在不相容的催化剂之间转变的方法与流程
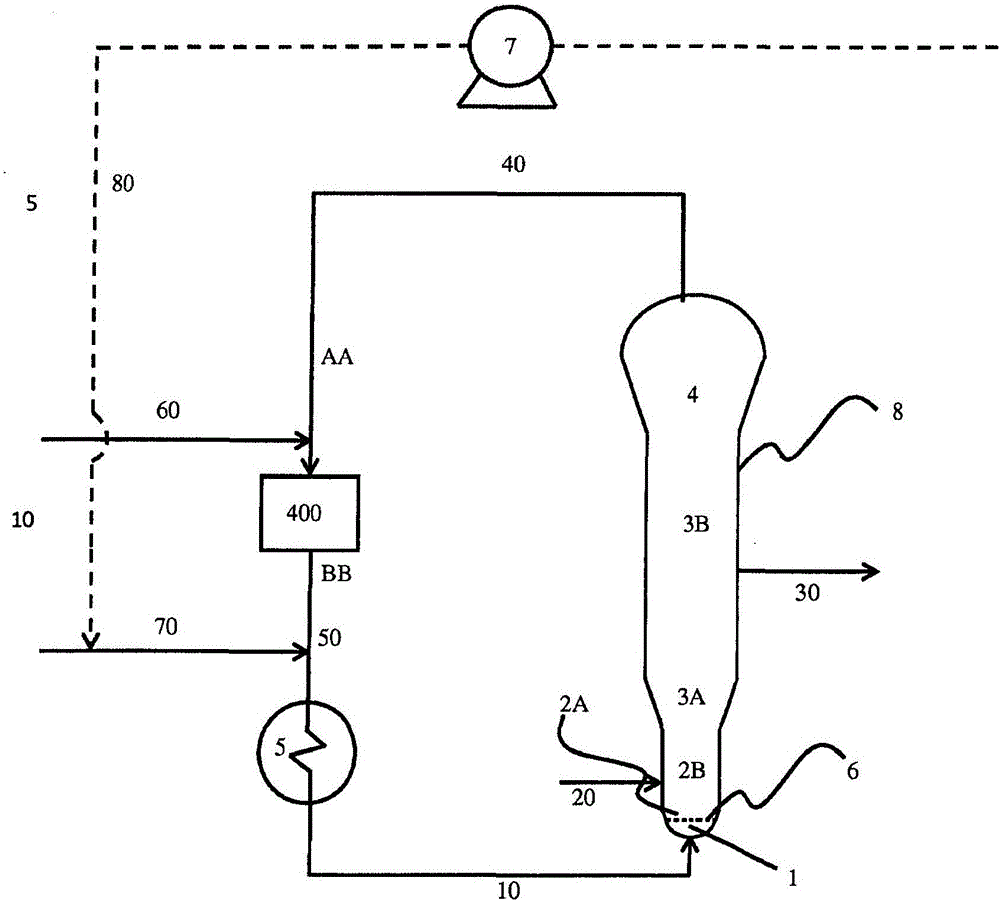
从生产具有某些特性和特征的聚合物的一种类型催化剂体系转变至能够生产不同的化学和/或物理属性的聚合物的另一催化剂体系是频繁需要的。在相似Ziegler-Natta催化剂体系或相容的催化剂体系之间转变通常容易发生。相容的催化剂是具有单体和共聚单体的终止和插入的相似动力学和/或不能与彼此有害地相互作用的那些催化剂。然而,当催化剂体系是不相容的或具有不同的类型时,该方法通常是复杂的。例如,当在两种不相容的催化剂体系例如Ziegler-Natta催化剂体系和基于铬的催化剂体系之间转变时,Ziegler-Natta催化剂体系的一些组分作为对基于铬的催化剂体系的毒物。因此,Ziegler-Natta催化剂体系的组分防止基于铬的催化剂体系促进聚合。此外,特别地,在连续转变方法中,在两种不相容的催化剂之间的相互作用可导致称为“细颗粒”的小于约120微米的小颗粒的高水平的生产。细颗粒可以引起在反应器中的可操作性问题和/或污垢和结皮事件。在过去,为了完成在不相容的催化剂之间的有效转变,第一催化的烯烃聚合方法通过本领域已知的各种技术停止。然后将反应器排空,再填充和将第二催化剂体系引入反应器。因为在转变期间需要使反应器停止一段延长的时间和等外材料,这样的催化剂转化是耗时且昂贵的。已经进行了多次尝试来改进在不相容的催化剂之间转变的方法。当然,为了抑制第一不相容的催化剂的聚合,需要中断催化剂注入反应器。停止第一催化剂进料至反应器不能立即停止在反应器内发生的聚合反应,因为流化床包含可以仍然聚合一段延长时间的催化剂颗粒。即使允许反应器内的聚合反应继续一段时间,但反应器内的催化剂不能在相当长的时间内完全失活。因此,为了基本上终止反应器内的这些聚合反应,已知使用聚合抑制剂或“催化剂杀伤剂”。存在两种通用类型的聚合抑制剂:可逆催化剂杀伤剂和不可逆催化剂杀伤剂。可逆催化剂杀伤剂通常初始抑制催化剂活性和聚合一段时间,但是不能不可逆地使催化剂失活。事实上,在一段时间后,在正常聚合条件下,催化剂再活化和继续聚合。这些可逆催化剂杀伤剂可以用于在过程中的引入的任何组合或顺序。不可逆催化剂杀伤剂不可逆地使催化剂聚合烯烃的能力失效。催化剂杀伤剂和/或减活剂的使用公开于Agapiou等人的美国专利号5,442,019,5,753,786,和6,949,612B2,Muhle等人的美国专利号5,672,666,和Burdett等人的美国专利号6,858,684B2。WO2006130310描述了用于从Ziegler类型催化剂转变至基于铬的催化剂的的方法的现有技术。从Ziegler类型催化剂至铬催化剂的一种转变程序使用二氧化硅注射以结合游离的三乙基铝(或其它烷基铝)—给电子体络合物,其通常存在于几乎所有的第二代Ziegler类型催化剂,和抑制铬催化剂活性。在二氧化硅上的表面硅烷醇理论上结合这些络合物,和该结合有效地清除烷基铝和给电子体络合物至有助于铬催化剂开始和良好的反应器可操作性的水平。然而,具有该复杂转变的商业经历不可再生,和周期性地,该转变机构导致反应器停车。当使用该程序时观察到的反应器可操作性问题的系统信号为转变失败,这是由于在二氧化硅注射期间静态引起的结皮,高水平的静态冷带化,导致床离析和结皮;由于二氧化硅引起的凝胶导致的额外等外品,和在反应开始时热电偶活性和板热电偶套管活性所证实的流化床内的温度偏离,导致结皮和聚集物形成。此外,使用二氧化硅作为TEA1/THF络合物的吸附剂不能完全结合反应体系中的所有烷基,和剩余的少量烷基铝可以导致极其高的铬活性,这也可以导致反应器可操作性问题。US4460755公开了通过Ziegler类型催化剂催化的烯烃聚合转化至通过基于铬的催化剂催化的聚合的方法,所述方法包括将含羟基的化合物加入包含Ziegler类型催化剂的反应器。这些转变主要通过以下来操作:停止将Ziegler催化剂体系的组分引入聚合反应器,和添加与Ziegler催化剂体系的组分反应和吸收该组合的含羟基的二氧化硅。这样的二氧化硅注射具有上述的问题。WO2006130310公开了通过Ziegler类型催化剂催化的连续烯烃聚合反应转变至通过基于铬的催化剂催化的聚合的方法。在该方法中,在停止引入Ziegler类型催化剂之后,将可逆毒物和/或不可逆毒物引入反应器,所述毒物在反应器内循环和用惰性气体吹扫反应器。有利的是,提供催化剂转变方法而不需要停止聚合反应,使用反应器以使其摆脱原始催化剂体系和使用另一催化剂体系重新开始聚合反应。此外,有利的是,转变方法可降低在转变方法期间产生的等外材料的量,降低转变时间,提高转变方法的稳健性和稳定性以及避免打开反应器以填充苗床的需要。另外,有利的是,提供催化剂转变方法,其不需要使用催化剂杀伤剂。本发明的目的是提供解决上述和/或其它问题的方法。因此,本发明提供从在气相反应器中在Ziegler-Natta催化剂的存在下进行的用于生产线型低密度聚乙烯的第一共聚单体和乙烯的第一连续聚合反应转变至在所述气相反应器中在基于铬的催化剂的存在下进行的用于生产高密度聚乙烯的第二共聚单体和乙烯的第二连续聚合反应的方法,所述方法包括:(i)减少所述第一共聚单体至所述反应器的进料直至所述反应器中所述第一共聚单体与乙烯的比例为最多0.1;(ii)停止引入Ziegler-Natta催化剂同时继续引入Ziegler-Natta催化剂的助催化剂,和随后停止引入所述助催化剂;(iii)维持所述反应器中的聚合条件和使聚合继续一段时间以便允许存在于所述反应器的Ziegler-Natta催化剂的组分在生产额外聚合物中消耗自身;(iv)停止引入至所述反应器的所有进料;(v)使所述反应器减压;(vi)流动吹扫所述反应器;(vii)降低所述反应器温度;(viii)将乙烯和H2引入所述反应器以获得用于第二聚合反应的乙烯分压和H2与乙烯的体积比例,其中将乙烯分压以这样的速度增加至用于第二聚合反应的压力,所述速度使得所述反应器温度维持在比第一聚合条件的温度更低的温度;(ix)将所述反应器温度升高至第二聚合条件的温度;(x)将第二催化剂引入所述反应器,以及(xi)将所述第二共聚单体引入所述反应器以获得用于第二聚合反应的反应器组合物。令人惊奇地发现本发明的转变方法可以在不使用催化剂杀伤剂的情况下进行。因此,成功的转变可以在短时间内进行。在本发明的方法中,首先在步骤(i)中降低第一共聚单体体积比例。第一共聚单体比例的降低导致产生的LLDPE的密度增加。发现了这对于避免床粘性的出现是必要的。在第一聚合期间第一共聚单体与乙烯的体积比例可为适合于获得希望的LLDPE的任何范围,例如0.3-0.5,更优选0.4-0.5。反应器中第一共聚单体与乙烯的比例降低至最多0.10,优选最多0.05。