的熔点69-70℃[16]);[实测:C,60.63;H,7.16;N,4.64.C15H21NO5需要C,61.00;H,7.17;N,4.74%];νmax/cm-1 3417,3356(NH和OH),1756(C=O,酯),1667(C=O,氨基甲酸),1523(酰胺II);1H NMR(300MHz,CDCl3)δH1.44(9H,s,C(CH3)3),2.24(1H,br,OH),3.91(1H,br d,J=11.1Hz,CHa-3),3.98(1H,br d,J=11.1Hz,CHb-3),4.41(1H,br,CH-2),5.19(1H,d,J=12.3Hz,COOCHa),5.24(1H,d,J=12.3Hz,COOCHb),5.44(1H,br,NH),7.35-7.37(5H,m,5x CHAr);13C NMR(75MHz,CDCl3)δC27.9(CH3,C(CH3)3),55.6(CH,C-2),63.3(CH2),67.1(CH2),80.1(quat.,C(CH3)3),127.8(2x CHAr),128.1(CHAr),128.3(2x CHAr),134.9(quat.,CAr),153.0(quat.,C=O),170.3(quat.,C=O);MS(ESI)m/z 318.3(MNa+)。
1.3制备tBoc-β-氯-L-丙氨酸苄基酯13
在氮气下将三氯乙腈(15.16g,10.5mL,105.0mmol)添加至tBoc-丝氨酸苄基酯12(15.58g,52.8mmol)在二氯甲烷(200mL)中的溶液,然后在室温下搅拌10分钟。在氮气氛下将三苯基膦(27.54g,105.0mmol)溶解在二氯甲烷(150mL)中,然后将该溶液滴加至经搅拌的反应混合物中。在室温下搅拌过夜后,反应物用盐水(250mL)中和;分离后,有机层用盐水萃取(3x 100mL)。有机层在无水硫酸钠上干燥,然后在低压下除去溶剂,得到粗产物。通过柱色谱进行纯制(70%石油醚,30%乙酸乙酯),形成白色固体形式的产物13(14.94g,47.6mmol,90%);熔点54-57℃;[实测:C,57.53;H,6.49;N,4.38.C15H20ClNO4需要C,57.42;H,6.42;N,4.46%];νmax/cm-1 3364(NH),1725(C=O,酯),1680(C=O,氨基甲酸),1518(酰胺II);1H NMR(300MHz,CDCl3)δH 1.45(9H,s,C(CH3)3),3.84(1H,dd,J=3.3和11.4Hz,CHa-3),4.00(1H,dd,J=3.3和11.4Hz,CHb-3),4.74(1H,m,CH-2),5.22(1H,d,J=12.3Hz,COOCHa),5.27(1H,d,J=12.3Hz,COOCHb),5.43(1H,d,J=7.2Hz,NH),7.36(5H,m,5x CHAr);13C NMR(75MHz,CDCl3)δC 28.3(CH3,C(CH3)3),45.5(CH2,C-3),54.6(CH,C-2),67.8(COOCH2),80.5(quat.,C(CH3)3),128.3(CH),128.6(2x CH),128.6(2x CH),134.9(quat.,CAr),155.0(quat.,C=O),169.0(quat.,C=O);HRMS(纳米喷雾电离)计算(C15H21NO4Cl)+314.1154,实测314.1159。
1.4制备β-氯-L-丙氨酸HBr苄基酯14
将tBoc-β-氯-L-丙氨酸苄基酯13(1.40g,4.46mmol)溶解在最小量的乙酸(5mL)中,然后添加在AcOH中的HBr(33%m/m)(5.53mL,30.7mmol的HBr),然后在室温下搅拌反应混合物10分钟。该反应物用二乙基醚(200mL)中和,然后该溶液在冷冻机中保持过夜。在静置时,沉淀出白色固体,过滤收集,然后用冷二乙基醚洗涤,得到产物14(1.10g,3.6mmol,81%);熔点131-134℃;[实测:C,40.56;H,4.51;N,4.68.C10H13BrClNO2需要C,40.77;H,4.45;N,4.75%];νmax/cm-1 2950,2875,2846(br NH3+),1750(C=O,酯),1489,1228,1208(C-O);1H NMR(300MHz,D2O)δH 4.17(1H,dd,J=3.3和12.6Hz,CHa-3),4.31(1H,dd,J=3.3和12.6Hz,CHb-3),4.81(1H,m,CH-2),5.31(1H,d,J=12.3Hz,COOCHa),5.39(1H,d,J=12.3Hz,COOCHb),7.55(5H,m,5x CHAr);13C NMR(75MHz,D2O)δC 41.8(CH2,C-3),53.9(CH,C-2),69.2(COOCH2),128.7(2x CH),128.9(2x CH),129.1(CH),134.5(quat.,CAr),167.0(quat.,C=O);HRMS(纳米喷雾电离)计算(C10H1335ClNO2)+214.0629,实测214.0630。
1.5制备化合物tBoc-β-氯-L-丙氨酸15
将tBoc-β-氯-L-丙氨酸苄基酯13(1.57g,5.0mmol)溶解在甲醇(50mL)中,然后添加在乙酸乙酯(20mL)中的10%钯炭(0.16g)。反应物在1.5bar H2压力下搅拌过夜。用Celite塞通过过滤除去催化剂,然后用甲醇(200mL)洗涤。低压除去甲醇后,粗产物通过柱色谱进行纯制(95%二氯甲烷,5%甲醇),得到白色固体形式的产物15(0.99g,4.4mmol,88%);熔点119-123℃(文献中的熔点123-125℃(13));νmax/cm-1 3434(NH),2975(OH),1752(C=O),1734(C=O),1677,1521(酰胺II),1370,1212;1H NMR(300MHz,CDCl3)δH1.47(9H,s,C(CH3)3),3.90(1H,dd,J=2.7和11.1Hz,CHa-3),4.05(1H,d,J=11.1Hz,CHb-3),4.78(1H,m,CH-2),5.47(1H,d,J=6.3Hz,NH);13C NMR(75MHz,CDCl3)δC 28.3(CH3,C(CH3)3),45.2(CH2,C-3),54.3(CH,C-2),80.9(quat.,C(CH3)3),155.3(quat.,C=O,C-4),173.2(quat.,C=O,C-1);HRMS(纳米喷雾电离)计算(C8H13NO435Cl)-222.0539,实测222.0541。
1.6制备tBoc-β-氯-L-丙氨酸五氟苯酚酯16
将tBoc-β-氯-L-丙氨酸15(1.16g,5.2mmol)和五氟苯酚(0.95g,5.7mmol)溶解在乙酸乙酯(25mL)中,然后在冰浴中冷却。添加二环己基碳二亚胺(1.05g,5.7mmol),然后该溶液搅拌3小时。沉淀出的副产物脲通过过滤除去。残留物在低压下浓缩,然后通过过滤除去任何额外的沉淀物。剩余的乙酸乙酯通过蒸发被除去。如此得到的油用石油醚研磨,通过过滤收集得到产物16白色固体形式的(3.35g,8.6mmol,69%);熔点128-131℃;[实测:C,43.53;H.3.49;N,3.63.C14H1335ClF5NO4需要C,43.15;H,3.36;N,3.59%];νmax/cm-1 3367(NH),1780(C=O),1681(C=O),1518(酰胺II);1H NMR(300MHz,CDCl3)δH 1.48(9H,s,C(CH3)3),3.94(1H,dd,J=3.6和11.4Hz,CHa-3),3.96(1H,dd,J=3.6和11.4Hz,CHb-3),5.10(1H,br t,J=3.6Hz,CH-2),5.46(1H,d,J=7.5Hz,NH);13C NMR(75MHz,CDCl3)δC 28.25(CH3,C(CH3)3),44.78(CH2,C-3),54.55(CH,C-2),81.26(quat.,C(CH3)3),118.94(m,C-FAr),154.74(quat.,C-1’),154.79(quat.,C=O,C-4),166.77(quat.,C=O,C-1);19F NMR(282MHz,CDCl3)δF-161.67(2F,t,J=19.8Hz,CF-3’和5’),-156.73(1F,t,J=23.1Hz,CF-4’),-151.63(2F,d,J=18.9Hz,CF-2’和6’);MS(ESI)m/z388.8(M-H)-。
1.7制备tBoc-β-氯-L-丙氨酰-L-fosfalin二乙基酯17
将L-fosfalin二乙基酯(1.10g,6.1mmol)溶解在无水DCM(50mL)中,然后在冰浴中冷却。分批添加tBoc-β-氯-丙氨酰五氟苯酚酯16(2.37g,6.1mmol),然后反应混合物在室温下搅拌直至反应完全。该反应物用水中和(150mL),然后用二氯甲烷(3x100mL)进行萃取。有机层在无水MgSO4上干燥,然后真空除去溶剂。残留物通过柱色谱进行纯制(50%石油醚,50%乙酸乙酯直至90%乙酸乙酯,10%甲醇),得到白色固体形式的产物17(1.45g,3.8mmol,62%);熔点80-83℃;[实测:C,43.46;H,7.27;N,7.21.C14H28ClN2O6P需要C,43.47;H,7.30;N,7.24%];νmax/cm-1 3303,3215,3065,2978(NH),1716(C=O),1667(C=O),1555,1518(酰胺II);1H NMR(300MHz,CDCl3)δH 1.25-1.42(9H,m,3x CH3),1.46(9H,s,C(CH3)3),3.73(1H,dd,J=4.2和11.1Hz,CHa-3),4.00(1H,dd,J=4.2和11.1Hz,CHb-3),4.07-4.18(4H,m,2x OCH2),4.39-4.54(2H,m,CH-2和2’),5.35(1H,d,J=8.4Hz,NH),6.77(1H,d,J=8.7Hz,NH);13C NMR(75MHz,CDCl3)δC15.8(CH3,C-3’),16.3-16.5(2x CH3,m,CH3CH2O),28.2(3x CH3,C(CH3)3),40.3(CH),42.4(CH),44.9(CH2,C-3),62.6(CH2,d,J=6.75Hz,OCH2CH3),62.9(CH2,d,J=6.6Hz,OCH2CH3),80.8(quat.,C(CH3)3),155.0(quat.,C=O),168.3(quat.,C=O);31P NMR(121.5MHz,CDCl3)δP 24.6(m);MS(ESI)m/z 387.3(MH+),409.3(MNa+),385,1(M-)。
1.8得到化合物β-氯-L-丙氨酰-L-fosfalin 18(图1中的化合物)
在33%m/m HBr/乙酸(17mL)中搅拌tBoc-β-氯-L-丙氨酰-L-fosfalin二乙基酯17(1.33g,3.4mmol)共24h。将反应混合物倾倒在二乙基醚(200mL)中,然后放置在冷冻机(-15℃)中过夜。滗析掉二乙基醚,而将残留的沉淀物放入最小量的甲醇(约10mL)中。大大过量地添加氧化丙烯(约250mL)。过滤吸水性的沉淀物,然后由水和丙酮中重结晶,得到白色固体形式的产物18(0.99g,3.2mmol,93%);熔点210-212℃;[实测:C,25.80;H,5.21;N,12.04.C5H12ClN2O4P需要C,26.04;H,5.25;N,12.15%]νmax/cm-13258(br NH),3100,2930(br)(OH),1654(C=O),1565,1514(酰胺II),1038;1H NMR(300MHz,D2O)δH 1.30(3H,dd,J=7.2和15Hz,CH3-3’),3.97-4.11(3H,m,CH2-3和CH-2’),4.39(1H,m,CH-2);13C NMR(75MHz,D2O)δC 15.2(CH3,C-3’),42.6(CH2,C-3),44.2(CH,d,J=147.9Hz,C-2’),54.2(CH,C-2),165.6(quat.,C=O,C-1);31P NMR(121.5MHz,CDCl3)δP 18.6(m);HRMS(纳米喷雾电离)计算(C5H11N2O4P35Cl)-229.0150,实测229.0154。
