杂环化合物以及使用该杂环化合物的有机发光器件的制作方法
本申请要求于2014年6月27日向韩国知识产权局提交的韩国专利申请第10-2014-0080226号的优先权和权益,其全部内容通过引用并入本文。
本申请涉及一种新型杂环化合物以及使用该新型杂环化合物的有机发光器件。
背景技术:
电致发光器件是一种自发光型的显示器件,其优点在于视角宽、对比度优异,以及响应速度快。
有机发光器件具有有机薄膜布置于两个电极之间的结构。当将电压施加到具有所述结构的有机发光器件时,从两个电极注入的电子和空穴在有机薄膜中彼此结合组成一对,且接着,在淬灭的同时发光。在必要时,有机薄膜可由单层或多层构成。
在必要时,用于有机薄膜的材料可具有发光功能。例如,作为用于有机薄膜的材料,还可以使用自身可以单独构成发光层的化合物,或者也可以使用可用作基于主体-掺杂剂的发光层的主体或掺杂剂的化合物。此外,作为用于有机薄膜的材料,还可以使用可执行如空穴注入、空穴传输、电子阻挡、空穴阻挡、电子传输或电子注入的功能的化合物。
为了改进有机发光器件的性能、使用寿命或效率,对于开发用于有机薄膜的材料存在持续的需求。
技术实现要素:
[技术问题]
本申请涉及一种新型杂环化合物以及使用该新型杂环化合物的有机发光器件。
[技术方案]
本申请提供一种下式1的化合物。
[式1]
在式1中,
Y为S或O,
X1和X2彼此相同或不同,并且各自独立地为N或CR10,以及
R1、R2和R4至R10彼此相同或不同,并且各自独立地选自氢;氘;卤素;取代或未取代的直链或支链C1至C60烷基;取代或未取代的直链或支链C2至C60烯基;取代或未取代的直链或支链C2至C60炔基;取代或未取代的直链或支链C1至C60烷氧基;取代或未取代的单环或多环C3至C60环烷基;取代或未取代的单环或多环C2至C60杂环烷基;取代或未取代的单环或多环C6至C60芳基;取代或未取代的单环或多环C2至C60杂芳基;以及未取代的或被C1至C20烷基、取代或未取代的单环或多环C6至C60芳基、或取代或未取代的单环或多环C2至C60杂芳基所取代的胺基。
此外,本申请提供一种有机发光器件,其包括阳极、阴极以及设置在所述阳极和所述阴极之间的一层或更多层有机材料层,其中所述有机材料层中的一层或多层包含所述式1的化合物。
[有益效果]
本申请中的化合物可被用作用于有机发光器件的有机材料层的材料。所述化合物可被用作有机发光器件中的空穴注入材料、空穴传输材料、发光材料、电子传输材料、电子注入材料等。特别是,式1的化合物可被用作用于有机发光器件的发光层的材料,尤其是,磷光主体。
附图说明
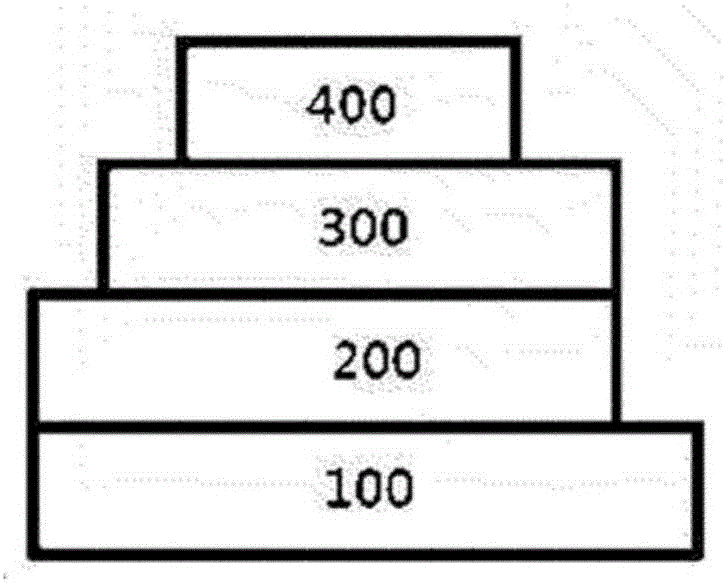
图1至图3图示根据本申请的示例性实施方式的有机发光器件的电极和有机材料层的堆叠次序。
图4图示化合物29的UV和PL的测量图。
图5和图6图示由测量化合物29的CV的结果导出的Eox值。
图7图示化合物42的UV和PL的测量图。
图8和图9图示由测量化合物42的CV的结果导出的Eox值。
图10图示化合物87的UV和PL的测量图。
图11和图12图示由测量化合物87的CV的结果导出的Eox值。
图13图示化合物88的UV和PL的测量图。
图14和图15图示由测量化合物88的CV的结果导出的Eox值。
图16图示化合物90的UV和PL的测量图。
图17和图18图示由测量化合物90的CV的结果导出的Eox值。
图19图示化合物91的UV和PL的测量图。
图20和图21图示由测量化合物91的CV的结果导出的Eox值。