在步骤(ii)中,停止引入Ziegler-Natta催化剂,同时继续引入Ziegler-Natta催化剂的助催化剂。在停止Ziegler-Natta催化剂之后的一段时间之后停止引入助催化剂。发现了这防止保留在反应器中的活性过度金属卤化物与后来引入的乙烯引起不可控的反应。助催化剂可在停止Ziegler-Natta催化剂之后的例如0.25-2小时或更优选0.5-1小时停止。在步骤(iii)中,在停止引入Ziegler-Natta催化剂和助催化剂之后,保持反应器中的聚合条件以便允许存在于所述反应器的Ziegler-Natta催化剂的组分在生产额外聚合物中消耗自身。反应器中保持的聚合条件可例如为83-88℃,例如85℃的反应器温度,15-25barg,例如18-22barg,例如20.7barg的反应器压力,5-10bara,例如7bara的乙烯分压,0.05-0.2,例如0.14的H2/乙烯比例。随后,在步骤(iv)中停止所有进料。可同时停止引入H2和乙烯,或可在乙烯之前停止引入H2或可在H2之前停止引入乙烯。优选在距彼此相对短的时间内,例如在距彼此5-10分钟内停止引入H2和停止引入乙烯。优选地,步骤(iv)通过停止引入乙烯同时继续引入H2和随后(例如在5-10分钟之后)停止引入H2来进行。这有助于在该阶段不具有聚合反应。在步骤(v)中,使反应器减压。可使反应器减压至例如5-9barg或6-8barg。在与Ziegler-Natta催化剂聚合的期间,在反应器内累积气体,其源自给电子体。这些气体通常对不相容的基于铬的催化剂是有毒的。用于传统的Ziegler-Natta催化剂的这些气体包括例如四氢呋喃(THF),乙醇,苯甲酸乙酯等。因此,在引入第二不相容的催化剂之前,反应器内容物经受本领域已知的压力吹扫。在步骤(vi)中,使反应器进行流动吹扫。当完成该程序时,将反应器体系中的气体组合物调节至第二聚合反应用于通过基于铬的催化剂获得希望的HDPE。在本发明的方法中,在将反应器中的气体组合物调节至第二聚合反应之前,采取以下的具体措施。在步骤(vii)中,在步骤(viii)中注射乙烯和氢之前使反应器温度降低。发现了将乙烯和氢注入反应器同时维持降低的温度是关键的。在较高的温度下注射乙烯导致床温度迅速增加。发现了床温度的迅速增加会导致在床中团聚或结块。优选地,使反应器温度降低至比步骤(iii)的温度低至少4℃的温度,更优选降低至比步骤(iii)的温度低至少5℃的温度。优选地,使反应器温度降低至小于81℃的温度。在步骤(viii)中,将乙烯和H2引入反应器以获得对于第二聚合反应希望的乙烯分压和H2与乙烯的体积比例。将乙烯分压以这样的速度增加至用于第二聚合反应的乙烯分压,所述速度使得反应器温度维持在比第一聚合条件的温度更低的温度。本文中用于第二聚合反应的乙烯分压指的是在第二聚合反应期间的希望的乙烯分压。本文中用于第二聚合反应的H2与乙烯的体积比例指的是在第二聚合期间的希望的H2与乙烯的体积比例。维持低温防止在床中形成团聚或结块。优选地,步骤(viii)通过以第一速度将乙烯分压增加至中间压力和以比第二速度更低的第二速度增加至用于第二聚合反应的压力来进行。优选地,中间压力比目标压力低5-7bara。优选地,中间压力为8-12bara。优选地,目标压力为13-17bara。第一速度可例如为10kg/hr流速。第二速度可例如为5kg/hr流速。这时,不将第二共聚单体引入反应器中。可开始将H2引入反应器同时提高乙烯分压。然而,优选地,在达到用于第二聚合反应的乙烯分压之后,开始将H2引入反应器。由此,将反应器中H2与乙烯的体积比例调节至用于第二聚合的希望的比例。用于第二聚合的反应器中的H2与乙烯的体积比例可例如为0.01-0.05,优选0.01-0.03。在步骤(ix)中,在达到目标乙烯分压和H2与乙烯比例之后,将温度逐渐升高至适合于第二聚合的温度。用于第二聚合的温度可为95-105℃,例如为100℃。温度升高速度可例如为2℃/小时。在步骤(x)中,将第二催化剂引入反应器。乙烯开始聚合。在步骤(xi)中,以相对于乙烯希望的比例将第二共聚单体引入反应器。在第二聚合期间第二共聚单体与乙烯的体积比例可为适合于获得希望的HDPE的任何范围,例如0.0010-0.0030。通过该步骤,将反应器组合物调节至适合于在基于铬的催化剂存在下获得希望的HDPE的组合物。发现了这些步骤的组合导致在不使用催化剂杀伤剂的情况下从使用Ziegler-Natta催化剂聚合至使用基于铬的催化剂聚合的成功转变。此外,本发明的方法有利地消除了使用基于铬的催化剂启动反应器的问题。为了使用基于铬的催化剂启动反应器,需要充分吹扫反应器回路以除去所有的O2和H2O至ppm水平的低限值。基于铬的催化剂对于O2和H2O极其敏感。甚至小量的O2和H2O的存在导致例如产生细颗粒和床离析的问题,这可使床变为不能流化的粉末。将O2和H2O除去至充足水平需要吹扫和加热一段长时间且消耗时间和成本。通过首先与Ziegler-Natta催化剂反应,消除了用于启动反应器的长的吹扫步骤。尽管不希望受限于任何理论,本发明人相信通过第一反应消耗存在于反应器中的O2和H2O和因此基于铬的催化剂不被O2和H2O中毒。通过所述方法获得的聚合物第一聚合反应导致线型低密度聚乙烯(LLDPE)。第一单体可为C3-C10α-烯烃共聚单体。适合的α-烯烃共聚单体包括1-丁烯,1-己烯,4-甲基戊烯和1-辛烯。优选共聚单体为1-丁烯和1-己烯,特别是1-丁烯。优选地,α-烯烃共聚单体的存在量为LLDPE的约5-约20重量%,更优选存在量为LLDPE的约7-约15重量%。优选地,线型低密度聚乙烯(LLDPE)通过ASTMD-1505确定的密度为915-930kg/m3。优选地,线型低密度聚乙烯通过ASTMD-1238-条件E(190℃,2.16kg)确定的熔体流动指数为0.1-4g/10min,例如0.3-3g/10min,例如0.2-2g/10min,例如0.5-1.5g/10min。LLDPE中未反应的催化剂优选很低。未反应的催化剂可以通过测量灰分含量确定。因此,通过第一聚合获得的LLDPE包含通过ASTMD5630确定的最多0.045wt%的灰分含量。第二聚合反应导致高密度聚乙烯(HDPE)。第二共聚单体可为具有4-8个碳原子的α-烯烃单体,例如1-丁烯,1-己烯或1-辛烯,优选1-己烯。优选地,HDPE通过ASTMD-1505确定的密度为940-965kg/m3。优选地,使用ASTMD-1238–条件F(190℃,21.6kg)确定的熔体流动指数为5-15g/10min。Ziegler-Natta催化剂在本领域中传统的Ziegler-Natta催化剂通常包含过度金属卤化物如钛或钒卤化物,和第1、2或3族金属的有机金属化合物,通常为三烷基铝化合物,其充当过度金属卤化物的活化剂或助催化剂。