1.9制备tBoc-β-氯-L-丙氨酰-β-氯-L-丙氨酸苄基酯19
将β-氯-L-丙氨酸苄基酯氢溴酸盐14(1.13g,3.9mmol)溶解在DMF(30mL),然后在0℃下添加至tBoc-β-氯-L-丙氨酸五氟苯酚酯16(1.50g,3.9mmol)在二氯甲烷(10mL)中的溶液内。在该溶液内滴加二异丙基乙基胺(0.50g,0.66mL,3.9mmol)。如此得到的混合物在室温下搅拌2小时,然后加热至35℃,反应的进展通过TLC(80%石油醚,20%乙酸乙酯)进行监测。一旦反应完全,用1M HCl溶液(50mL)进行中和,并用水(100mL)和盐水(100mL)洗涤有机层。有机层在无水硫酸钠上干燥,然后真空蒸发溶剂。粗产物通过梯度柱色谱进行纯制(由80%石油醚,20%乙酸乙酯;至50%石油醚,50%乙酸乙酯),得到白色固体形式的产物19(1.16g,2.8mmol,70%);熔点89-91℃;[实测:C,51.83;H,5.78;N,6.78.C18H2435Cl2N2O5需要C,51.56;H,5.77;N,6.68%];νmax/cm-1 3336(NH),3321(NH),1739(C=O),1655(m,C=O),1508(酰胺II);1H NMR(300MHz,CDCl3)δH 1.48(9H,s,C(CH3)3),3.73(1H,dd,J=4.8和11.1Hz,CH),3.89-4.06(3H,m,3x CH),4.55(1H,br,CH),4.98(1H,m,CH),5.24(2H,m,COOCH2),5.35(1H,br,NH),7.25(1H,br,NH),7.37(5H,br s,5x CHAr);13C NMR(75MHz,CDCl3)δC 28.24(CH3,C(CH3)3),33.90(CH2Ph),44.5(CH),44.60(CH2),53.61(CH),68.13(CH2),81.37(quat.,C(CH3)3),128.22(CHAr),128.41(CHAr),128.69(CHAr),134.71(quat.),168.19(2x quat.,C=O),168.90(quat.,C=O);HRMS(纳米喷雾电离)计算(C18H25N2O535Cl)+419.1135,实测419.1139。
1.10制备化合物tBoc-β-氯-L-丙氨酰-β-氯-L-丙氨酸20
将tBoc-β-氯-L-丙氨酸-β-氯-L-丙氨酸苄基酯19(1.00g,2.4mmol)溶解在甲醇(50mL)中,然后添加在乙酸乙酯(20mL)中的10%钯炭(0.10g)。反应混合物在H2气氛(1.8bar)中搅拌24h,然后通过Celite塞进行过滤,并用甲醇(200mL)洗涤。真空除去溶剂,并在通过柱色谱进行纯制(95%DCM,5%甲醇)后,用石油醚研磨,得到白色固体形式的产物20(0.52g,1.6mmol,66%);熔点72-74℃;νmax/cm-1 3320(NH),2978(NH),1724(C=O),1665(C=O),1530(酰胺II);1H NMR(300MHz,DMSO-d6)δH 1.39(9H,s,C(CH3)3),3.67(1H,dd,J=8.4和11.1Hz,CH),3.79-3.95(3H,m,CH2和CH),4.36(1H,br,CH),4.61-4.67(1H,m,CH),7.16(1H,d,J=8.1Hz,NH),8.38(1H,d,J=7.5Hz,NH);HRMS(纳米喷雾电离)计算(C11H1735Cl2N2O5)-327.0520,实测327.0517。
1.11制备tBoc-β-氯-L-丙氨酰--氯-L-丙氨酸五氟苯酚酯22
将tBoc-β-氯-L-丙氨酰-β-氯-L-丙氨酸20(0.48g,1.46mmol)溶解在乙酸乙酯(50mL),并在冰浴中冷却,然后添加五氟苯酚(0.29g,1.56mmol)和二环己基碳二亚胺(0.33g,1.61mmol)。该溶液在0℃下搅拌2小时,然后过滤除去沉淀出的脲。残留物在真空下浓缩,并通过过滤除去任何新的沉淀物。真空除去乙酸乙酯,如此得到的油用石油醚研磨,得到白色固体形式的产物22(0.65g,1.3mmol,89%),其通过过滤进行收集,并在未纯制的情况下用于下一步;νmax/cm-1 3334(NH),3006,2970,2939(NH),1787(C=O),1688(C=O),1664(C=O),1514(酰胺II);1H NMR(300MHz,CDCl3)δH 1.46(9H,s,C(CH3)3),3.76(1H,dd,J=4.7和11.3Hz,CHa),3.98(1H,dd,J=3.5和11.7Hz,CHc),4.06(1H,dd,J=4.4和11.3Hz,CHb),4.16(1H,dd,J=3.2和11.7Hz,CHd),4.53(1H,br,CH),5.32-5.37(2H,m,CH和NH),7.31(1H,d,J=7.1Hz,NH)。
1.12制备tBoc-β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-fosfalin二乙基酯23
在0℃下将tBoc-β-氯-L-丙氨酰-β-氯-L-丙氨酸五氟苯酚酯22(0.95g,1.9mmol)和二乙基1-氨基乙基膦酸3(0.32g,1.9mmol)溶解在二氯甲烷(25mL),然后在室温下搅拌直至通过TLC监测反应完全。用水(100mL)萃取后,有机层在无水MgSO4上干燥,然后低压除去挥发性的成分。粗产物固体通过柱色谱进行纯制(97%DCM,3%MeOH),得到白色固体形式的产物23(0.62g,1.3mmol,66%);熔点154.6-155.9℃;νmax/cm-1 3291,3265,2962,2848(NH),1680(C=O),1639(C=O),1523(酰胺II);1H NMR(300MHz,CDCl3)δH 1.35-1.41(9H,m,3x CH3),1.48(9H,s,C(CH3)3),3.73-3.80(2H,m,CHa-3和CHa-3’),4.01-4.2(6H,m,CHb-3,CHb-3’和2x OCH2),4.43-4.54(2H,m,CH-2”和CH-2),4.79-4.84(1H,m,CH-2’),5.38(1H,d,J=6.9Hz,NH),7.05(1H,d,J=9.0Hz,NH),7.19(1H,d,J=7.8Hz,NH);13C NMR(75MHz,CDCl3)δC15.7(CH3,C-3’),16.6(CH3,OCH2CH3),16.7(CH3,OCH2CH3),28.4(3x CH3,C(CH3)3),40.7(CH),44.5(CH2),44.6(CH2),49.4(CH),54.1(CH),62.8(CH2,d,J=15.5Hz,OCH2CH3),63.1(CH2,d,J=15.7Hz,OCH2CH3),81.6(quat.,C(CH3)3),156.9(quat.,C=O),168.9(quat.,C=O),177.3(quat.,C=O);MS m/z 514.3,515.2,516.2(MNa+);HRMS(纳米喷雾电离)计算(C17H33N2O7P35Cl2)+492.1428,实测492.1422。
1.13得到β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-fosfalin 24(图1中的化合物E)
将tBoc-β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-fosfalin二乙基酯23(0.36g,0.7mmol)溶解在乙酸(5mL)中,然后添加HBr/AcOH(33%m/m(4mL)。该溶液在室温下搅拌过夜,然后该反应物用二乙基醚(200mL)中和。在冷冻机(-15℃)中放置4h后,滗析掉二乙基醚,由此分离如此得到的棕色油。粗产物用冷二乙基醚(5x 50mL)洗涤。残留物放入甲醇(3mL)中,添加氧化丙烯(150mL),形成白色沉淀。随后滗析掉液体,而残留物用冷二乙基醚(5x 30mL)研磨,得到白色固体形式的产物24(0.18g,0.4mmol,59%);熔点157-159℃;νmax/cm-1 3285,3258(br NH),1646(br)(C=O),1539(large酰胺II),1152,1044;1H NMR(300MHz,D2O)δH 1.37(3H,dd,J=7.2和15.3Hz,CH3-3”),3.98-4.2(5H,m,CH2-3和CH2-3’和CH),4.62(1H,t,J=4.8Hz,CH),4.87-4.90(1H,m,CH);13C NMR(75MHz,D2O)δC 15.4(CH3,C-3”),42.4(CH2),43.4(CH2),45.1(CH,C-2”),53.8(CH),55.1(CH),166.8(quat.,C=O),168.9(quat.,C=O);HRMS(纳米喷雾电离)计算(C8H15N3O5P35Cl2)-336.0102,实测336.0096。
实施例2:评估根据实施例1合成的抗微生物化合物C和E的抗菌活性
2.1介绍
该实施例2显示了以下研究的结果:
-评估在实施例1中合成的根据本发明的化合物,即图1中表示的化合物C和E的抗菌活性,以及
-对比该抗菌活性和用现有技术的三种模拟肽(图1中的化合物A、B和D)以及磷霉素(化合物F)得到的抗菌活性。
5个化合物A-E的抗菌活性是用大规模采集297种细菌进行评估的,这些细菌包括主要的多抗药性菌株,其包括产碳青霉烯酶的肠细菌(n=128)、抗甲氧西林的金黄色葡萄球菌(n=37)、以及抗糖肽的肠道球菌(n=43)。为进行比较还包括磷霉素(F),其是一种天然存在的也包含膦酸基的抗生素。
作为提醒,化合物A-F如下:
A:L-丙氨酰-L-1-氨基乙基膦酸(阿拉磷);
B:L-丙氨酰-L-丙氨酰-L-1-氨基乙基膦酸(二-丙氨酰fosfalin);
C:β-氯-L-丙氨酰-L-1-氨基乙基膦酸(β-Cl-阿拉磷);
D:β-氯-L-丙氨酰-β-氯-L-丙氨酸(β-Cl-Ala-β-Cl-Ala);
E:β-氯-L-丙氨酰-β-氯-L-丙氨酰-L-1-氨基乙基膦酸(β-Cl-Ala-β-Cl-阿拉磷);
F:[(2R,3S)-3-甲基环氧乙烷-2-基]膦酸二钠(磷霉素)。
2.2材料和方法
抗菌剂和培养基:磷霉素、阿拉磷、葡萄糖-6-磷酸以及无拮抗剂的琼脂培养基的所有成分都购自Sigma Chemical Company,Poole,United Kingdom。琼脂培养基IsoSensitest购自Oxoid,Basingstoke,United Kingdom。
细菌分离菌:由各种国际来源得到肠细菌(n=197),并且所有都具有β-内酰胺酶,这些酶是由著名实验室和/或本领域著名专家在分子水平上定义的。这些包括弗氏柠檬酸杆菌(Citrobacter freundii)(n=5)、其它种的柠檬酸杆菌(Citrobacter)(n=4)、产气肠杆菌(Enterobacter aerogenes)(n=1)、阴沟肠杆菌(n=27)、大肠埃希氏菌(n=53)、产酸克雷伯氏菌(Klebsiella oxytoca)(n=5)、肺炎克雷伯氏菌(n=87)、克吕沃尔氏菌属(Kluyvera spp)(n=1)、奇异变形菌(Proteus mirabilis)(n=8)、雷氏普罗威登斯菌(Providencia rettgeri)(n=2)、沙门氏菌属(n=3)、以及粘质沙雷氏菌(n=1)。在这些197种分离菌中,有128(65%)种是产碳青霉烯酶的细菌,包括87种具有NDM-1,9种具有IMP,11种具有KPC,14种具有OXA-48,以及7种具有VIM。绝大部分的产碳青霉烯酶的细菌联合产生广谱β-内酰胺酶(BSBL)或头孢菌素酶(AmpCβ-内酰胺酶),但后者由于清楚的原因未被记录。在其余的分离菌中,47种具有BSBL(20种具有CTX-M,19种具有SHV型,以及8种具有TEM型),以及22种具有AmpC(3种具有ACC-1,6种具有CMY型,6种具有DHA-1,3种具有FOX型,以及4种具有LAT型)。