图22图示化合物92的UV和PL的测量图。
图23和图24图示由测量化合物92的CV的结果导出的Eox值。
图25图示化合物93的UV和PL的测量图。
图26和图27图示由测量化合物93的CV的结果导出的Eox值。
图28图示由测量化合物73的CV的结果导出的Eox值。
图29图示化合物73的UVPL的测量图。
图30图示化合物85的LTPL的测量图。
图31图示化合物85的UVPL的测量图。
图32图示化合物86的LTPL的测量图。
图33图示化合物86的UVPL的测量图。
图34图示化合物87的LTPL的测量图。
图35图示化合物87的UVPL的测量图。
图36图示化合物88的LTPL的测量图。
图37图示化合物88的UVPL的测量图。
图38图示化合物90的LTPL的测量图。
图39图示化合物90的UVPL的测量图。
图40图示化合物91的LTPL的测量图。
图41图示化合物91的UVPL的测量图。
图42图示化合物92的LTPL的测量图。
图43图示化合物92的UVPL的测量图。
图44图示化合物93的LTPL的测量图。
图45图示化合物93的UVPL的测量图。
图46图示化合物245的LTPL的测量图。
图47图示化合物245的UVPL的测量图。
图48图示化合物246的LTPL的测量图。
图49图示化合物246的UVPL的测量图。
图50图示化合物250的LTPL的测量图。
图51图示化合物250的UVPL的测量图。
图52图示化合物253的LTPL的测量图。
图53图示化合物253的UVPL的测量图。
图54图示化合物259的LTPL的测量图。
图55图示化合物259的UVPL的测量图。
图56图示化合物260的LTPL的测量图。
图57图示化合物260的UVPL的测量图。
图58图示化合物409的LTPL的测量图。
图59图示化合物409的UVPL的测量图。
图60图示化合物420的LTPL的测量图。
图61图示化合物420的UVPL的测量图。
图62图示化合物425的LTPL的测量图。
图63图示化合物425的UVPL的测量图。
图64图示化合物427的LTPL的测量图。
图65图示化合物427的UVPL的测量图。
图66图示化合物434的LTPL的测量图。
图67图示化合物434的UVPL的测量图。
<附图标记和符号说明>
100:基板
200:阳极
300:有机材料层
301:空穴注入层
302:空穴传输层
303:发光层
304:空穴阻挡层
305:电子传输层
306:电子注入层
400:阴极
具体实施方式
下文中,将详细描述本申请。
本说明书中描述的化合物可由式1表示。具体来讲,式1的化合物由于以上所述的母核结构和取代基的结构特征,可被用作用于有机发光器件的有机材料层的材料。
在本说明书中,“取代或未取代的”意指未取代的或被一个或更多个取代基取代,所述一个或更多个取代基选自氘;-CN;直链或支链C1至C60烷基;直链或支链C2至C60烯基;直链或支链C2至C60炔基;单环或多环C3至C60环烷基;单环或多环C2至C60杂环烷基;单环或多环C6至C60芳基;单环或多环C2至C60杂芳基;-SiRR’R”;-P(=O)RR’;以及-NRR’,或者意指未取代的或被这些取代基当中两个或多个取代基连接而成的取代基取代。例如,“所述两个或多个取代基连接而成的取代基”可为联苯基团。也就是,所述联苯基团还可以是芳基,并且可被理解为两个苯基连接而成的取代基。R、R'、以及R"彼此相同或不同,并且各自独立地为直链或支链C1至C60烷基;单环或多环C6至C60芳基;或单环或多环C2至C60杂芳基。