一些Ziegler-Natta催化剂体系包含络合至烷基铝或过度金属的内给电子体。过度金属卤化物可负载在卤化镁上或络合至卤化镁。该Ziegler-Natta催化剂也可以浸渍在无机载体例如二氧化硅或氧化铝上。反应器中Ziegler-Natta催化剂的完全活化通过添加助催化剂来实现,所述助催化剂通常为上述的有机金属化合物。对于关于传统的Ziegler-Natta催化剂的更详细描述,参见例如,美国专利号3,687,920,4,086,408,4,376,191,5,019,633,4,482,687,4,101,445,4,560,671,4,719,193,4,755,495,5,070,055,其全部通过参考引入本文。优选地,Ziegler-Natta催化剂通过以下制备:(a)使具有羟基的脱水载体与具有通式MgR1R2的镁化合物接触,其中R1和R2相同或不同和独立地选自烷基、烯基、二烯基、芳基、烷芳基、烯基芳基和二烯基芳基;(b)使步骤(a)中获得的产物与改性化合物(A)、(B)和(C)接触,其中:(A)为选自羧酸、羧酸酯、酮、酰基卤、醛和醇的至少一种化合物;(B)为具有通式R11f(R12O)gSiXh的化合物,其中f、g和h各自为0-4的整数且f、g和h之和等于4,条件是当h等于4时,则改性化合物(A)不为醇,Si为硅原子,O为氧原子,X为卤原子和R11和R12相同或不同和独立地选自烷基、烯基、二烯基、芳基、烷芳基、烯基芳基和二烯基芳基;(C)为具有通式(R13O)4M的化合物,其中M为钛原子、锆原子或钒原子,O为氧原子和R13选自烷基、烯基、二烯基、芳基、烷芳基、烯基芳基和二烯基芳基;和(c)使步骤(b)中获得的产物与具有通式TiX4的卤化钛化合物接触,其中Ti为钛原子和X为卤原子。该催化剂描述于WO2012069157,其通过参考引入本文。用于制备Ziegler-Natta催化剂组分的方法包括使具有羟基(OH)的脱水固体载体与镁化合物接触以形成包含固体镁的载体材料的第一步骤(a)。固体载体为包含羟基的任何材料。这样的材料的适合例子包括无机氧化物,例如二氧化硅,氧化铝,氧化镁,氧化钍,氧化锆和这样的氧化物的混合物。优选地,多孔二氧化硅用作载体,因为由其获得较高的堆积密度和较高的催化剂产生力。二氧化硅可为颗粒形式,所述颗粒具有约1微米-约500微米,优选5微米-150微米和最优选10微米-100微米的平均粒径。较低的平均粒径产生较高水平的聚合物细颗粒和较高的平均粒径降低聚合物堆积密度。二氧化硅可具有约5m2/g-约1500m2/g,优选50m2/g-1000m2/g的表面积和约0.1cm3/g-约10.0cm3/g,优选0.3cm3/g-3.5cm3/g的孔隙体积,因为在这个范围内获得较高的催化剂生产力。脱水固体载体可以通过以下获得:干燥固体载体以便物理除去结合水和降低羟基含量至可为约0.1mmol-约5.0mmol羟基/g载体,优选约0.2mmol-约2.0mmol羟基/g载体的水平,因为该范围允许将活性催化剂组分充分并入载体,其通过J.J.Fripiat和J.Uytterhoeven,J.Phys.Chem.66,800,1962中所述的方法或通过使用1HNMR光谱来确定。该范围内的羟基含量可通过在氮气或空气流下在约150℃-约900℃的温度下加热和流化载体约1小时-约15小时的时间来实现。脱水载体可以优选通过搅拌在适合的烃溶剂中成浆,在所述烃溶剂中各个催化剂组分至少部分可溶。适合的烃溶剂的包括正戊烷,异戊烷,环戊烷,正己烷,异己烷,环己烷,正庚烷,异庚烷,正辛烷,异辛烷和正癸烷。所用溶剂的量不是关键的,尽管溶剂应该以提供催化剂组分的良好混合的量使用。镁化合物由通式MgR1R2表示,其中R1和R2相同或不同和独立地选自烷基、烯基、二烯基、芳基、烷芳基、烯基芳基和二烯基芳基和可具有1-20碳原子。镁化合物的适合例子包括二甲基镁,二乙基镁,乙基甲基镁,二正丙基镁,二异丙基镁,正丙基乙基镁,异丙基乙基镁,二正丁基镁,二异丁基镁,正丁基乙基镁,正丁基正丙基镁,正丁基异丙基镁,异丁基乙基镁,异丁基正丙基镁,异丁基异丙基镁,二正戊基镁,二异戊基镁,正戊基乙基镁,正戊基正丙基镁,正戊基异丙基镁,正戊基正丁基镁,正戊基异丁基镁,二正己基镁,二异己基镁,正己基乙基镁,正己基正丙基镁,正己基异丙基镁,正己基正丁基镁,正己基异丁基镁,异己基乙基镁,异己基正丙基镁,异己基异丙基镁,异己基正丁基镁,异己基异丁基镁,二正辛基镁,二异辛基镁,正辛基乙基镁,正辛基正丙基镁,正辛基异丙基镁,正辛基正丁基镁,正辛基异丁基镁,异辛基乙基镁,异辛基正丙基镁,异辛基异丙基镁,异辛基正丁基镁,异辛基异丁基镁,二环戊基镁,环戊基乙基镁,环戊基正丙基镁,环戊基异丙基镁,环戊基正丁基镁,环戊基异丁基镁,二环己基镁,环己基乙基镁,环己基正丙基镁,环己基异丙基镁,环己基正丁基镁,环己基异丁基镁,二苯基镁,苯基乙基镁,苯基正丙基镁,苯基正丁基镁及其混合物。优选地,镁化合物选自二正丁基镁、正丁基乙基镁和正辛基正丁基镁。镁化合物的使用量可以为约0.01-约10.0mmol/g固体载体,优选约0.1-约3.5mmol/g载体和更优选0.3-2.5mmol/g载体,因为通过使用该范围,降低产物的聚合物细颗粒水平和获得较高的催化剂生产力。镁化合物可优选通过搅拌与载体在约15℃-约140℃的温度下在约5分钟-约150分钟的期间,优选在约20℃-80℃的温度下在10分钟-100分钟的持续时间内反应。Mg与固体载体中的OH基团的摩尔比例可以为约0.01-约10.0,优选为约0.1-约5.0和更优选为约0.1-约3.5,因为降低产物的聚合物细颗粒水平和获得较高的催化剂生产力。改性化合物(A)为选自羧酸、羧酸酯、酮、酰基卤、醛和醇的至少一种化合物。改性化合物(A)可由通式R3COOH,R4COOR5,R6COR7,R8COX,R9COH或R10OH表示,其中X为卤原子和R3,R4,R5,R6,R7,R8,R9和R10独立地选自烷基、烯基、二烯基、芳基、烷芳基、烯基芳基和二烯基芳基和可具有1-20个碳原子。羧酸的适合例子包括乙酸,丙酸,异丙酸,丁酸,异丁酸,戊酸,异戊酸,己酸,异己酸,庚酸,异庚酸,辛酸,异辛酸,壬酸,异壬酸,癸酸,异癸酸,环戊烷羧酸,苯甲酸及其混合物。羧酸酯的适合例子包括乙酸甲酯,乙酸乙酯,正丙基乙酸酯,异丙基乙酸酯,正丁基乙酸酯,异丁基乙酸酯,异戊基乙酸酯,乙基丁酸酯,正丁基丁酸酯和/或异丁基丁酸酯。酮的适合例子包括二甲基酮,二乙基酮,甲基乙基酮,二正丙基酮,二正丁基酮,甲基正丙基酮,甲基异丁基酮,环己酮,甲基苯基酮,乙基苯基酮,正丙基苯基酮,正丁基苯基酮,异丁基苯基酮,二苯基酮及其混合物。酰基卤的适合例子包括乙酰氯,丙酰氯,异丙酰氯,正丁酰氯,异丁酰氯,苯甲酰氯及其混合物。