采集的50株金黄色葡萄球菌分离菌包括在欧洲经常遇见的36株抗甲氧西林的金黄色葡萄球菌(MRSA),包括在比利时、芬兰、法国、德国以及英国分离的菌株。另一个菌株MRSA,NCTC 11939是作为对照包括在内的,还包括甲氧西林敏感对照(NCTC 6571)。还包括最近由血液培养基中采集的12种其他的甲氧西林敏感金黄色葡萄球菌(MSSA)的分离菌。最后,50种肠道球菌的分离菌包括2种对照菌株(粪肠球菌NCTC 755和屎肠球菌NCTC 7171)和48种得自于至少3所不同医院的临床样品的分离菌。这些临床分离菌包括粪肠球菌(E.faecalis)(n=10)、屎肠球菌(E.faecium)(n=33)、铅黄肠球菌(Enterococcus casseliflavus)(n=3)、鹑鸡肠球菌(Enterococcus gallinarum)(n=2)。在这50种分离菌中,如MIC值以及由PCR证实的抗性基因所表明的,43种是抗万古霉素的。
测定最小抑制浓度(MIC):所有的MIC都是用琼脂稀释法[16]测定的。这需要使用如前所述制备的没有明确拮抗剂的培养基,其包含2%用皂苷溶解的马血、25.0μg/mL的NAD和25.0μg/m的氯高铁血红素[17]。所测试的氯高铁血红素合物溶解在无菌去离子水中,然后以0.0031至8.0μg/mL(对于革兰氏阳性菌为0.016至32μg/mL)的浓度加入琼脂培养基中。所有的分离菌以相当于无菌去离子水中0.5McFarland单位的密度进行制备,其中使用密度计进行测量(约1.5x 108CFU/mL),然后按1:15稀释。接着用多点接种器将1份1μL每种经稀释的悬浮液放置在板上,达到最终推荐的接种10 000CFU/点[16]。磷霉素的MIC是用相同的方法测定的,但使用IsoSensitest培养基(Oxoid,Basingstoke,United Kingdom),再加上25.0μg/mL的葡萄糖-6-磷酸(Sigma,Poole,United Kingdom)以及宽范围浓度的磷霉素。所有的盘(包括无抗微生物的对照组)在37℃下培养22小时。所有这些测试都至少独立充分2次,以检查可重复性。
2.3结果
计算5个化合物A-E的最小抑制浓度(MIC)50和90,并示于下表1中。这些值分别相应于足以在活体外抑制50%和90%细菌菌株生长的最小抗生素浓度,分别为MIC50和MIC90。
更具体而言,下表1示出了6种抗微生物剂对所测试的主要细菌组的MIC。阿拉磷对大多数的肠细菌分离菌具有良好的活性,虽然不同的种类表现出不同程度敏感性。对53种大肠埃希氏菌分离菌观察到高活性,其中35种分离菌(66%)是产碳青霉烯酶的细菌。对大肠埃希氏菌的MIC90为0.25μg/mL,而且在2μg/mL时抑制了所有分离菌的生长。发现阿拉磷的活性约为磷霉素活性的4倍。
虽然阿拉磷对大肠埃希氏菌的抗菌活性是令人满意的,但是如前所述,已证明β-Cl-阿拉磷对大肠埃希氏菌的活性至少是阿拉磷活性的2倍,并且至少是磷霉素活性的8倍。MIC90是0.125μg/mL,而且在0.5μg/mL时看见所有分离菌的生长都被抑制。
与大肠埃希氏菌相比,肺炎克雷伯氏菌(K.pneumoniae)于所有测试的化合物都更为不敏感,但多种分离菌表现出较低的MIC。例如,87%的肺炎克雷伯氏菌分离菌被8μg/mL阿拉磷抑制,而93%被8μg/mLβ-Cl-阿拉磷抑制。
阴沟肠杆菌(E.cloacae)(n=27)被4μg/mL阿拉磷抑制,其对该菌种的活性通常为磷霉素的32倍。如前所述,已证实β-Cl-阿拉磷是活性最高的化合物,其中在1μg/mL时抑制所有的分离菌。其它种的肠细菌不在表1中,这是因为只有低于10种测试的分离菌。
对于分类群柠檬酸杆菌(n=9)、产气肠杆菌(E.aerogenes)(n=1)、克吕沃尔氏菌属(n=1)和粘质沙雷氏菌(S.marcescens)(n=1),所有分离菌对于≤4μg/mL阿拉磷和≤2μg/mLβ-Cl-阿拉磷都是敏感的。5种产酸克雷伯氏菌(K.oxytoca)分离菌中的一种需要MIC>8μg/mL阿拉磷,但是所有都被≤2μg/mLβ-Cl-阿拉磷抑制,再一次表明后者具有高度令人满意的抗菌活性。
8种奇异变形菌(P.mirabilis)和2种雷氏普罗威登斯菌(P.rettgeri)分离菌对于所有的测试药剂(包括磷霉素)都需要MIC≥8μg/mL。
3种沙门氏菌属分离菌对于阿拉磷表现出MIC≥8.0μg/mL,但是对于β-Cl-阿拉磷仅为2.0-4.0μg/mL。
β-Cl-阿拉磷和β-Cl-Ala-β-Cl-Ala对金黄色葡萄球菌具有最高的活性,但是在5种肽抗微生物剂之间则具有略微总的差异。90%的源自各种地理源的MRSA分离菌被8.0μg/mL阿拉磷抑制,而所有的分离菌被2.0μg/mLβ-Cl-阿拉磷抑制,再一次表明其具有比阿拉磷更高的抗菌活性。
对于肠球菌,最显著的观察结果是二丙氨酰fosfalin的高活性,对此MIC(平均)比阿拉磷的活性低16倍,而且在某些情况下,低256倍。在34种屎肠球菌分离菌(包括31种抗万古霉素分离菌)中,所有都被32μg/mL阿拉磷或4μg/mL二-丙氨酰fosfalin抑制。在任何情况下,在肠球菌中,β-Cl-Ala-β-Cl-阿拉磷(根据本发明的化合物)的抗菌活性证实比阿拉磷的活性高。
2.4结论
如我们在此研究中所显示的,根据本发明的抗微生物化合物,特别是β-Cl-阿拉磷,对于绝大多数的细菌、特别是绝大多数的多抗药性细菌都有活体外抗菌活性(非常常见地高于阿拉磷的活性)。因此,例如,阿拉磷对于CPE的MIC50和MIC90分别是1μg/mL和4μg/mL,但是根据本发明的磷酸肽,β-Cl-阿拉磷,呈现0.5μg/mL和2μg/mL两个值之一。阿拉磷对MRSA仅具有中等活性,其MIC90为8μg/mL,而β-Cl-阿拉磷则活性更高,其MIC90为2μg/mL。
与阿拉磷相比,β-Cl-Ala-β-Cl-阿拉磷针对某些菌种能够获得抑制作用。这对于抑制一个菌种但不改变微生物菌群之余者是有用的,例如抑制大肠埃希氏菌但不抑制其它肠细菌,或抑制粪肠球菌但不抑制屎肠球菌。这对于通过抑制其它细菌而特异性地分离一个菌种是特别有利的,例如寻找金黄色葡萄球菌。
表1:各种抗微生物剂对多种细菌的最小抑制浓度,包括具有明确抗药机理的分离菌
缩写:CPE:产碳青霉烯酶的肠杆菌科;BSBL:具有广谱β-内酰胺酶的肠杆菌科;MRSA:抗甲氧西林的金黄色葡萄球菌;MSSA:甲氧西林敏感的金黄色葡萄球菌;GRE:抗糖肽的肠球菌。
实施例3:用于检测沙门氏菌属细菌的根据本发明的反应培养基
3.1培养基的组成
本实施例3的目的在于对比根据本发明的反应培养基的检测特异性,意欲检测沙门氏菌属的细菌,即、“改良chromID Salmonella”培养基对用于相同细菌的参比检测培养基,即、“chromID Salmonella”培养基。
根据本发明的反应培养基和参比反应培养基的相应组成示于下表2中。
表2:根据本发明反应培养基和参比培养基的组成
3.2结果
所得结果示于以下表3中。
表3:结果
3.3结论
在chromID Salmonella培养基中添加β-Cl-阿拉磷使得能够选择性地抑制大肠埃希氏菌菌株的osidase活性。因为与在蓝色菌落的培养基中相比,在无色菌落培养基中更容易检测一个或多个淡紫色菌落。如果沙门氏菌株以低浓度存在于与一个或多个大肠埃希氏菌菌株的混合物中,则能够明显更容易地在本发明的反应培养基中检测到前者。
实施例4:根据本发明的抗微生物化合物G的合成方法(图2):L-正缬氨酰-β–氯-L-丙氨酰-D/L-fosfalin
L-正缬氨酰-β–氯-L-丙氨酰-D/L-fosfalin(L-Nva-β-Cl-L-Ala-D/L-fosfalin)由下式代表:
抗微生物化合物的合成详细说明如下。该相应的合成方法另外也示于图4中。
如图4中所示,根据本发明的上述化合物是由tBoc-L-正缬氨酸(市售可得的)、β-氯-L-丙氨酸O-苄基酯(其合成示于图5中)和D/L-fosfalin二乙基酯(其合成示于图6中)合成的。
4.1由化合物tBoc-L-丝氨酸41(如图5所示)合成反应中间体β-氯-L-丙氨酸O-苄基酯32
4.1.1合成(S)-苄基2-((tert-丁氧基羰基)氨基)-3-羟基丙酸酯(tBoc-L-丝氨酸O-苄基酯)43[9]
将tBoc-L-丝氨酸(市售可得的)41(30mmol,6.16g)溶解在无水苯(100mL)中,然后向其中添加1,8-二氮杂二环[5,4,0]十一烷-7-烯(DBU)(36mmol,5.5mL)和苄基溴42(36mmol,4.4mL)。该溶液在室温和氮气下搅拌过夜,然后在低压下除去溶剂,得到米色液体残留物。添加乙酸乙酯(200mL),烧瓶中的内容物进行超声处理,然后用1M HCl溶液(2x 50mL)、10%w/v K2CO3水溶液(2x 50mL)和盐水(2x 50mL)洗涤。有机层在MgSO4上干燥,过滤,真空浓缩并通过柱色谱纯制[石油溶剂油/乙酸乙酯(1:1)],得到白色固体形式的产物43(8.05g,27.3mmol,91%);m.p.61–66℃(lit.m.p.[10]59–60℃);[α]21D-18.5°(c 1.0,CH3OH);νmax/cm-1 3419(NH),3361(OH),2978(CH),1758(C=O),1668(C=O),1524(NH bend),1155(C-O),1068(C-O);1H NMR(300MHz,DMSO)δH 1.38(9H,s,C(CH3)3),3.68(2H,t,J=6.0Hz,CH2OH),4.10-4.16(1H,m,CH-2),4.91(1H,t,J=6.0Hz,OH),5.10(1H,d,J=12.0Hz,OCH2aAr),5.17(1H,d,J=12.0Hz,OCH2bAr),6.97(1H,d,J=9.0Hz,NH),7.32-7.38(5H,m,5x CHAr);13C NMR(75MHz,DMSO)δC 28.6(C(CH3)3),57.0(CH-2),61.8(CH2-3),66.2(OCH2Ar),78.8(C(CH3)3),128.0(CHAr),128.4(CHAr),128.8(CHAr),136.5(CHAr quat),155.8(C-4,quat),171.4(C-1,quat)。
4.1.2合成(R)-苄基2-((tert-丁氧基羰基)氨基)-3-氯丙酸酯(tBoc-β-氯-L-丙氨酸O-苄基酯)44[11]
将上述步骤4.1.1中得到的((S)-苄基2-(((tert-丁氧基羰基)氨基)-3-羟基丙酸酯43(25mmol,7.39g)溶解在无水DCM(100mL)中,然后添加三氯-乙腈(50mmol,5.0mL)。该溶液在室温下搅拌2小时。向该溶液丈夫缓慢添加在无水DCM(50mL)中的三苯基膦(50mmol,13.15g)。如此得到的溶液在室温和氮气下搅拌过夜。添加盐水(100mL),以使反应停止。分离后,有机层用盐水(3x 60mL)洗涤,在MgSO4上干燥,过滤并真空浓缩,得到橙色液体残留物。残留物通过柱色谱进行纯制[石油溶剂油/乙酸乙酯(7:3)],得到米色固体形式的产物44(7.41g,23.6mmol,95%);m.p.53–58℃;[α]22D-23.0°(c 1.0,CH3OH);νmax/cm-1 3365(NH),2917(CH),1727(C=O),1679(C=O),1522(NH bend),1181(C-O),1158(C-O);1H NMR(300MHz,CDCl3)δH 1.45(9H,s,C(CH3)3),3.85(1H,dd,J=12.0Hz,3.0Hz,CH2a-3),3.99(1H,dd,J=12.0Hz,3.0Hz,CH2b-3),4.74(1H,m,CH-2),5.20(1H,d,J=12.0Hz,OCH2aAr),5.25(1H,d,J=12.0Hz,OCH2bAr),5.44(1H,d,J=6.0Hz,NH),7.33-7.38(5H,m,5x CHAr);13C NMR(75MHz,CDCl3)δC 27.3(C(CH3)3),44.5(CH2-3),53.5(CH-2),66.8(OCH2Ar),79.5(C(CH3)3),127.3(CHAr),127.6(CHAr),127.6(CHAr),133.9(CHAr quat),154.0(C-4,quat),168.0(C-1,quat);CHN[实测:C,57.71;H,6.46;N,4.18.C15H20ClNO4需要C,57.42;H,6.42;N,4.46%].