在本说明书中,所述烷基包括具有1至60个碳原子的直链或支链,并且可额外地被其它取代基取代。所述烷基的碳原子的数目可为1至60个,特别地为1至40个,并且更特别地为1至20个。
在本说明书中,所述烯基包括具有2至60个碳原子的直链或支链,并且可额外地被其它取代基取代。所述烯基的碳原子的数目可为2至60个,特别地为2至40个,并且更特别地为2至20个。
在本说明书中,所述炔基包括具有2至60个碳原子的直链或支链,并且可额外地被其它取代基取代。所述炔基的碳原子的数目可为2至60个,特别地为2至40个,并且更特别地为2至20个。
在本说明书中,所述环烷基包括具有3至60个碳原子的单环或多环,并且可额外地被其它取代基取代。此处,所述多环意指环烷基直接连接到另一个环基团或与另一个环基团稠合的基团。此处,另一个环基团还可以是环烷基,但还可为另一种环基团,例如,杂环烷基、芳基、杂芳基等。环烷基的碳原子的数目可为3至60个,特别地为3至40个,并且更特别地为5至20个。
在本说明书中,所述杂环烷基包括O、S、Se、N以及Si中的一种或多种作为杂原子,包括具有2至60个碳原子的单环或多环,并且可额外地被其它取代基取代。此处,所述多环意指杂环烷基直接连接到另一个环基团或与另一个环基团稠合的基团。此处,另一个环基团还可以是杂环烷基,但还可为另一种环基团,例如,环烷基、芳基、杂芳基等。所述杂环烷基的碳原子的数目可为2至60个,特别地为2至40个,并且更特别地为3至20个。
在本说明书中,所述芳基包括具有6至60个碳原子的单环或多环,并且可额外地被其它取代基取代。此处,所述多环意指芳基直接连接到另一个环基团或与另一个环基团稠合的基团。此处,另一个环基团还可以是芳基,但还可为另一种环基团,例如,环烷基、杂环烷基、杂芳基等。所述芳基包括螺环基团。所述芳基的碳原子的数目可为6至60个,特别地为6至40个,并且更特别地为6至25个。所述芳基的具体实例包括苯基、联苯基、三苯基、萘基、蒽基、基(chrysenyl)、菲基、苝基、荧蒽基、苯并[9,10]菲基(triphenylenyl)、迫苯并萘基(phenalenyl)、芘基、并四苯基、并五苯基、芴基、茚基、苊基、芴基、苯并芴基、螺双芴基等,或它们稠合的环,但不限于此。
在本说明书中,所述螺环基团是包含螺环结构的基团,并且可具有15至60个碳原子。例如,所述螺环基团可以包括2,3-二氢-1H-茚基团或环己烷螺-键合到芴基团的结构。具体来讲,所述螺环基团包括一组以下结构式:
在本说明书中,所述杂芳基包括S、O、Se、N以及Si中的一种或更多种作为杂原子,包括具有2至60个碳原子的单环或多环,并且可额外地被其它取代基取代。此处,所述多环意指杂芳基直接连接到另一个环基团或与另一个环基团稠合的基团。此处,另一个环基团还可以是杂芳基,但还可为另一种环基团,例如,环烷基、杂环烷基、芳基等。所述杂芳基的碳原子的数目可为2至60个,特别地为2至40个,并且更特别地为3至25个。所述杂芳基的具体实例包括吡啶基、吡咯基、嘧啶基、哒嗪基、呋喃基、噻吩基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、异噻唑基、三唑基、呋吖基、噁二唑基、噻二唑基、二噻唑基、四唑基、吡喃基、噻喃基、二嗪基、噁嗪基、噻嗪基、二噁烯基(dioxynyl)、三嗪基、四嗪基、喹啉基、异喹啉基、喹唑啉基、异喹唑啉基、萘啶基、吖啶基、菲啶基、咪唑并吡啶基、二氮杂萘基、三氮杂茚基、吲哚基、吲哚嗪基(indolyzinyl)、苯并噻唑基、苯并噁唑基、苯并咪唑基、苯并噻吩基、苯并呋喃基、二苯并噻吩基、二苯并呋喃基、咔唑基、苯并咔唑基、二苯并咔唑基、吩嗪基、二苯并噻咯、螺二(二苯并噻咯)、二氢吩嗪基、吩噁嗪基、菲啶基等、或它们稠合的环、但不限于此。