醛的适合例子包括乙醛,丙醛,正丁醛,异丁醛,正戊醛,异戊醛,正己醛,异己醛,正庚醛,苯甲醛及其混合物。醇的适合例子包括甲醇,乙醇,正丙醇,异丙醇,正丁醇,异丁醇,仲丁醇,叔丁醇,环丁醇,正戊醇,异戊醇,环戊醇,正己醇,异己醇,环己醇,正辛醇,异辛醇,2-乙基己醇,苯酚,甲酚,乙二醇,丙二醇及其混合物。优选地,改性化合物(A)为选自甲基正丙基酮,乙酸乙酯,正丁基乙酸酯,乙酸,异丁酸,异丁醛,乙酰氯,乙醇和仲丁醇的至少一种化合物,和更优选选自甲基正丙基酮,正丁基乙酸酯,异丁酸和乙酰氯,因为获得产物的较高堆积密度和较高的催化剂生产力和这些化合物可以用于改变产物的分子量分布。改性化合物(A)与固体载体中镁的摩尔比例可以为约0.01-约10.0,优选为约0.1-约5.0,更优选为约0.1-约3.5和最优选为0.3-2.5,因为获得产物的较高堆积密度和较高的催化剂生产力。改性化合物(A)可优选通过搅拌在约15℃-约140℃的温度下在约5分钟-约150分钟的持续时间内,优选在20℃-80℃的温度下在10分钟-100分钟的持续时间内加入步骤(f)获得的反应产物。改性化合物(B)为由通式R11f(R12O)gSiXh表示的硅化合物,其中f、g和h各自为0-4的整数且f、g和h之和等于4,条件是当h等于4时,则改性化合物(A)不为醇,Si为硅原子,O为氧原子,X为卤原子和R11和R12相同或不同。R11和R12独立地选自烷基、烯基、二烯基、芳基、烷芳基、烯基芳基和二烯基芳基。R11和R12可具有1-20个碳原子。适合的硅化合物包括四甲氧基硅烷,四乙氧基硅烷,四正丙氧基硅烷,四异丙氧基硅烷,四正丁氧基硅烷,四异丁氧基硅烷,甲基三甲氧基硅烷,乙基三甲氧基硅烷,正丙基三甲氧基硅烷,异丙基三甲氧基硅烷,正丁基三甲氧基硅烷,异丁基三甲氧基硅烷,正戊基三甲氧基硅烷,正己基三甲氧基硅烷,正辛基三甲氧基硅烷,异辛基三甲氧基硅烷,乙烯基三甲氧基硅烷,苯基三甲氧基硅烷,二甲基二甲氧基硅烷,二乙基二甲氧基硅烷,异丁基甲基二甲氧基硅烷,二异丙基二甲氧基硅烷,二异丁基二甲氧基硅烷,异丁基异丙基二甲氧基硅烷,二环戊基二甲氧基硅烷,环己基甲基二甲氧基硅烷,苯基甲基二甲氧基硅烷,二苯基二甲氧基硅烷,三甲基甲氧基硅烷,三乙基甲氧基硅烷,甲基三乙氧基硅烷,乙基三乙氧基硅烷,正丙基三乙氧基硅烷,异丙基三乙氧基硅烷,正丁基三乙氧基硅烷,异丁基三乙氧基硅烷,正戊基三乙氧基硅烷,正己基三乙氧基硅烷,正辛基三乙氧基硅烷,异辛基三乙氧基硅烷,乙烯基三乙氧基硅烷,苯基三乙氧基硅烷,二甲基二乙氧基硅烷,二乙基二乙氧基硅烷,异丁基甲基二乙氧基硅烷,二异丙基二乙氧基硅烷,二异丁基二乙氧基硅烷,异丁基异丙基二乙氧基硅烷,二环戊基二乙氧基硅烷,环己基甲基二乙氧基硅烷,苯基甲基二乙氧基硅烷,二苯基二乙氧基硅烷,三甲基乙氧基硅烷,三乙基乙氧基硅烷,四氯化硅,甲基三氯硅烷,乙基三氯硅烷,正丙基三氯硅烷,异丙基三氯硅烷,正丁基三氯硅烷,异丁基三氯硅烷,正戊基三氯硅烷,正己基三氯硅烷,正辛基三氯硅烷,异辛基三氯硅烷,乙烯基三氯硅烷,苯基三氯硅烷,二甲基二氯硅烷,二乙基二氯硅烷,异丁基甲基二氯硅烷,二异丙基二氯硅烷,二异丁基二氯硅烷,异丁基异丙基二氯硅烷,二环戊基二氯硅烷,环己基甲基二氯硅烷,苯基甲基二氯硅烷,二苯基二氯硅烷,三甲基氯硅烷,三乙基氯硅烷,氯三甲氧基硅烷,二氯二甲氧基硅烷,三氯甲氧基硅烷,氯三乙氧基硅烷,二氯二乙氧基硅烷和/或三氯乙氧基硅烷。优选地,使用的改性化合物(B)为四乙氧基硅烷,正丙基三乙氧基硅烷,异丁基三甲氧基硅烷,二甲基二氯硅烷,正丁基三氯硅烷和四氯化硅,和更优选异丁基三甲氧基硅烷,四乙氧基硅烷,正丙基三乙氧基硅烷,正丁基三氯硅烷和四氯化硅,因为获得较高的催化剂生产力和较高的堆积密度,具有通过使用这些优选化合物改变产物的分子量分布的能力。改性化合物(B)与镁的摩尔比例可为约0.01-约5.0,优选约0.01-约3.0,更优选约0.01-约1.0和最优选约0.01-约0.3,因为获得较高的催化剂生产力和较高的堆积密度。改性化合物(B)可优选通过搅拌在约15℃-约140℃的温度下在约5分钟-约150分钟的期间,优选在20℃-80℃的温度下在10分钟-100分钟的期间加入步骤(f)获得的反应产物。改性化合物(C)为由通式(R13O)4M表示的过度金属烷氧化物,其中M为钛原子、锆原子或钒原子,O为氧原子和R13为选自烷基、烯基、二烯基、芳基、烷芳基、烯基芳基和二烯基芳基的化合物。R13可具有1-20个碳原子。适合的过度金属烷氧化物化合物包括四甲氧基钛,四乙氧基钛,四正丙氧基钛,四异丙氧基钛,四正丁氧基钛,四异丁氧基钛,四正戊氧基钛,四异戊氧基钛,四正己氧基钛,四正庚氧基钛,四正辛氧基钛,四环己氧基钛,四苯甲氧基钛,四苯氧基钛,四甲氧基锆,四乙氧基锆,四正丙氧基锆,四异丙氧基锆,四正丁氧基锆,四异丁氧基锆,四正戊氧基锆,四异戊氧基锆,四正己氧基锆,四正庚氧基锆,四正辛氧基锆,四环己氧基锆,四苯甲氧基锆,四苯氧基锆,四甲氧基钒,四乙氧基钒,四正丙氧基钒,四异丙氧基钒,四正丁氧基钒,四异丁氧基钒,四正戊氧基钒,四异戊氧基钒,四正己氧基钒,四正庚氧基钒,四正辛氧基钒,四环己氧基钒,四苯甲氧基钒,四苯氧基钒或其混合物。优选地,使用四乙氧基钛、四正丁氧基钛和四正丁氧基锆,因为获得较高的催化剂生产力和较高的堆积密度,具有通过使用这些优选化合物改变产物的分子量分布的能力。改性化合物(C)与镁的摩尔比例可为约0.01-约5.0,优选约0.01-约3.0,更优选约0.01-约1.0和最优选约0.01-约0.3,因为获得较高的催化剂生产力,较高的堆积密度和聚合中改进的氢响应。改性化合物(C)可优选通过搅拌在约15℃-约140℃的温度下在约5分钟-约150分钟的持续时间内,优选在20℃-80℃的温度下在10分钟-100分钟的持续时间内与步骤(f)获得的产物反应。改性化合物(A)、(B)和(C)可以以任何顺序或同时与在步骤(a)获得的包含固体镁的载体接触。优选地,(A)首先加入在步骤(a)中获得的反应产物,然后添加(B),之后添加(C),因为通过使用该改性化合物添加顺序获得较高的催化剂生产力和较高的产物堆积密度。也可以有效使用各个催化剂组分的预混合物。优选地,当改性化合物(A)为甲基正丙基酮和改性化合物(C)为四乙氧基钛时,当改性化合物(B)以下面顺序以与卤化钛化合物相同的水平选自异丁基三甲氧基硅烷、正丙基三乙氧基硅烷、四乙氧基硅烷、正丁基三氯硅烷和四氯化硅时,获得进一步增加的分子量分布。在改性化合物(B)为四氯化硅和改性化合物(C)为四乙氧基钛的优选情况下,当改性化合物(A)以下面顺序以与卤化钛化合物相同的水平选自异丁醛、乙酸乙酯、正丁基乙酸酯、甲基正丙基酮和异丁酸时,获得催化剂生产力和堆积密度的进一步改进的组合卤化钛化合物由通式TiX4表示,其中Ti为钛原子和X为卤原子。