4.1.3合成(R)-1-(苄基氧基)-3-氯-1氧代丙烷-2-氯化铵(β-氯-L-丙氨酸盐酸盐)32
将在上述步骤4.1.2.中得到的化合物44(10mmol,3.14g)溶解在2M HCl之乙醚溶液(200mL)中。该溶液在室温下搅拌过夜。由此得到的固体过滤并用二乙基醚洗涤,得到白色固体形式的产物32(2.36g,9.5mmol,95%);m.p.145℃(secondary);νmax/cm-12848(CH),1749(C=O),1593(Ar C-C),1490(Ar C-C),1231(C-O);1H NMR(300MHz,DMSO)δH 4.16(1H,dd,J=12.0Hz,3.0Hz,CH2a-3),4.22(1H,dd,J=12.0Hz,3.0Hz,CH2b-3),4.77(1H,t,J=3.0Hz,CH-2),5.26(1H,d,J=12.0Hz,OCH2aAr),5.31(1H,d,J=15.0Hz,OCH2bAr),7.33-7.46(5H,m,5x CHAr),9.09(3H,br,NH3+);13C NMR(75MHz,DMSO)δC 43.3(CH2-3),53.5(CH-2),68.0(OCH2Ar),128.6(CHAr),128.8(CHAr),128.9(CHAr),135.4(CHAr quat),167.0(C-1,quat);CHN[实测:C,47.16;H,5.43;N,5.43.C10H13Cl2NO2·0,2H2O需要C,47.34;H,5.32;N,5.52%].
4.2合成(R)-苄基2-((S)-2-((tert-丁氧基羰基)氨基)戊酰胺基)-3-氯丙酸酯(tBoc-L-正缬氨酰-β-氯-L-丙氨酸O-苄基酯)33
将tBoc-L-正缬氨酸(市售可得的)31(6.0mmol,1.31g)溶解在无水THF(50mL)中,然后添加N-甲基吗啉(6.0mmol,0.66mL)。该溶液接着冷却至0℃,然后滴加氯甲酸异丁基酯(6.0mmol,0.78mL)。混合物在0℃和氮气下搅拌1小时。于0℃下向该搅拌的溶液中添加在上述步骤4.1.3中得到的一些(R)-1-(苄基氧基)-3-氯-1-氧代丙烷-2-氯化铵(β-Cl-L-丙氨酸苄基酯的盐酸盐)32(5.4mmol,1.36g),其事先在无水DCM(30mL)中用N-甲基吗啉(5.4mmol,0.59mL)中和。该如此得到溶液在室温和氮气下搅拌过夜。该溶液过滤并真空浓缩。残留物溶解在DCM(60mL)中并用10%w/v柠檬酸溶液(2x 25mL)、接着用水(25mL)洗涤。有机层在MgSO4上干燥,过滤并真空浓缩,得到一黄色液体,其通过柱色谱进行纯制[石油溶剂油/乙酸乙酯(7:3)],得到白色固体形式的产物33(1.74g,4.2mmol,78%);m.p.95–98℃;[α]25D-25.0°(c 1.0,CH3OH);νmax/cm-1 3327(NH),2960(CH),1738(C=O),1668(C=O),1518(NH bend),1169(C-O);1H NMR(300MHz,CDCl3)δH 0.92(3H,t,J=9.0Hz,CH3-8),1.35-1.45(11H,[m,CH2-7],[s,C(CH3)3]),1.52-1.65(1H,m,CH2a-6),1.75-1.82(1H,m,CH2b-6),3.89(1H,dd,J=12.0Hz,3.0Hz,CH2a-3),3.99(1H,dd,J=12.0Hz,3.0Hz,CH2b-3),4.11-4.15(1H,m,CH-5),4.96-5.00(2H,[m,CH-2],[m,HNCO2]),5.20(1H,d,J=12.0Hz,OCH2a Ar),5.25(1H,d,J=12.0Hz,OCH2a-Ar),6.97(1H,d,J=6.0Hz,HNCO),7.33-7.37(5H,m,5x CHAr);13C NMR(75MHz,CDCl3)δC 12.7(CH3-8),17.8(CH2-7),27.3(C(CH3)3),33.4(CH2-6),43.8(CH2-3),52.2(CH-2),53.4(CH-5),67.0(OCH2Ar),79.3(C(CH3)3),127.4(CHAr),127.6(CHAr),127.7(CHAr),133.8(CHAr quat),154.5(C-9,quat),167.5(C-1,quat),171.2(C-4,quat);CHN[实测:C,58.49;H,7.22;N,6.81.C20H29ClN2O5需要C,58.18;H,7.08;N,6.78%];HRMS(纳米喷雾电离)计算(C20H30ClN2O5)+413.1838,实测MH+413.1837.
4.3合成(R)-2-((S)-2-((tert-丁氧基羰基)氨基)戊酰胺基)-3-氯丙酸(tBoc-L-正缬氨酰-β-氯-L-丙氨酸)34
将在上述步骤4.2中得到的产物33(1.8mmol,0.75g)溶解在甲醇(60mL)中,然后添加至不锈钢压力容器中。添加10%钯炭(0.0941g),如此得到的溶液在3.5bar H2压和室温下搅拌72小时。在Celite上过滤除去催化剂,用甲醇洗涤并真空浓缩,得到亮黄色固体形式的产物34(0.573g,1.78mmol,99%);m.p.59-63℃;[α]25D-12.0°(c 1.0,CH3OH);νmax/cm-13313(NH),2964(CH),1655(C=O),1509(NH bend),1161(C-O);1H NMR(300MHz,CDCl3)δH0.87(3H,t,J=4.2Hz,CH3-8),1.38(11H,[m,CH2-7],[s,C(CH3)3]),1.51-1.62(1H,m,CH2a-6),1.67-1.79(1H,m,CH2b-6),3.91(2H,m,CH2-3),4.19(1H,m,CH-5),4.85(1H,m,CH-2),5.20(1H,m,NH氨基甲酸),6.45(1H,br,OH),7.25(1H,m,NH酰胺);13C NMR(75MHz,CDCl3)δC12.7(CH3-8),17.9(CH3-7),27.3(C(CH3)3),33.6(CH2-6),43.4(CH-3),52.4(CH-2),53.2(CH-5),79.7(C(CH3)3),155.1(C-9,quat),170.7(C-1/4,quat),172.0(C-1/4,quat);CHN[实测:C,48.77;H,7.61;N,8.22.C13H23ClN2O5需要C,48.37;H,7.18;N,8.68%].
4.4合成反应中间体D/L-fosfalin二乙基酯35
如图6所示,D/L-fosfalin二乙基酯35是根据Kudzin和Stec法[9]由N-苯基硫脲53、三苯基亚磷酸酯52和乙醛51在酸性条件下合成的。该反应中间体的合成详细说明如下。
4.4.1合成(β)-(1-氨基乙基)膦酸(D/L-fosfalin)54[12]
将N-苯基硫脲53(40mmol,6.10g)溶解在冰乙酸(20mL)中。滴加乙醛51(60mmol,3.40mL),然后添加三苯基亚磷酸酯52(40mmol,11mL)。该溶液在室温下搅拌5分钟,然后在85℃下加热至回流1小时。添加冰乙酸(2mL)和盐酸(37%,20mL)的混合物,然后将反应物在145℃下加热至回流过夜。该溶液在室温下冷却,转移至500mL圆底烧瓶中并用乙醇洗涤。通过过滤得到少量的fosfalin盐酸盐,而滤液进行真空浓缩,得到暗橙色的液体残留物。将该残留物溶解在最小量的乙醇(20mL)中,然后添加氧化丙烯(120mL),产生白色沉淀物。该白色固体在氮气中过滤,并在干燥器(在五氧化磷上)中干燥3天,然后由热水/乙醇中重结晶,得到白色固体形式的两性离子产物54(4.39g,35mmol,88%);m.p.265–268℃(s)(lit.m.p.[13]271–275℃);νmax/cm-12910(br OH),1616(P-OH),1532(NH bend),1143(P=O),1035(P-O),930(P-O);1H NMR(500MHz,D2O)δH 1.47(3H,dd,JP-H=14.9Hz和JH-H=7.3Hz,CH3),3.40(1H,m,CH);13C NMR(125MHz,D2O)δC 13.5(CH3,d,JP-C=2.7Hz),44.7(CH,d,JP-C=144.2Hz).
4.4.2合成(1-(2,2,2-三氟-乙酰胺基)乙基)膦酸二乙基酯56[14]
将在上述步骤4.4.1中得到的1-氨基乙基膦酸44(40mmol,6.47g)添加至三氟乙酸(5mL)和三氟乙酸酐(25mL)的混合物中。该溶液搅拌并在60℃加热至回流。1小时后,该溶液冷却,然后缓慢地添加原甲酸三乙基酯(150mL)。该溶液在110℃加热至回流共2小时,然后冷却至室温。真空除去溶剂,得到棕色固体。将该固体重新溶解在DCM中,然后通过柱色谱纯制[DCM/MeOH(9:1)],得到米色固体形式的产物56(11.00g,39.6mmol,99%);m.p.98–103℃(s)(lit.m.p.3 101–102℃);νmax/cm-1 3202(NH),1715(C=O),1565(NH bend),1210(P=O),1011(C-F),968(P-O);1H NMR(300MHz,CDCl3)δH 1.23(3H,t,J=6.0Hz,OCH2CH3-a),1.28(3H,t,J=9.0Hz,OCH2CH3-b),1.38(3H,dd,JP-H=15.0Hz和JH-H=6.0Hz,CH3-2),3.98-4.13(4H,m,2x OCH2CH3),4.32-4.47(1H,m,CH-1),8.11(1H,d,J=9.0Hz,NH);13C NMR(75MHz,CDCl3)δC 13.7(CH3-2),15.2(OCH2CH3,d,JP-C=2.3Hz),15.3(OCH2CH3,d,JP-C=1.5Hz),40.8(CH-1,d,JP-C=159.0Hz),61.8(OCH2CH3,d,JP-C=6.8Hz),62.2(OCH2CH3,d,JP-C=7.5Hz),114.9(CF3,q,JF-C=285.8Hz),156.0(C=O,q,JF-C=6.0Hz);31P-1Hdecoup NMR(121MHz,CDCl3)δP 23.0;19F-1Hdecoup NMR(282MHz,CDCl3)δP-75.5.
4.4.3合成(β)-二乙基(1-氨基乙基)膦酸酯(D/L-fosfalin二乙基酯)35[15]
将在上述步骤4.4.2.中得到的(1-(2,2,2-三氟-乙酰胺基)乙基)膦酸二乙基酯56(20mmol,5.55g)溶解在乙醇(200mL)中,然后缓慢添加硼氢化钠(200mmol,7.57g)。如此得到的混合物在室温下搅拌1小时,然后加热至回流(90℃)3小时。该溶液冷却至室温,然后在低压下除去溶剂,得到白色固体残留物。该残留物用NaHCO3(96g/L)饱和溶液(150mL)处理,然后在DCM(6x 50mL)中萃取。有机层在MgSO4上干燥并过滤。滤液进行真空浓缩,得到淡黄色液体,然后通过柱色谱纯制[DCM/MeOH(9.0:1.0)],得到黄色液体形式的产物35(2.52g,13.9mmol,70%);νmax/cm-1 3431(NH),2980(CH),1215(P=O),1020(P-O),957(P-O);1H NMR(300MHz,CDCl3)δH 1.19-1.30(9H,[dd,JP-H=17.4Hz,7.2Hz,CH3-2],[t,J=7.2Hz,2x OCH2CH3),1.65(2H,br,NH2),2.99-3.09(1H,m,CH-1),4.02-4.14(4H,m,2x OCH2CH3);13C NMR(75MHz,CDCl3)δC 15.5(OCH2CH3-a),15.6(OCH2CH3-b),16.3(CH3-2),43.3(CH-1,d,JP-C=148.5Hz),61.1(OCH2CH3-a,d,JP-C=1.5Hz),61.2(OCH2CH3-b,d,JP-C=1.5Hz);31P-1HdecoupNMR(121MHz,CDCl3)δP 29.6.
4.5合成((2S-1-(((2R)-3-氯-1-((1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-基)氨基)-1-氧代戊烷-2-基)氨基甲酸叔丁酯(tBoc-L-正缬氨酰-β-氯-L-丙氨酰-D/L-fosfalin二乙基酯)36
将在上述步骤4.3中得到的(R)-2-((S)-2-((tert-丁氧基羰基)氨基)戊酰胺基)-3-氯丙酸(tBoc-L-正缬氨酰-β-氯-L-丙氨酸)34(1.8mmol,0.58g)溶解在无水THF(35mL)中,然后添加N-甲基吗啉(1.9mmol,0.21mL)。该溶液冷却至0℃,然后滴加异丁基氯甲酸酯(1.9mmol,0.25mL)。混合物在0℃下搅拌1小时。将在无水THF(10mL)中的于上述步骤4.4.3.中得到的1-氨基乙基膦酸二乙基酯(D/L-fosfalin二乙基酯)35(1.8mmol,0.33g)添加至上述搅拌溶液中,如此得到的溶液在氮气和室温下搅拌过夜。该溶液过滤并真空浓缩。残留物溶解在DCM(60mL)中,然后用10%w/v柠檬酸溶液(2x30mL)、10%w/v碳酸钾(30mL)和水(30mL)洗涤。有机层在MgSO4上干燥,过滤并真空浓缩,得到亮黄色液体残留物。残留物通过柱色谱进行纯制[乙酸乙酯/甲醇(96:4)],得到粘稠白色固体形式的产物36(0.45g,0.93mmol,52%);m.p.196℃(分解);[α]24D-22.5°(c 1.0,CH3OH);νmax/cm-1 3272(NH),2977(CH),1709(C=O),1644(C=O),1530(NH bend),1229(C-O),1165(P-O),1019(P-O),972(P-O);1H NMR(300MHz,CDCl3)δH 0.86(3H,t,J=6.0Hz,CH3-12),1.17-1.38(20H,[m,2x OCH2CH3],[m,CH3-2],[s,C(CH3)3],[m,CH2-11]),1.53-1.59(1H,m,CH2a-10),1.70-1.86(1H,m,CH2b-10),3.69(1H,dd,J=12.0Hz,3.0Hz,CH2a-6),3.91(1H,dd,J=12.0Hz,3.0Hz,CH2a-6),3.97-4.13(5H,[m,2x OCH2CH3],[m,CH-9]),4.35-4.46(1H,m,CH-1),4.73-4.79(1H,m,CH-5),4.97-5.03(1H,m,NH-13),7.00-7.10(1H,2x d,J=9.0Hz,9.0Hz,NH-7,非对映-异构体L,L,L和L,L,D),7.23-7.34(1H,2x d,J=9.0Hz,9.0Hz,NH-3,非对映-异构体L,L,L和L,L,D);13C NMR(75MHz,CDCl3)δC 12.7(CH3-12),14.5(CH3-2),15.3,15.5(2x OCH2CH3),17.9(CH2-11),27.3(C(CH3)3),33.2(CH2-10),40.4(d,JP-C=157.5Hz,CH-1),43.4(CH2-6),52.7(CH-5),61.4,61.9(2x OCH2CH3),79.4(C(CH3)3),155.0(C-14,quat),166.8(C-4,quat),171.4(C-8,quat);31P-1Hdecoup NMR(121MHz,CDCl3)δP 24.8;HRMS(纳米喷雾电离)计算(C19H38ClN3O7P)+486.2130,实测486.2124;CHN[实测:C,46.51;H,7.76;N,8.21.C19H37ClN3O7P需要C,46.96;H,7.67;N,8.65%].
4.6合成氢合(1-((R)-2-((S)-2-铵基戊酰胺基)-3-氯丙酰胺基)乙基)膦酸(L-正缬氨酰-β-氯-L-丙氨酰-D/L-fosfalin)37
将在上述步骤4.5中得到的((2S)-1-(((2R)-3-氯-1-((1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-基)氨基)-1-氧代戊烷-2-基)氨基甲酸叔丁酯(tBoc-L-正缬氨酰-β-氯-L-丙氨酰-D/L-fosfalin二乙基酯)36(2.0mmol,0.99g)溶解在HBr和乙酸(33%)(3.0mL)中。该溶液在室温下搅拌过夜。添加无水二乙基醚(150mL),然后该混合物在-20℃下保存过夜。滗析掉溶剂,而油状棕色粗产物用无水二乙基醚(5x 60mL)研磨。将橙色-棕色吸水性残留物溶解在无水甲醇(5mL)中,然后添加过量的氧化丙烯。该溶液过滤并用二乙基醚洗涤,得到淡绿色固体,其随后进行过滤,得到淡绿色固体形式的最终产物37(0.64g,1.94mmol,97%);m.p.175℃(secondary)[α]21D-2.50°(c 1.0,H2O+DIEA,9.9:0.1);νmax/cm-13294(NH+),2963(CH),1668(C=O),1645(C=O),1538(NH bend),1132(P-O),1039(P-O),998(P-O);1H NMR(300MHz,D2O)δH 1.01(3H,t,J=9.0Hz,CH3-12),1.30-1.37(3H,m,CH3-2),1.44-1.54(2H,m CH3-11),1.90-1.98(2H,m,CH2-10),3.91-4.15(4H,[m,CH2-6],[m,CH-9],[m,CH-1]),4.79(1H,m,CH-5);13C NMR(75MHz,D2O)δC 12.9(CH3-12),15.7(CH3-2),17.6(CH2-11),33.0(CH2-10),43.3(CH2-6),53.1(CH-1,CH-9),55.0(CH-5),170.4(C-4,C-8,quat);31P-1Hdecoup NMR(121MHz,CDCl3)δP 18.5;HRMS(纳米喷雾电离)计算(C10H20ClN3O5P)-328.0835,实测328.0833.
实施例5:根据本发明的抗微生物化合物H的合成方法(图2):L-甲硫氨酰-β-氯-L-丙氨酰-D/L-fosfalin
L-甲硫氨酰-β-氯-L-丙氨酰-D/L-fosfalin(L-Met-β-Cl-L-Ala-D/L-fosfalin)由以下式表示:
以下详细说明抗微生物化合物的合成。
5.1.合成反应中间体((2R)-3-氯-1-((1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-基)氨基甲酸叔丁酯(tBoc-β-Cl-L-Ala-D/L-Fos二乙基酯)
tBoc-β-Cl-L-Ala-D/L-Fos二乙基酯由以下式代表:
在-5℃下将N-甲基吗啉(7.8mmol,0.90mL)添加至tBoc-β-Cl-L-Ala-OH(7.8mmol,1.74g)在无水THF(60mL)中的悬浮液内。缓慢添加异氯甲酸丁基酯(7.8mmol,1.00mL),并在-5℃下搅拌所得的混合物1小时。在-5℃下向该搅拌的混合物中添加在无水THF(20mL)中的1-氨基乙基膦酸二乙基酯(8.6mmol,1.57g)。所得的混合物在氮气和-5℃下搅拌30分钟,然后在室温下搅拌过夜。该混合物过滤并真空除去溶剂,得到淡黄色液体,其用10%w/v柠檬酸溶液(2x 25mL)、10%w/v碳酸钾(25mL)和水(25mL)洗涤。合并的有机层在硫酸镁上干燥,过滤并真空浓缩,得到淡黄色液体。该液体通过柱色谱进行纯制,其中使用100%DCM,然后逐渐增加至90:10DCM/MeOH,得到黄色糖浆形式的产物,其由2个非对映异构体组成,tBoc-β-Cl-L-Ala-L-Fos二乙基酯和tBoc-β-Cl-L-Ala-D-Fos二乙基酯(2.70g,7.0mmol,90%);νmax/cm-1 3261(NH),1713(C=O),1670(C=O),1517(NH bend),1225(P=O),1164(P-O),1020(P-O),970(P-O);1H NMR(300MHz,CDCl3)δH 1.15-1.39(18H,[s,C(CH3)3],[m,CH3-2],[m,2x OCH2CH3]),3.64-3.71(1H,m,CHa/b-6),3.89-3.96(1H,m,CHa/b-6),4.01-4.12(4H,m,2x OCH2CH3),4.42-4.49(2H,[m,CH-1],[m,CH-5]),5.40(1H,d,J=3.0Hz,NH-3或NH-7-A),5.43(1H,d,J=6.0Hz,NH-3或NH-7-A),7.00(1H,d,J=9.0Hz,NH-3或NH-7-A),7.07(1H,d,J=9.0Hz,NH-3或NH-7-B);13C NMR(75MHz,CDCl3)δC 15.6(d,JP-C=5.3Hz,CH3-2),16.3(d,JP-C=2.3Hz,OCH2CH3),16.5(d,JP-C=2.3Hz,OCH2CH3),28.3(C(CH3)3)41.2(d,JP-C=157.5Hz,CH-1-A),41.3(d,JP-C=156.8Hz,CH-1-B),44.9(CH2-6-A),45.0(CH2-6-B),55.3(CH-5),62.5(d,JP-C=3.0Hz,OCH2CH3-A),62.6(d,JP-C=3.8Hz,OCH2CH3-B),62.9(d,JP-C=3.0Hz,OCH2CH3-A),63.0(d,JP-C=3.0Hz,OCH2CH3-B),80.7(C(CH3)3),154.9(C=O-8),168.3(C=O-4);31P-1Hdecoup NMR(121MHz,CDCl3)δP 24.8.