适合的卤化钛化合物包括四氯化钛,四溴化钛,四氟化钛或其混合物。优选卤化钛化合物为四氯化钛,因为获得较高的催化剂生产力。卤化钛化合物与镁的摩尔比例可为约0.01-约10.0,优选约0.01-约5.0和更优选约0.05-约1.0,因为获得高催化剂生产力和高堆积密度的较好平衡。卤化钛化合物可以以任何常规方式,例如通过搅拌,在约15℃-约140℃的温度下在约5分钟-约150分钟的持续时间内,优选在20℃-80℃的温度下在10分钟-100分钟的持续时间内加入使用步骤(a)和步骤(b)获得的反应混合物。然后可使用氮吹扫和/或通过真空在约15℃-约140℃,优选30℃-100℃和最优选50℃-80℃的温度下干燥反应混合物以产生最终固体催化剂组分。改性化合物(C)和卤化钛化合物与镁的总摩尔比例可为约0.01-约10.0,优选为约0.01-约5.0和更优选为约0.05-约1.0,因为获得高催化剂生产力和高堆积密度的较好平衡。改性化合物(C)和卤化钛化合物与脱水之后载体中的羟基(OH)的总摩尔比例可为约0.01-约10.0,优选为约0.01-约5.0和更优选为约0.05-约1.0,因为获得高催化剂生产力和高堆积密度的较好平衡。较高的水平将产生高催化剂生产力,尽管具有降低堆积密度,尤其是在气相聚合方法中。此外,使用这些量消除催化剂制备中的进行溶剂倾析、溶剂过滤、溶剂洗涤步骤的要求和因此消除了高度有害溶剂废物材料的产生。基于铬的催化剂基于铬的催化剂可以选自例如基于氧化铬的催化剂和基于甲硅烷基铬酸酯的催化剂。这些基于铬的催化剂描述于WO2006130310,其通过参考引入本文。氧化铬催化剂可为CrO3,或在使用的活化条件下可转化为CrO3的任何化合物。可转化为CrO3化合物为例如公开于US2,825,721,US3,023,203,US3,622,251和US4,011,382和包括乙酰丙酮铬,氯化铬,硝酸铬,乙酸铬,硫酸铬,铬酸铵,二铬酸铵,或其它可溶的铬酸盐。基于甲硅烷基铬酸酯的催化剂的特征在于存在下式I的至少一种基团:其中在每种情况下R为具有1-14个碳原子的烃基。在其中优选基于甲硅烷基铬酸酯的催化剂为下式II的双(三烃基甲硅烷基)铬酸酯:其中R为具有1-14个碳原子的烃基,例如烷基,烷芳基,芳烷基或芳基,其包含1-14个碳原子,优选3-10碳原子。其说明性例子为甲基,乙基,丙基,异丙基,正丁基,异丁基,正戊基,异戊基,叔戊基,己基,2-甲基戊基,庚基,辛基,2-乙基己基,壬基,癸基,十一烷基,十二烷基,十三烷基,十四烷基,苯甲基,苯乙基,对甲基苯甲基,苯基,甲苯基,二甲苯基,萘基,乙基苯基,甲基萘基,二甲基萘基。说明性优选的甲硅烷基铬酸酯为这样的化合物,例如双(三甲基甲硅烷基)铬酸酯,双(三乙基甲硅烷基)铬酸酯,双(三丁基甲硅烷基)铬酸酯,双(三异戊基甲硅烷基)铬酸酯,双(三-2-乙基己基甲硅烷基)铬酸酯,双(三癸基甲硅烷基)铬酸酯,双(三(十四烷基)甲硅烷基)铬酸酯,双(三苯甲基甲硅烷基)铬酸酯,双(三苯乙基甲硅烷基)铬酸酯,双(三苯基甲硅烷基)铬酸酯,双(三甲苯基甲硅烷基)铬酸酯,双(三(二甲苯基)甲硅烷基)铬酸酯,双(三萘基甲硅烷基)铬酸酯,双(三乙基苯基甲硅烷基)铬酸酯,双(三甲基萘基甲硅烷基)铬酸酯,聚二苯基甲硅烷基铬酸酯,聚二乙基甲硅烷基铬酸酯。这样的催化剂的例子公开于例如US3,324,101,US3,704,287和US4,100,105。本发明的基于铬的催化剂可沉积在常规催化剂载体如无机氧化物材料上。可以用作基于铬的催化剂的载体的无机氧化物材料为具有高表面积如50-1000平方米/克的表面积和20-200微米的粒度的多孔材料。可使用的无机氧化物包括二氧化硅,氧化铝,氧化钍,氧化锆,磷酸铝和其它可比的无机氧化物,以及这样的氧化物的混合物。当一起使用基于氧化铬的催化剂和基于甲硅烷基铬酸酯的催化剂时,每种催化剂沉积在单独的载体上。基于氧化铬的催化剂将铬物质沉积在载体上的方法是本领域已知的,和可见于前述的美国专利中。铬化合物通常由其溶液以这样的量沉积在载体上,从而该量使得在活化步骤之后提供催化剂中希望水平的铬。改性材料如钛和氟化物通常在活化之前添加。在将化合物置于载体上和被活化之后,这产生粉末状自由流动的颗粒材料。通常,基于铬的催化剂通过使用可商购的二氧化硅来制备,向其中添加了铬源。在沉积Cr化合物之后,或在该沉积之前,可用钛酯(通常使用四异丙氧基钛或四乙氧基钛)处理二氧化硅基材。载体通常在150-200℃下预干燥以物理除去吸附的水。可将钛酸酯以溶液形式添加至二氧化硅在异戊烷溶剂中的浆料或直接加入载体的流化床。如果以浆料形式添加,则使浆料干燥。通常,可转化为Cr+6的Cr化合物已经加入载体。然后,在至多1000℃的温度下通过在空气中煅烧使载体转化成活性催化剂。在活化期间,将钛转化为一些类型的表面氧化物。将铬化合物(通常为乙酸铬(III))转化为一些类型的Cr+6氧化物。在活化过程期间也可以添加氟化剂以选择性坍塌载体中的一些孔隙,改性分子量响应催化剂。还可以在使用之前使用还原剂如流化床中的一氧化碳,或其它还原剂如烷基铝、烷基硼、烷基锂处理活化催化剂。该类型的催化剂描述于多个专利中,例如WO2004/094489,EP0640625,US4,100,105,和这些参考文献内引用的参考文献。例如,有用的催化剂为负载的铬-钛催化剂(或钛酸化的氧化铬催化剂),其为基本上非球形或不规则形状,和具有宽的粒度分布,其中其孔隙体积的至少75%的孔径为负载的氧化铬催化剂的活化可以在至多约其烧结温度的几乎任何温度下完成。在活化期间使干空气或氧体流通过负载的催化剂有助于从载体驱替水和将铬物质至少部分转化为Cr+6。300℃-900℃的活化温度达大于1小时至高达48小时的时间是可接收的。使用干燥好的空气或氧气和将温度维持在载体的烧结温度以下。优选条件使用300℃-900℃,优选700℃-850℃的温度达至少两小时,优选5小时-15小时。如果使用的话,将铬化合物、钛化合物和氟化合物以这样的量沉积在载体上,所述量使得在活化步骤之后提供催化剂中希望水平的铬、钛和氟。可使用的优选铬化合物包括CrO3,或在使用的活化条件下可燃为CrO3的铬的任何化合物。可使用的除了CrO3的铬化合物公开于US2,825,721和US3,622,521和包括乙酰丙酮铬,硝酸铬,乙酸铬,氯化铬,硫酸铬,和铬酸铵。水溶性的铬化合物如CrO3是用于将铬化合物由该化合物的溶液沉积在载体上的优选化合物。也可以使用有机溶剂溶性的铬化合物。可使用的钛化合物包括在使用的活化条件下可燃为TiO2的所有那些化合物,尤其是公开于US3,622,521和US4,011,382的那些化合物。这些化合物包括具有以下结构的那些化合物(R’)nTi(OR')m(RO)mTi(OR')n和TiY4其中m为1,2,3或4;n为0,1,2或3;和m+n=4;R为C1-C12烷基、芳基或环烷基或它们的组合,例如芳烷基,烷芳基;R'为R、环戊二烯基或C2-C12烯基,例如乙烯基,丙烯基,异丙烯基,丁烯基;和Y为氯、溴、氟或碘。因此,钛化合物包括四氯化钛、四异丙氧基钛和四丁氧基钛。将钛化合物由其烃溶剂溶液方便地沉积在载体上。钛(以Ti计)相对于Cr(以Cr计)以0-180和优选为4-35的摩尔比例存在于催化剂中。可使用的氟化合物包括HF,或在使用的活化条件下产生HF的氟的任何化合物。可使用的除了HF以外的氟化合物公开于US4,011,382。这些化合物包括六氟硅酸铵,四氟硼酸铵,和六氟钛酸铵。将氟化合物由其含水溶液方便地沉积在载体上,或在活化之前通过干共混固体氟化合物与催化剂的其它组分沉积在载体上。催化剂所使用的载体为多孔的无机氧化物材料,具有高表面积,即50-1000平方米/克的表面积,和10-200微米的平均粒度。可使用的无机氧化物包括二氧化硅,氧化铝,氧化钍,氧化锆和其它可比的无机氧化物,以及这样的氧化物的混合物。二氧化硅、二氧化硅氧化铝、磷酸铝、二氧化硅二氧化钛和二氧化硅磷酸铝为优选的载体化合物。特别优选的载体为表面积为200-500平方米/克、孔径为和平均粒度为20-100微米的微球状颗粒(例如,获自DavisonChemicalDivision,W.R.GraceandCompany的级别952MS,957HS,957二氧化硅,和获自IneosCorporation的IneosEP30X,获自IneosCorporation的IneosEP30XA,以及获自PhiladelphiaQuartz的相似二氧化硅等级)。甲硅烷基铬酸酯催化剂甲硅烷基铬酸酯催化剂的特征在于存在下式I的至少一种基团:其中在每种情况下,R为具有1-14个碳原子的烃基。在其中,具有式I的基团的优选化合物为下式II的双(三烃基甲硅烷基)铬酸酯:其中R为具有1-14个碳原子的烃基,例如烷基,烷芳基,芳烷基或芳基,其包含1-14个碳原子,优选3-10个碳原子。其说明性例子为甲基,乙基,丙基,异丙基,正丁基,异丁基,正戊基,异戊基,叔戊基,己基,2-甲基戊基,庚基,辛基,2-乙基己基,壬基,癸基,十一烷基,十二烷基,十三烷基,十四烷基,苯甲基,苯乙基,对甲基苯甲基,苯基,甲苯基,二甲苯基,萘基,乙基苯基,甲基萘基,二甲基萘基。说明性优选甲硅烷基铬酸酯为这样的化合物,例如双(三甲基甲硅烷基)铬酸酯,双(三乙基甲硅烷基)铬酸酯,双(三丁基甲硅烷基)铬酸酯,双(三异戊基甲硅烷基)铬酸酯,双(三-2-乙基己基甲硅烷基)铬酸酯,双(三癸基甲硅烷基)铬酸酯,双(三(十四烷基)甲硅烷基)铬酸酯,双(三苯甲基甲硅烷基)铬酸酯,双(三苯乙基甲硅烷基)铬酸酯,双(三苯基甲硅烷基)铬酸酯,双(三甲苯基甲硅烷基)铬酸酯,双(三(二甲苯基)甲硅烷基)铬酸酯,双(三萘基甲硅烷基)铬酸酯,双(三乙基苯基甲硅烷基)铬酸酯,双(三甲基萘基甲硅烷基)铬酸酯,聚二苯基甲硅烷基铬酸酯,聚二乙基甲硅烷基铬酸酯。尤其是优选化合物为双(三苯基甲硅烷基)铬酸酯,双(三甲苯基甲硅烷基)铬酸酯和双(三金刚烷基甲硅烷基)铬酸酯。为了成为活性聚合催化剂,必须将甲硅烷基铬酸酯负载在无机载体上,之后进行任选但是优选的还原反应以将一些(或所有)的Cr+6物质转化为更低的价态。催化剂所使用的载体为多孔的无机氧化物材料,具有高表面积,即50-1000平方米/克的表面积,和10-200微米的平均粒度。可使用的无机氧化物包括二氧化硅,氧化铝,氧化钍,氧化锆和其它可比的无机氧化物,以及这样的氧化物的混合物。二氧化硅、二氧化硅氧化铝、磷酸铝、二氧化硅二氧化钛和二氧化硅磷酸铝为优选的载体化合物。特别优选的载体为表面积为200-500平方米/克、孔径为和平均粒度为20-100微米的微球状颗粒(例如,获自DavisonChemicalDivision,W.R.GraceandCompany的级别952MS,955二氧化硅,和获自IneosCorporation的IneosEP30,以及获自PhiladelphiaQuartz的相似二氧化硅等级)。催化剂所使用的载体必须在尝试负载甲硅烷基铬酸酯之前部分脱水。部分脱水通常在流化床脱水器中使用氮气或空气作为流化气体进行。可使用300℃-800℃的脱水温度。脱水在1-48小时,通常1.5-8小时内进行。优选的脱水温度为350-600℃。然后经由适合的方法将铬酸酯化合物沉积在脱水载体材料上。最典型的方法是在使得溶剂保持液体的温度和压力下将脱水载体悬浮置于惰性芳族或脂肪族烃(例如异戊烷,己烷,庚烷,甲苯,烃的混合物)中,将铬酸酯化合物加入悬浮的载体中,和允许铬酸酯化合物沉积在载体上。理论上,铬酸酯化合物与载体表面上的剩余表面羟基反应,导致负载的cupport-铬酸酯材料。典型的反应条件包括10-100℃的反应温度和1-48小时的反应持续时间。优选的温度为35-80℃,最优选35-60℃。优选的反应时间为2-24小时,更优选4-20小时,最优选6-10小时。优选的溶剂为异戊烷,混合的戊烷异构体,正己烷,己烷异构体和庚烷和庚烷异构体。最优选的溶剂为饱和的C5和C6烃。在完成沉积反应之后,反应产物进一步用烷基铝化合物处理以将Cr+6物质部分或完全还原至更低的价态。这可在沉积反应之后立即进行或在较晚时间进行,尽管顺序反应是高度优选的。适合的烷基铝化合物由下式描述:Al(R14)(3-x)(OR15)x其中x为0-2;R14和R15可相同或不同,和为包含1-25个碳的烷基、烷芳基或芳族基团。优选的R14或R15基团为甲基,乙基,正丙基,正丁基,异丁基,正戊基,正己基,正辛基,甲苯基,和金刚烷基。X优选为0-1.5,最优选为1.0。最优选R14或R15基团为乙基。如上所指,还原反应在可与沉积所述的溶剂相同或不同的溶剂中进行。优选地,使用相同的溶剂和反应是顺序的。温度和压力受控以维持溶剂处于液态,直到开始干燥悬浮液。典型的反应温度和时间分别为35-90℃和1-48小时。优选的反应时间在12和24小时之间,温度在45和75℃之间。在完成还原反应之后,催化剂在氮气或真空下干燥至自由流动的固体,和在基本上无毒的氮气中储存直到使用。不相容性用于第一聚合的Ziegler-Natta催化剂和用于第二聚合的基于铬的催化剂是不相容的。出于该专利说明书和所附的权利要求的目的,术语“不相容的催化剂”被理解为满足以下的一种或多种的那些催化剂:1)在彼此存在下减低至少一种催化剂的生产力大于50%的那些催化剂;2)在相同反应性条件下催化剂之一生产具有比体系中任何其它催化剂高两倍以上的分子量(Mw)的聚合物的那些催化剂;和3)在相同条件下在共聚单体并入或反应性比例方面不同大于约30%的那些催化剂。生产力在本文中被理解为在某些时间段内每kg催化剂的kg产物。