5.2.合成反应中间体(2R)-3-氯-1-((1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-氯化铵(β-Cl-L-Ala-D/L-Fos二乙基酯盐酸盐)
β-Cl-L-Ala-D/L-Fos二乙基酯盐酸盐由下式代表:
在氮气和室温下搅拌tBoc-β-Cl-L-Ala-D/L-Fos二乙基酯(步骤5.1.中得到的;6.7mmol,2.59g)于2M HCl之二乙基醚溶液(100mL)中的溶液过夜。该混合物接着过滤,而米色吸水性固体用二乙基醚洗涤。该固体在包含氧化磷(V)的干燥器中干燥过夜,然后用石油溶剂油研磨,得到淡绿色固体形式的产物,其由2种非对映异构体组成,β-Cl-L-Ala-L-Fos二乙基酯盐酸盐和β-Cl-L-Ala-D-Fos二乙基酯盐酸盐(1.51g,4.7mmol,70%);m.p.127–131℃(分解);νmax/cm-1 3204(NH),1687(C=O),1562(NH bend),1204(P=O),1010(P-O),961(P-O);1H NMR(300MHz,D2O)δH 1.28(3H,t,J=6.0Hz,OCH2CH3),1.29(3H,t,J=6.0Hz,OCH2CH3),1.37(3H,dd,3JP-H=18.0Hz,3JH-H=6.0Hz,CH3-2),3.92-4.04(2H,m,CH2-6),4.07-4.21(4H,m,2x OCH2CH3),4.38-4.48(2H,[m,CH-1],[m,CH-5]);13C NMR(75MHz,D2O)δC 13.7(CH3-2),14.0(CH3-2),15.7(OCH2CH3),15.7(OCH2CH3),41.7(d,JP-C=158.3Hz,CH-1),42.0(d,JP-C=157.5Hz,CH-1),42.4(CH2-6),53.7(CH-5),53.8(CH-5),64.3(d,JP-C=6.8Hz,OCH2CH3),64.5(d,JP-C=6.8Hz,OCH2CH3-B),165.7(C=O-4-A),165.8(C=O-4-B);31P-1Hdecoup NMR(121MHz,CDCl3)δP 26.1.
5.3.合成反应中间体((2S)-1-(((2R)-3-氯-1-((1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-基)氨基)-4-(甲硫基)-1-氧代丁烷-2-基)氨基甲酸叔丁酯(tBoc-L-Met-β-Cl-L-Ala-D/L-Fos二乙基酯)
tBoc-L-Met-β-Cl-L-Ala-D/L-Fos二乙基酯由下式代表:
将N-甲基吗啉(3.4mmol,0.40mL)添加至tBoc-L-Met-OH(3.4mmol,0.85g)在无水THF(60mL)中的溶液内。该溶液冷却至-5℃,然后滴加氯甲酸异丁基酯(3.4mmol,0.45mL)。该混合物在-5℃下搅拌1小时。在-5℃下向该搅拌的混合物中滴加在步骤5.2.中得到的β-Cl-L-Ala-D/L-Fos二乙基酯盐酸盐(3.4mmol,1.10g)在无水DCM(20mL)中的溶液,该溶液已用N-甲基吗啉(3.4mmol,0.40mL)中和过。所得的混合物在氮气和-5℃下搅拌30分钟,然后在室温下搅拌过夜。该混合物过滤并真空浓缩,然后用10%w/v柠檬酸溶液(2x 25mL)、10%w/v碳酸钾(25mL)、水(25mL)和盐水(30mL)洗涤。有机层在MgSO4上干燥,过滤并真空除去溶剂,得到黄色液体,其通过柱色谱[DCM/MeOH(95:5)]进行纯制,得到无色液体。由二乙基醚/石油溶剂油中重结晶,形成白色固体形式的产物,其由2个非对映异构体组成,tBoc-L-Met-β-Cl-L-Ala-L-Fos二乙基酯和tBoc-L-Met-β-Cl-L-Ala-D-Fos二乙基酯(0.88g,1.7mmol,50%);m.p.96-99℃;νmax/cm-1 3278(NH),1709(C=O),1639(C=O),1523(NH bend),1228(P=O),1018(P-O),970(P-O);1H NMR(300MHz,CDCl3)δH 1.17-1.36(9H,[m,CH3-2],[m,2x OCH2CH3]),1.38(9H,s,C(CH3)3),1.87-2.07(5H,[s,CH3-12],[m,CH2-10]),2.48-2.54(2H,m,CH2-11),3.71(1H,dd,J=12.0Hz,6.0Hz,CHa/b-6),3.88(1H,dd,J=12.0Hz,6.0Hz,CHa/b-6),3.99-4.13(4H,m,2x OCH2CH3),4.20(1H,m,CH-9),4.37-4.47(1H,m,CH-1),4.78-4.84(1H,m,CH-5),5.39(1H,d,J=6.0Hz,NH-13-A),5.41(1H,d,J=6.0Hz,NH-13-B),7.15(1H,d,J=6.0Hz,NH-7-A),7.24(1H,d,J=6.0Hz,NH-7-B),7.52(1H,m,NH-3);13C NMR(75MHz,CDCl3)δC 14.4(CH3-2或CH3-12),14.5(CH3-2或CH3-12),15.4(OCH2CH3),15.5(OCH2CH3),27.3(C(CH3)3),29.2(CH2-11-A),29.3(CH2-11-B),30.2(CH2-10-A),30.4(CH2-10-B),40.3(d,JP-C=159.0Hz,CH-1),43.5(CH2-6-A),43.7(CH2-6-B),52.7(CH-5),53.1(CH-9),61.6(d,JP-C=6.8Hz,OCH2CH3-A),61.7(d,JP-C=6.0Hz,OCH2CH3-B),62.0(d,JP-C=6.8Hz,OCH2CH3-A),62.1(d,JP-C=7.5Hz,OCH2CH3-B),79.6(C(CH3)3),154.8(C=O-14),166.7(C=O-4-A),166.8(C=O-4-B),170.7(C=O-8-A),170.8(C=O-8-B);31P-1Hdecoup NMR(121MHz,CDCl3)δP 24.8;CHN[实测:C,44.08;H,7.47;N,8.18.C19H37ClN3O7PS需要C,44.06;H,7.20;N,8.11%].
5.4.合成抗微生物化合物H(图2),即、化合物氢合(1-((R)-2-((S)-2-铵基-4-(甲硫基)丁酰胺基)-3-氯丙酰胺基)乙基)膦酸(L-Met-β-Cl-L-Ala-D/L-Fos)
抗微生物化合物L-Met-β-Cl-L-Ala-D/L-Fos由下式代表:
在步骤5.3.中得到的tBoc-L-Met-β-Cl-L-Ala-D/L-Fos二乙基酯(1.4mmol,0.71g)在溴化氢/冰乙酸(33%)(8.0mL)中的溶液在室温下搅拌过夜。添加无水二乙基醚(70mL),然后将该混合物放置在冷冻机中过夜。笔析掉溶剂,而粗产物用无水二乙基醚研磨(5x 50mL)。将该棕色-橙色粗产物溶解在甲醇(5mL)中,然后添加过量的氧化丙烯。该混合物过滤并用无水二乙基醚洗涤,得到绿色固体,其在包含氧化磷(V)的干燥器中干燥,然后由热水/乙醇中重结晶,得到淡绿色固体形式的产物,其由2种非对映异构体组成L-Met-β-Cl-L-Ala-L-Fos和L-Met-β-Cl-L-Ala-D-Fos(0.17g,0.48mmol,35%);m.p.175–179℃(分解);νmax/cm-13264(NH+),2829(large OH),1641(C=O),1546(NH bend),1149(P-O),1041(P-O),921(P-O);1H NMR(300MHz,D2O)δH 1.10-1.50(3H,m,CH3-2),2.08-2.32(5H,[m,CH3-12],[m,CH2-10])2.61(2H,CH2-11),3.37-4.16(4H,[m,CH2-6],[m,CH-1],[m,CH-9]),4.48(1H,m,CH-5);13C NMR(75MHz,D2O.)δC 14.0(CH3-12),15.5(CH3-2),28.2(CH2-11),30.0(CH2-10),43.3(CH2-6),44.8(CH-1),52.3(CH-9),55.0(CH-5),169.4(C=O-4和C=O-8);31P-1Hdecoup NMR(121MHz,CDCl3)δP 18.7.
实施例6:评估分别根据实施例4和5合成的L-正缬氨酰-β-氯-丙氨酰-D/L-fosfalin(化合物G;参考图2)和L-甲硫氨酰-β-氯-丙氨酰-D/L-fosfalin(化合物H;参考图2)的抗菌活性
6.1.介绍
在使用如实施例2所述的琼脂稀释法(特别是参考“2.2材料和方法”)温育22小时后,测定化合物G和H对12株革兰氏阴性菌和6株革兰氏阳性菌的最小抑制浓度(MIC)。
6.2结果
所得结果示于下表4中。
表4:抗微生物化合物G和H对各种细菌的最小抑制浓度
根据上述表4中所示的数据,其清楚地表明,化合物G和H对某些革兰氏阴性菌种和某些革兰氏阳性菌种的测试菌株具有强烈的抑制作用,特别是对于大肠埃希氏菌、肺炎克雷伯氏菌、粘质沙雷氏菌、小肠结肠炎耶尔森氏菌、粪肠球菌,但对于其他的革兰氏阴性和革兰氏阳性菌种不太抑制,特别是对于鲍氏不动杆菌、洋葱伯克霍尔德氏菌、铜绿假单胞菌、鼠伤寒沙门氏菌、肠炎沙门氏菌、单核细胞增生利斯特氏菌。
6.3结论
在各种细菌之间观察到的抑制浓度具有非常显著的差异,这使得化合物G和H特别适合作为掺入反应培养基的化合物,其能够选择性地寻找和/或分离生物样品中的某些细菌,特别是可以选择性地寻找:
‐革兰氏阴性菌(革兰氏阴性靶细菌),例如鲍氏不动杆菌、洋葱伯克霍尔德氏菌、铜绿假单胞菌、鼠伤寒沙门氏菌、肠炎沙门氏菌;或
‐革兰氏阳性菌(革兰氏阳性靶细菌),例如单核细胞增生利斯特氏菌。
实施例7:根据本发明的抗微生物化合物I的合成方法(图2):L-正缬氨酰-L-丙氨酰-D/L-fosfalin
L-正缬氨酰-L-丙氨酰-D/L-fosfalin(L-Nva-L-Ala-D/L-fosfalin)由下式代表:
以下详细描述该抗微生物化合物的合成。
7.1.合成反应中间体(S)-苄基2-((S)-2-((tert-丁氧基羰基)氨基)戊酰胺基)丙酸酯(tBoc-L-Nva-L-Ala-OBzl)
化合物tBoc-L-Nva-L-Ala-OBzl由下式代表:
将N-甲基吗啉(15.0mmol,1.65mL)添加至tBoc-L-Nva-OH(10.0mmol,2.17g)在无水THF(60mL)中的溶液内。该溶液冷却至-5℃,然后滴加氯甲酸异丁基酯(15.0mmol,1.95mL)。该混合物在-5℃下搅拌1小时。向该搅拌的混合物中滴加一些在无水DCM(30mL)中的L-丙氨酸苄基酯p-甲苯磺酸盐(10.0mmol,3.52g),其已在-5℃下用二异丙基乙基胺(15.0mmol,2.60mL)中和。所得的溶液在氮气和-5℃下搅拌30分钟,然后在室温下搅拌过夜。该溶液过滤并真空浓缩,接着用10%w/v柠檬酸(2x 25mL)、10%w/v碳酸钾(25mL)和水(25mL)洗涤。有机层在MgSO4上干燥,过滤并真空浓缩,得到黄色吸水性液体,该液体通过柱色谱进行纯制[40-60石油溶剂油/乙酸乙酯(7:3)],得到米色固体形式的产物(2.40g,6,3mmol,63%);m.p.60-63℃;νmax/cm-1 3299(NH),1743(C=O),1655(C=O),1527(NH bend),1245(C-O),1162(C-O);1H NMR(300MHz,CDCl3)δH 0.83(3H,t,J=9.0Hz,CH3-8),1.25-1.36(14H,[d,J=6.0Hz,CH3-3],[m,CH2-7],[s,C(CH3)3]),1.42-1.54(1H,m,CHa/b-6),1.64-1.73(1H,m,CHa/b-6),4.02(1H,m,CH-5),4.54(1H,pentet,J=6.0Hz,CH-2),4.96(1H,d,J=9.0Hz,NHCO2),5.07(1H,d,J=12.0Hz,OCHa/bAr),5.12(1H,d,J=12.0Hz,OCHa/bAr),6.56(1H,d,J=6.0Hz,NHCO),7.27(5H,m,5xCHAr);13C NMR(75MHz,CDCl3)δC 12.7(CH3-8),17.3(CH3-3),17.8(CH2-7),27.3(C(CH3)3),33.7(CH2-6),47.1(CH-2),53.4(CH-5),66.1(OCH2Ar),79.0(C(CH3)3),127.1-127.6(5xCHAr),134.3(CHAr quat.),154.6(C=O-9),170.8(C=O-4),171.5(C=O-1);CHN[实测:C,63.75;H,8.37;N,7.86.C20H30N2O5需要C,63,47;H,7.99;N,7.40%];HRMS(纳米喷雾电离)计算(C20H31N2O5)+379.2227,实测379.2222.