Mw在本文被理解为使用SEC(尺寸排阻色谱法)使用1,2,4-三氯苯作为洗提液测量和使用线型聚乙烯标准样校准的重均分子量。共聚单体并入通过根据美国专利号4,798,081和Wilde,L.;RyIe,T.R.;Knobeloch,D.C;Peat,LR.;DeterminationofBranchingDistributionsinPolyethyleneandEthyleneCopolymers,J.Polym.ScL,20,441-455(1982)中描述的方法进行的分析升温洗脱分级(aTREF)进行测量,所述文献通过参考以其全部引入本文。待分析的组合物溶于经由0.2μm过滤器过滤的分析级的1,2-二氯苯和通过以0.1℃/min的冷却速率缓慢降低温度至20℃使在包含惰性载体(填充有150μm不锈钢珠的柱(体积2500μL))的柱中结晶。该柱装配有红外检测器。然后,通过从柱中洗脱结晶的聚合物样品通过以1℃/min的速率将洗脱溶剂(1,2-二氯苯)的温度从20℃缓慢增加至130℃产生ATREF色谱曲线。使用的器械可为PolymerCharCrystaf-TREF300。稳定剂:1g/LTopanol+1g/LIrgafos168样品:在20mL中大约70mg样品体积:0.3mL泵流量:0.50mL/minPolymerCharCrystaf-TREF-300的软件可以用于产生光谱。出于该专利说明书和所附的权利要求的目的,术语“催化剂”和“催化剂体系”是可交换使用的。“乙烯和共聚单体的连续聚合反应”在本文中指的是将乙烯和共聚单体进料至反应器和因此产生的聚烯烃通过连接至反应器的聚合物排出体系(半)连续抽取。流化床本发明的方法可以用于在气相反应器中的任何聚合方法。气相反应器可为适合于气相聚合的任何反应器和可例如为垂直地、水平地机械搅拌的反应器或流化床反应器。流化床反应器中的气相聚合方法是优选的。在典型的用于由单体生产聚合物的连续气体流化床聚合方法中,在催化剂存在下在反应性条件下使包含单体的气态物流通过流化床反应器。流化床反应器包括在聚合区域中生长聚合物颗粒的床。将聚合催化剂连续或间歇地引入聚合区域,同时进料单体和抽取聚合物。气体流化床聚合设备通常使用连续气体循环。在循环的一部分中,在反应器中通过聚合的热量加热循环气体物流。该热量通过反应器外部的冷却体系在循环的另一部分中除去。在一个实施方案中,使循环气体物冷却以形成气体和液体相混合物,然后引入反应器。从反应器抽取聚合物产物。对于气相方法的详述,参见在本文通过参考完全引入本文的美国专利号4,543,399和4,588,790。使用流化床聚合方法相比于其它聚合方法基本上降低了能量需要和最重要地降低了运行这样的聚合方法所要求的资本投资。在优选的实施方案中,在本发明的转变方法期间使流化床保持在流化条件下。存在多种类型的流化床反应器,其中鼓泡流化床反应器,循环流化床反应器,环形流化床反应器,多区域流化床反应器和闪蒸反应器。如本文所用的“流化床”指的是在固体/流体混合物中的一定量的固体颗粒(在这种情况下优选固体催化剂和/或附着单体的固体催化剂)起到流体的作用。这可以通过将所述量的固体颗粒置于恰当的条件下来实现,例如通过以足够高的速度将流体引入通过固体颗粒以使固体颗粒悬浮和引起他们表现为流体。气相流化床反应器(FBR)的例子示于图1。图1示出多区域气相流化床反应器(FBR)、压缩机和冷却装置。反应器的底部包括用于反应组合物如乙烯、氮气(N2)、氢气(H2)、共聚单体的连接至进料器的入口。在反应器中在分布板上方的的中间区域包括可以与氮气(N2)组合进料至反应器的聚合催化剂的入口。反应器的中间区域还包括至产物排出罐的出口。反应器的顶部区域包括顶部循环物流的出口,其中顶部循环物流的出口连接至压缩机的入口。压缩机包括经压缩的流体的出口和压缩机的出口连接至冷却装置的经压缩的流体的入口。冷却装置包括提供底部循环物流的出口,冷却装置的底部循环物流的出口连接至在反应器的底部处的入口。本发明的方法可在多区域流化床反应器中进行。多区域流化床反应器(FBR)体系的例子示于图2。该例子的多区域反应器为以冷凝模式操作的多区域反应器,所述多区域反应器包括第一区域、第二区域、第三区域、第四区域和分布板;其中第一区域通过分布板与第二区域分开;其中多区域反应器在垂直方向上延伸;其中多区域反应器的第二区域位于第一区域的上方,和其中多区域反应器的第三区域位于第二区域的上方,和其中多区域反应器的第四区域位于第三区域的上方其中第二区域包括内壁,其中第二区域内壁的至少部分为逐渐增加的内径或者连续开口圆锥的形式,其中直径或开口以垂直方向朝多区域反应器的顶部增加;其中第三区域包括内壁,其中第三区域内壁的至少部分为逐渐增加的内径或者连续开口圆锥的形式,其中直径或开孔以垂直方向朝多区域反应器的顶部增加其中第三区域内壁的最大直径大于第二区域内壁的最大直径。该例子的多区域反应器可以以所谓的“冷凝模式”或“冷凝的模式”操作,所述模式对于除去在放热聚合期间产生的热是有效的。在该模式中,热除去通过将气态循环物流冷却至其露点以下的温度来实现,导致循环物流的至少一部分冷凝以形成包含液体和气体的底部循环物流。然后将因此形成的底部循环物流引入流化床聚合反应器,其中液体部分在暴露于反应器的热时蒸发,所述蒸发从反应器除去热和能够进料一种或多种非常高度活性催化剂。以冷凝模式操作的FBR的详述进一步描述于申请EP13195141.0,其通过参考引入本文。图2说明了包括多区域反应器(8)、压缩机(400)和冷却装置(5)的FBR体系。该例子的多区域反应器(8)以垂直方向延伸和包括四个反应区域(1)、(2)、(3)和(4)。第一区域(1)包括接收底部循环物流(10)的第一入口和第一区域(1)通过分布板(6)与第二区域(2)分开。第二区域(2)包括接收固体聚合催化剂(20)的第一入口。第三区域(3)包括提供聚烯烃(30)的第一出口。该出口还可以位于第二区域(2)的上方。第四区域(4)包括顶部循环物流(40)的第一出口,其经由第一连接构件(AA)连接至压缩机(400)的第一入口。连接构件(AA)包括接收进料(60)的第一入口。压缩机(400)包括经压缩的流体(50)的第一出口,其经由第二连接构件(BB)连接至冷却装置(5)的经压缩的流体的第一入口。第二连接构件(BB)包括接收进料(70)的入口。冷却装置(5)包括提供底部循环物流(10)的第一出口,其连接至第一区域(1)的第一入口。FBR体系可进一步包括聚合物抽取体系、聚合物脱气体系和排出气体回收体系(未示于图2)。来自排出气体回收体系的回收组分(为液体形式)(80)的出口可通过泵(7)输送至第二连接构件(BB)的第一入口(70)。