7.2.合成反应中间体(S)-2-((S)-2-((tert-丁氧基羰基)氨基)戊酰胺基)丙酸(tBoc-L-Nva-L-Ala-OH)
化合物tBoc-L-Nva-L-Ala-OH由下式代表:
将在步骤7.1.中得到的tBoc-L-Nva-L-Ala-OBzl(6.0mmol,2.27g)溶解在甲醇(60mL)中,然后在3.5bar H2和室温下于5%钯炭(0.23g)存在时进行氢化过夜。催化剂通过用Celite过滤而被除去,然后用甲醇洗涤。该溶液进行真空浓缩,得到白色固体形式的产物(1.66g,5.7mmol,96.0%);m.p.53-58℃(分解);νmax/cm-1 3500-2500(br,OH),3300(NH),2961(NH),1688(C=O),1655(C=O),1522(NH bend),1245(C-O),1164(C-O);1H NMR(300MHz,CDCl3)δH 0.85(3H,t,J=9.0Hz,CH3-8),1.27-1.39(14H,[m,CH3-3],[m,CH2-7],[s,C(CH3)3]),1.48-1.53(1H,m,CHa/b-6),1.67-1.71(1H,m,CHa/b-6),4.10(1H,m,CH-5),4.50(1H,m,CH-2),5.27(1H,m,NHCO2),6.93(1H,m,NHCO),8.87(1H,br,OH);13C NMR(75MHz,CDCl3)δC 13.7(CH3-8),18.0(CH3-3),18.8(CH2-7),28.3(C(CH3)3),34.5(CH2-6),48.1(CH-2),54.3(CH-5),80.4(C(CH3)3),156.0(C=O-9),172.5(C=O-4),175.5(C=O-1);CHN[实测:C,54.18;H,8.78;N,9.62.C13H24N2O5需要C,54.15;H,8.39;N,9.72%];HRMS(纳米喷雾电离)计算(C13H25N2O5)+289.1758,实测289.1758.
7.3.合成反应中间体((2S)-1-(((2S)-1-((1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-基)氨基)-1-氧代戊烷-2-基)氨基甲酸叔丁酯(tBoc-L-Nva-L-Ala-D/L-Fos二乙基酯)
tBoc-L-Nva-L-Ala-D/L-Fos二乙基酯由下式代表:
在-5℃下将N-甲基吗啉(5.3mmol,0.58mL)添加至tBoc-L-Nva-L-Ala-OH(5.0mmol,1.45g;得自于步骤7.2.)在无水THF(50mL)中的溶液内。缓慢添加氯甲酸异丁基酯(5.3mmol,0.70mL),而所得的混合物在-5℃下搅拌1小时。在-5℃下向该搅拌的混合物中添加在无水THF(15mL)中的1-氨基乙基膦酸二乙基酯(4.8mmol,0.87g;其合成如实施例4.4中所述,并示于图6中)。所得的混合物在氮气和-5℃下搅拌30分钟,接着在室温下搅拌过夜。混合物过滤并真空除去溶剂,得到白色固体,其重新溶解在DCM(50mL)中,并用10%w/v柠檬酸(2x 25mL)、10%w/v碳酸钾(25mL)和水(25mL)洗涤。有机层在硫酸镁上干燥,过滤并真空浓缩,得到白色固体,其通过柱色谱进行纯制,其中使用100%DCM,并增加至90:10DCM/甲醇,得到白色固体,其由2种非对映异构体组成,tBoc-L-Nva-L-Ala-L-Fos二乙基酯和tBoc-L-Nva-L-Ala-D-Fos二乙基酯(1.70g,3.8mmol,78%);m.p.165-168℃;νmax/cm-1 3267(NH),1708(C=O),1638(C=O),1537(NH bend),1227(P=O),1019(P-O),966(P-O);1H NMR(300MHz,CDCl3)δH 0.85(3H,t,J=9.0Hz,CH3-12),1.18-1.34(14H,[m,2x OCH2CH3],[CH3-2],[m,CH3-6],[m,CH2-11]),1.37(s,C(CH3)3),1.47-1.54(1H,m,CHa/b-10),1.65-1.73(1H,m,CHa/b-10),3.98-4.12(5H,[m,2x OCH2CH3],[m,CH3-5或CH3-9]),4.33-4.44(1H,m,CH-1),4.48-4.54(1H,m,CH-5或CH-9),5.18-5.23(1H,d,J=6.0Hz,NH-7或NH-13),5.18-5.23(1H,d,J=6.0Hz,NH-7或NH-13),6.77-6.88(1H,d,J=6.0Hz,NH-7或NH-13),6.77-6.88(1H,d,J=6.0Hz,NH-7或NH-13),7.13-7.24(1H,d,J=9.0Hz,9.0Hz,NH-3),7.13-7.24(1H,d,J=9.0Hz,9.0Hz,NH-3);13C NMR(75MHz,CDCl3)δC 12.7(CH3-12),14.5(CH3-2,d,JP-C=6.0Hz),15.3(OCH2CH3-A),15.4(OCH2CH3-A),15.5(OCH2CH3-B),15.6(OCH2CH3-B),17.6(CH3-6-A),17.7(CH3-6-B),17.8(CH2-11-A),17.9(CH2-11-B),27.3(C(CH3)3),33.8(CH2-10-A),33.9(CH2-10-B),39.9(CH-1-A,d,JP-C=157.5Hz),40.0(CH-1-B,d,JP-C=156.8Hz),47.7(CH-5或CH-9-A),47.9(CH-5或CH-9-B),53.5(CH-5或CH-9-A),53.5(CH-5或CH-9-B),61.4-62.0(2x OCH2CH3-A和B,4x d,J=7.5Hz,6.8Hz,7.5Hz,7.5Hz),78.9(C(CH3)3),154.7(C=O-14),170.6(C=O-4或C=O-8-A),170.7(C=O-4或C=O-8-B),171.0(C=O-4或C=O-8-A),171.1(C=O-4或C=O-8-B);31P-1Hdecoup NMR(121MHz,CDCl3)δP 24.8;CHN[实测:C,50.74;H,8.55;N,9.51.C19H38N3O7P需要C,50.54;H,8.48;N,9.31%];HRMS(纳米喷雾电离)计算(C19H39N3O7P)+452.2520,实测452.2518.
7.4.合成化合物I,即、化合物氢合(1-((S)-2-((S)-2-铵基戊酰胺基)丙酰胺基)乙基)膦酸(L-Nva-L-Ala-D/L-Fos)
如前所述,化合物L-Nva-L-Ala-D/L-Fos由下式代表:
将在步骤7.3中得到的tBoc-L-Nva-L-Ala-D/L-Fos二乙基酯(1.6mmol,0.72g)溶解在冰乙酸(30mL)中,然后添加溴化氢/乙酸(33%)(25mL)。该溶液在室温下搅拌过夜。添加无水二乙基醚(150mL),然后将该混合物放置在冷冻机中过夜。笔析出溶剂,而粗产物用无水二乙基醚研磨(5x 100mL)。橙黄色的粗产物溶解在甲醇(5mL)中,然后添加过量的氧化丙烯。该溶液过滤并用二乙基醚洗涤,得到淡绿色固体,其由热水/丙酮中重结晶,得到淡绿色固体形式的产物,其由2种非对映异构体组成,L-Nva-L-Ala-L-Fos和L-Nva-L-Ala-D-Fos(0.22g,0.75mmol,47%);m.p.200–210℃(分解);νmax/cm-1 3280(NH+),1643(C=O),1552(NH bend),1149(P-O),1037(P-O),922(P-O);1H NMR(300MHz,D2O)δH 0.96(3H,t,CH3-12),1.29(3H,m,CH3-6)1.42-1.40(5H,[m,CH3-2],[m,CH2-11]),1.88-1.86(2H,m,CH2-10),4.02-4.00(2H,[m,CH-5],[m,CH-9]),4.34-4.39(1H,m,CH-1);13C NMR(75MHz,D2O.)δC13.4(CH3-12),16.0(CH3-2),17.1(CH3-6),17.2(CH3-6),18.1(CH2-11),33.5(CH2-10),50.7(d,JP-C=22.5Hz,CH-1),53.5(CH-5或CH-9),170.4(C=O-8),174.7(C=O-4);HRMS(纳米喷雾电离)计算(C10H23N3O5P)+296.1370,实测296.1373;31P-1Hdecoup NMR(121MHz,CDCl3)P18.5;CHN[实测:C,37.61;H,7.51;N,12.91.C10H22N3O5P·1.4H2O需要C,37.48;H,7.80;N,13.11%].
实施例8:抗微生物化合物J的合成方法(图2):L-甲硫氨酰-L-丙氨酰-D/L fosfalin
化合物L-甲硫氨酰-L-丙氨酰-D/L fosfalin由下式代表:
以下详细描述该抗微生物化合物的合成。
8.1.合成反应中间体((S)-1-(((R)-1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-基)氨基甲酸叔丁酯(tBoc-L-Ala-D/L-Fos二乙基酯)
tBoc-L-Ala-D/L-Fos二乙基酯由下式代表:
在-5℃下将N-甲基吗啉(15.0mmol,1.65mL)添加至tBoc-L-Ala-OH(10.0mmol,1.90g)在无水THF(60mL)中的溶液内。添加氯甲酸异丁基酯(15.0mmol,1.90mL),所得的混合物在-5℃下搅拌1小时。在-5℃下向该搅拌的混合物中添加在无水THF(20mL)中的1-氨基乙基膦酸二乙基酯(10.0mmol,1.84g)。所得的混合物在氮气和-5℃下搅拌30分钟,接着在室温下搅拌过夜。该溶液过滤并真空浓缩,得到淡黄色的糖浆,将其重新溶解在DCM(60mL)中,然后用10%w/v柠檬酸(2x 25mL)、10%w/v碳酸钾(25mL)和水(25mL)洗涤。合并的有机层在硫酸镁上干燥,过滤并真空浓缩,得到淡绿色糖浆,其通过柱色谱进行纯制,其中初始使用100%DCM,然后增加至95:5DCM/甲醇,得到米色固体形式的产物,其由2种非对映异构体组成,tBoc-L-Ala-L-Fos二乙基酯和tBoc-L-Ala-D-Fos二乙基酯(2.49g,7.1mmol,71%);m.p.102-105℃;νmax/cm-1 3280(NH),1710(C=O),1652(C=O),1556(NH bend),1229(P=O),1173(P-O),1013(P-O),973(P-O);1H NMR(300MHz,CDCl3)δH 1.23-1.44(21H,[s,C(CH3)3],[m,CH3-2],[m,CH3-6],[m,2x OCH2CH3]),4.06-4.23(5H,[m,2x OCH2CH3],[m,CH-5]),4.40-4.52(1H,m,CH-1),5.11-5.15(1H,2x d,J=1.5Hz,1.5Hz,NH-7),6.70-6.78(1H,2x d,J=2.3Hz,2.3Hz,NH-3);13C NMR(75MHz,CDCl3)δC 15.6(CH3-2),16.3(d,J=3.0Hz,OCH2CH3),16.4(d,J=2.3Hz,OCH2CH3),16.4(d,J=3.8Hz,OCH2CH3),16.5(d,J=1.5Hz,OCH2CH3),18.4(CH3-6),28.3(C(CH3)3)40.8(d,JP-C=156.8Hz,CH-1),41.0(d,JP-C=156.8Hz,CH-1),50.0(CH-5),62.4-62.8(4x d,JP-C=6.8Hz,6.8Hz,6.8Hz,6.8Hz,2x OCH2CH3),80.0(C(CH3)3),155.2(C=O-8),172.1(C=O-4);CHN[实测:C,48.22;H,8.58;N,7.87.C14H29N2O6P需要C,47.72;H,8.30;N,7.95%].
8.2.合成反应中间体(S)-1-(((R)-1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-氯化铵(L-Ala-D/L-Fos二乙基酯盐酸盐)
L-Ala-D/L-Fos二乙基酯盐酸盐由下式代表:
在氮气和室温下搅拌tBoc-L-Ala-D/L-Fos二乙基酯(步骤8.1中得到的;6.0mmol,2.13g)在2M HCl之二乙基醚溶液(100mL)中的溶液过夜。过滤收集所得的固体,并用无水二乙基醚洗涤。米色吸水性固体在包含氧化磷(V)的干燥器中干燥过夜,然后用石油溶剂油洗涤,得到淡绿色固体形式的产物,其由2种非对映异构体组成,L-Ala-L-Fos二乙基酯盐酸盐和L-Ala-D-Fos二乙基酯盐酸盐(1.46g,5.1mmol,84%);m.p.102-105℃;νmax/cm-1 2986(NH+),1673(C=O),1555(NH bend),1017(P-O),950(P-O);1H NMR(300MHz,d4-CH3OH)δH 1.29-1.44(9H,[m,2x OCH2CH3],[m,CH3-2],1.51(3H,d,J=6.0Hz,CH3-6),3.90-3.98(1H,m,CH-5),4.08-4.22(4H,m,2x OCH2CH3),4.28-4.47(1H,m,CH-1);13C NMR(75MHz,d4-CH3OH)δC13.7(CH3-2-A),14.0(CH3-2-B),15.4(2x OCH2CH3),16.3(CH3-6),41.1(d,JP-C=158.3Hz,CH-1),41.4(d,JP-C=158.3Hz,CH-1),48.8(CH-5),48.9(CH-5),62.7-63.0(2x OCH2CH3),169.0(C=O-4).
8.3.合成反应中间体Tert-丁基((2S)-1-(((2S)-1-((1-(二乙氧基磷酰基)乙基)氨基)-1-氧代丙烷-2-基)氨基-4-(甲硫基)-1-氧代丁烷-2-基)氨基甲酸(tBoc-L-Met-L-Ala-D/L-Fos二乙基酯)
化合物tBoc-L-Met-L-Ala-D/L-Fos二乙基酯由下式代表:
将N-甲基吗啉(5.1mmol,0.60mL)添加至tBoc-L-Met-OH(3.4mmol,0.88g)在无水THF(50mL)中的溶液内。该溶液冷却至-5℃,然后滴加氯甲酸异丁基酯(5.1mmol,0.70mL)。该混合物在-5℃下搅拌1小时。向该搅拌的溶液内滴加在无水THF(15mL)中的由步骤8.2中得到的L-Ala-D/L-Fos二乙基酯盐酸盐(3.4mmol,0.97g),其已在-5℃下用二异丙基乙基胺(5.1mmol,1,00mL)进行中和。所得的溶液在氮气和-5℃下搅拌30分钟,然后在室温下搅拌过夜。该溶液过滤并真空浓缩,接着用10%w/v柠檬酸(2x 25mL)、10%w/v碳酸钾(25mL)和水(25mL)洗涤。有机层在MgSO4上干燥,过滤并真空浓缩,得到黄色固体,其通过柱色谱进行纯制[DCM/MeOH(95:5)],得到米色固体形式的产物,其由2种非对映异构体组成,tBoc-L-Met-L-Ala-L-Fos二乙基酯和tBoc-L-Met-L-Ala-D-Fos二乙基酯(0.53g,1.1mmol,32%);m.p.172-176℃;νmax/cm-1 3272(NH),1708(C=O),1637(C=O),1530(NH bend),1226(P=O),1165(P-O),1020(P-O),966(P-O);1H NMR(300MHz,CDCl3)δH 1.16-1.36(12H,[m,CH3-2],[m,CH3-6],[m,2x OCH2CH3]),1.36(9H,s,C(CH3)3),1.82-2.01(2H,m,CH2-10),2.04(3H,s,CH3-12),2.49(2H,t,J=9.0Hz,CH2-11),4.00-4.12(4H,m,2x OCH2CH3),4.21(1H,m,CH-9),4.33-4.43(1H,m,CH-1),4.45-4.53(1H,m,CH-5),5.40(1H,d,J=9.0Hz,NH-13-A),5.44(1H,d,J=6.0Hz,NH-13-B),6.85(1H,d,J=6.0Hz,NH-7-A),6.92(1H,d,J=6.0Hz,NH-7-B),7.07(1H,d,J=9.0Hz,NH-3-A),7.16(1H,d,J=9.0Hz,NH-3-B);13C NMR(75MHz,CDCl3)δC 14.3(CH3-2-A),14.3(CH3-2-B),15.4-15.5(2x OCH2CH3),17.7(CH3-6),27.3(C(CH3)3),29.2(CH2-11-A),29.3(CH2-11-B),30.8(CH2-10-A),30.8(CH2-10-B),39.9(d,JP-C=156.8Hz,CH-1-A),40.0(d,JP-C=156.8Hz,CH-1-B),47.9(CH-5-A),48.0(CH-5-B),52.6(CH-9),61.5(d,JP-C=4.5Hz,OCH2CH3-A),61.6(d,JP-C=4.5Hz,OCH2CH3-B),61.7(d,JP-C=6.8Hz,OCH2CH3-A),61.9(d,JP-C=6.8Hz,OCH2CH3-B),79.1(C(CH3)3),154.6(C=O-14),170.3(C=O-4或C=O-8-A),170.4(C=O-4或C=O-8-B),170.5(C=O-4或C=O-8-A),170.6(C=O-4或C=O-8-B).
8.4.合成根据本发明的化合物J,即、化合物氢合(1-((S)-2-((S)-2-铵基-4-(甲硫基)丁酰胺基)丙酰胺基)乙基)膦酸(L-甲硫氨酰-L-丙氨酰-D/L fosfalin)
如前所述,化合物L-甲硫氨酰-L-丙氨酰-D/L-fosfalin由以下结构式代表:
在室温下搅拌tBoc-L-Met-L-Ala-D/L-Fos二乙基酯(步骤8.3中得到的;0.9mmol,0.43g)在溴化氢/冰乙酸(33%)(10mL)中的溶液过夜。添加无水二乙基醚(150mL),然后该混合物放置在冷冻机中过夜。笔析出溶剂,而粗产物用无水二乙基醚研磨(5x100mL)。棕黄色的粗固体溶解在甲醇(5mL)中,然后添加过量的氧化丙烯。该溶液过滤并用洗涤二乙基醚,得到绿色固体,其由热乙醇重结晶,然后在包含氧化磷(V)的干燥器中进一步干燥,得到淡绿色固体形式的产物,其由2种非对映异构体组成,L-Met-L-Ala-L-Fos和L-Met-L-Ala-D-Fos(0.13g,0.41mmol,46%);m.p.213–217℃(分解);νmax/cm-13263(NH+),2834(large OH),1641(C=O),1552(NH bend),1150(P-O),1041(P-O),919(P-O);1H NMR(300MHz,D2O)δH1.12-1.29(3H,m,CH3-2),1.38(3H,d,J=6.0Hz,CH3-6),2.11-2.34(5H,[s,CH3-12],[m,CH2-10]),2.62(2H,m,CH2-11),3.95-4.11(2H,[m,CH-1],[m,CH-9]),4.32-4.46(1H,m,CH-5);13C NMR(75MHz,D2O.)δC14.2(CH3-12),15.5(CH3-2),16.6(CH3-6),28.5(CH2-11),29.5(CH2-10),49.9(CH-1,CH-5和CH-9),169.4(C=O-4和C=O-8);31P-1Hdecoup NMR(121MHz,CDCl3)δP 20.7.
实施例9:评估分别在实施例7和8中合成的L-正缬氨酰-L-丙氨酰-D/L-fosfalin(图2中的化合物I)和L-甲硫氨酰-L-丙氨酰-D/L-fosfalin(图2中的化合物J)的抗菌活性
9.1.介绍
如实施例6,在使用如实施例2所述的琼脂稀释法(特别是参考“2.2材料和方法”)温育22小时后,测定化合物I和J对12株革兰氏阴性细菌和6株革兰氏阳性细菌的最小抑制浓度(MIC)。
用于本实施例测试的18株细菌与实施例6中所用的相同。
9.2结果
所得结果示于以下表5中。
表5:抗微生物化合物I和J对各种细菌的最小抑制浓度
根据上述表5中所示的数据,其清楚地表明,化合物I和J对某些革兰氏阴性菌种和某些革兰氏阳性菌种的测试菌株具有强烈的抑制作用,特别是对于大肠埃希氏菌、肺炎克雷伯氏菌、粘质沙雷氏菌、小肠结肠炎耶尔森氏菌、粪肠球菌,但对于其他的革兰氏阴性和革兰氏阳性菌种不太抑制,特别是对于鲍氏不动杆菌、洋葱伯克霍尔德氏菌、铜绿假单胞菌、鼠伤寒沙门氏菌、肠炎沙门氏菌、单核细胞增生利斯特氏菌。
参考文献
[1]Rao J,Lahiri J,Weis RM,Whitesides GM.2000.Design,synthesis,and characterization of a high-affinity trivalent system derived from vancomycin and L-Lys-D-Ala-D-Ala.J.Am.Chem.Soc.122:2698-2710.
[2]Kametani T,Suzuki Y,Kigasawa K,Hiiragi M,Wakisaka K,Sugi H,Tanigawa K,Fukawa K,Irino O,Saita O,Yamabe S.1982.Studies on the synthesis of chemotherapeutics.Part XIII.Synthesis and biological studies on phosphonopeptides having alkyl-,phenyl-,and heterocyclic substituents.Heterocycles 18:295-319.
[3]J.Bacteriol.1984,160(1),122-130Gibson et al
[4]Cheung et al,Chloroalanyl and Propargylglycyl Diperptides.Suicide Substrate Containing Antibacterials.J.Med.Chem.1983,26,1733-1741
[5]Cheung KS,Boisvert W,Lerner SA,Johnston M.1986.Chloroalanyl antibiotic peptides:antagonism of their antimicrobial effects by L-alanine and L-alanyl peptides in gram-negative bacteria.J.Med.Chem.29:2060-2068.
[6]Atherton et al.,Synthesis and Structure-Activity Relationships of Antibacterial Phosphonopeptides Incorporating(l-Aminoethyl)phosphonic Acid and(Aminomethyl)phosphonic Acid,Chemistry Department,Roche Products Limited,1984
[7]Arfin et al.,Inhibition of Growth of Salmonella typhimurium and of Threonine Deaminase and Transamine B by β-Chloroalanine
[8]Orenga et al.,2009;J.Microbiol.Methods;79(2:139-55)
[9]Lavielle,S.;Ling,N.C.;Saltman,R.;Guillemn,R.C.Carbohydrate Research,1981,89(2),229
[10]Nakata,T.;Nakatani,M.;Takahashi,M.;Okai,J.;Kawaoka,Y.;Kouge,K.;Okai,H.Bulletin of Chemical Society of Japan,1996,69(4),1099
[11]Doctoral thesis of Varadi,L.,University of Sunderland,2012
[12]Kudzin,Z.H.;Stec,W.J.Synthesis,1978,469.
[13]Boduszek,B.;Soroka,M.Polish Journal of Chemistry,2002,76(8),1105
[14]Kudzin,Z.H.;Luczak,J.Synthesis,1995,1995(5),509.
[15]Yuan,C.;Xu,C.;Zhang,Y.Tetrahedron,2003,59(32),6095
[16]Andrews JM.2001.Determination of minimum inhibitory concentrations.J.Antimicrob.Chemother.48Suppl 1:5-16.
[17]Atherton FR,Hall MJ,Hassall CH,Lambert RW,Ringrose PS.1979.Phosphonopeptides as antibacterial agents:rationale,chemistry,and structure-activity relationships.Antimicrob.Agents Chemother.15:677-683.
- 还没有人留言评论。精彩留言会获得点赞!