该体系可以适合地用于包括以下的连续聚合方法:-使用接收固体聚合催化剂(20)的第一入口将固体聚合催化剂供应第二区域(2);-将包含α-烯烃单体的进料(60)供应至第一连接构件(AA);-任选将包含可冷凝惰性组分的进料(70)供应至第二连接构件(BB);-使用第二区域(2)和/或第三区域(3)的第一出口抽取聚烯烃(30);和-将流体从第四区域(4)的第一出口循环至第一区域的第一入口;其中流体通过以下流通:-使用压缩机(400)压缩进料(60)和顶部循环物流(40)以形成经压缩的流体(50);-随后使用冷却装置(5)冷却经压缩的流体(50)至经压缩的流体的露点以下以形成底部循环物流(10);和-经由用于接收第一区域的底部循环物流的入口将底部循环物流(10)进料至多区域反应器(8)的第一区域;和-其中该方法中的表观气体速度为0.5-5m/s。进料(60)包括链转移剂如氢气和可进一步包含气态α-烯烃单体和惰性(insert)气态组分如氮气。进料(70)包括可冷凝惰性组分,例如选自具有4-20个碳原子、优选4-8个碳原子的烷烃及它们的混合物,例如丙烷,正丁烷,异丁烯,正戊烷,异戊烷,新戊烷,正己烷,异己烷或具有6个C原子的其它饱和烃,正庚烷,正辛烷和具有7或8个C原子的其它饱和烃和它们的任何混合物的可冷凝惰性组分;和可进一步包含可冷凝α-烯烃单体、α-烯烃共聚单体和/或它们的混合物。上述FBR体系的优点在于允许引入较高量的的液体而不会引起流化床的不稳定。尽管出于说明的目的详细描述了本发明,但应理解的是,这样的详述仅用于上述目的和本领域技术人员可以进行变型而不会脱离权利要求所定义的本发明的精神和范围。还要注意的是,本发明涉及本文所述的特征的所有可能的组合,优选特别是存在于权利要求中的特征的那些组合。还要注意的是,术语“包含”和“包括”不排除其它要素的存在。然而,还要理解的是,对包含某些组分的产物的描述还公开了由这些组分组成的产物。相似地,还要理解的是,对包括某些步骤的方法的描述还公开了由这些步骤组成的方法。现在通过以下的实施例描述本发明,然而不限于此。实施例将示意性示于图1的气相反应器体系用于转变方法。气态进料物流在混合三通设置中混合在一起和从底部进入反应器,和通过穿孔的分布板。未反应的气体物流与夹带的聚合物颗粒分离,和然后压缩、冷却和循环回反应器。通过调节反应条件(温度、压力、流速等)来控制产物特性。聚合在具有45cm的内径和228cm的反应区域高度的连续气相流化床反应器中进行。流化床由聚合物颗粒制成。反应器填充有通过高速度气体物流强烈搅拌的约50kg的干聚合物颗粒的床。反应区域中聚合物颗粒的床通过作为流化介质以及用于吸收反应区域内产生的放热的热消散剂工作的循环物流保持流化状态。控制乙烯、氢气和共聚单体的各自的流速以保持固定的组成目标。控制乙烯浓度以保持恒定的乙烯分压。很好地控制氢气/乙烯流量比例以保持最终树脂的相对稳定的熔体指数。所有气体的浓度通过在线气相色谱来测量以确保循环气体物流中相对恒定的组成。使用纯氮气作为载体将固体催化剂直接注入流化床。调节其速率以保持恒定的约10-15kg/hr的生产速率。生长聚合物颗粒的反应床通过使补充进料和循环气体连续流过反应区域而保持流化状态。0.40m/sec的表观气体速度用于实现该状态。反应器在下表所示的压力和温度下操作。为了保持恒定的反应器温度,将循环气体的温度连续向上或向下调节以适应由于聚合导致的热产生速率的任何改变。通过以等于微粒产物形成速率的速率抽取一部分流化床而将流化床保持在恒定高度。经由一系列阀门半连续地除去产物进入固定体积室。将由此获得的产物吹扫以除去夹带的烃和使用少量蒸汽(steam)的潮湿氮气处理以使任何痕量的残留催化剂失活。聚合物的特性通过以下测试方法来确定:表1Ziegler-Natta(ZN)催化剂用于生产具有918kg/m3的密度和1.0的熔体指数的1-丁烯共聚的LLDPE。获得的LLDPE适合于通过吹膜挤出方法来处理。ZN催化剂为根据WO2012069157的实施例1制备的催化剂:将在氮气流下在600℃下脱水4小时的2.5g的Sylopol955二氧化硅置于40cm3烧瓶中。将15cm3的异戊烷加入浆液二氧化硅中,然后将2.5mmol的二正丁基镁加入烧瓶中和所得混合物在35℃的温度下搅拌60分钟。然后,将3.5mmol的甲基正丙基酮加入烧瓶中和所得混合物在35℃的温度下搅拌60分钟。然后,将0.25mmol的四乙氧基硅烷加入烧瓶中和所得混合物在35℃的温度下搅拌30分钟。接来下,将0.25mmol的四乙氧基钛加入烧瓶中和所得混合物在35℃的温度下搅拌30分钟。随后,将1.50mmol的四氯化钛加入烧瓶中和所得混合物在35℃的温度下搅拌30分钟。最后,使用氮气吹扫在70℃下使浆液干燥60分钟以产生自由流动的固体产物。该第一聚合的条件示于表2中。表2反应器条件床温度(℃)85反应器压力(barg)20.7C2分压(bara)7.0床水平(mbar)35表观速度(m/s)0.40H2/C2体积比例0.140C4/C2体积比例0.40TEAL流量(kg/h)0.08在以稳态运行反应器以生产LLDPE之后,停止至反应器的1-丁烯流进料直至C4/C2体积比例降至0.1或更低。随后停止ZN催化剂的进料。在停止ZN催化剂进料之后,助催化剂TEAL的进料在其停止之前保持额外30分钟。维持反应器条件,即除了C4/C2体积比例和TEAL流量为零的表2的条件直至反应结束。当通过首先停止乙烯进料和随后在停止乙烯进料之后的5-10分钟内停止H2进料而使反应结束时停止所有进料。使反应器减压至7barg。随后在85℃的反应器温度下用N2流动吹扫反应器4小时。随后将床温度减少至80℃。在两个步骤中将C2分压增加至目标压力。通过进料10kg/hr流速的C2将C2分压逐渐增加至10bara,同时维持床温度至80℃。在第二步骤中,通过进料5kg/hr流速的C2将C2分压逐渐增加至15bara,同时维持床温度至80℃。当达到15bara的目标C2分压时,引入H2以达到0.02的H2/C2体积比例。随后,通过以2℃/小时增加而使床温度逐渐升高至95℃。将基于铬的催化剂填充至反应器。基于铬的催化剂包括二氧化硅载体上的氧化铬,其通过在催化剂活化之前干燥和然后用四异丙氧基钛处理而钛酸化。这之后引入1-己烯。使用基于铬的催化剂用于聚合的最终反应器组成列于下表3中。表3反应器条件目标床温度(℃)100反应器压力(barg)20.7C2分压(bara)15床水平(mbar)35表观速度(m/s)0.40H2/C2体积比例0.02C6/C2体积比例0.0015TEAL流量(kg/h)0成功获得了密度为0.952g/cm3和流动指数(在21.6kg下测量)为10g/10min的HDPE。实现了从Ziegler-Natta催化剂至基于铬的催化剂的成功转变。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1