根据本申请的示例性实施方式,式1中的Y为S。
根据本申请的示例性实施方式,式1中的Y为O。
根据本申请的示例性实施方式,式1中的X1和X2中一个为N而另一个为CR10。
根据本申请的示例性实施方式,式1中的X1和X2中一个为N而另一个为CR10,并且R1、R2和R10中至少一个为–(L)m-(Z)n,
L为取代或未取代的直链或支链C2至C60亚烷基;取代或未取代的单环或多环C6至C60亚芳基;或取代或未取代的单环或多环C2至C60亚杂芳基,
m为0至3的整数,
n为1或2的整数,以及
Z选自取代或未取代的单环或多环C6至C60芳基;取代或未取代的单环或多环C2至C60杂芳基;-SiR11R12R13;-P(=O)R14R15;以及未取代的或被C1至C20烷基、取代或未取代的单环或多环C6至C60芳基、或取代或未取代的单环或多环C2至C60杂芳基所取代的胺基,并且R11至R15彼此相同或不同,并且各自独立地为取代或未取代的直链或支链C1至C60烷基;单环或多环C6至C60芳基;或单环或多环C2至C60杂芳基。
根据本申请的示例性实施方式,m可为0,或1、2或3的整数,当m为2或更大的整数时,各L可以彼此相同或不同。
根据本申请的示例性实施方式,当n为2的整数时,各Z可以彼此相同或不同。
根据本申请的示例性实施方式,L为C2至C60亚烷基;或C6至C60亚芳基;
根据本申请的示例性实施方式,L为亚苯基、亚萘基或亚蒽基。
根据本申请的示例性实施方式,Z为取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的三苯基、取代或未取代的萘基、取代或未取代的蒽基、取代或未取代的菲基、取代或未取代的茚基、取代或未取代的苝基、取代或未取代的芘基、取代或未取代的苊烯基(acenaphthalenyl)、取代或未取代的芴基、取代或未取代的荧蒽基、取代或未取代的苯并[9,10]菲基、取代或未取代的迫苯并萘基、取代或未取代的吡咯基、取代或未取代的吡啶基、取代或未取代的嘧啶基、取代或未取代的哒嗪基、取代或未取代的三嗪基、取代或未取代的噻吩基、取代或未取代的呋喃基、取代或未取代的苯并呋喃基、取代或未取代的二苯并呋喃基、取代或未取代的苯并噻唑基、取代或未取代的苯并噁唑基、取代或未取代的吲哚基、取代或未取代的咔唑基、取代或未取代的苯并咔唑基、取代或未取代的二苯并咔唑基、取代或未取代的吲哚[2,3-a]咔唑基、取代或未取代的吲哚[2,3-b]咔唑基、取代或未取代的喹啉基、取代或未取代的异喹啉基、取代或未取代的苯硫基(thiophenyl)、取代或未取代的苯并噻吩基、取代或未取代的二苯并噻吩基、取代或未取代的芴基、取代或未取代的二氢吲哚基、取代或未取代的10,11-二氢-二苯并[b,f]氮杂基团、取代或未取代的9,10-二氢吖啶基团、取代或未取代的螺环基团(其中,2,3-二氢-1H-茚或环己烷螺-键合(spiro-bonded)到芴)、取代或未取代的二烷基胺、取代或未取代的二芳基胺、取代或未取代的烷基芳基胺、取代或未取代的乙酰苯基团、取代或未取代的二苯甲酮基团、-SiR11R12R13或-P(=O)R14R15,R11至R15彼此相同或不同,并且各自独立地为直链或支链C1至C60烷基、单环或多环C6至C60芳基、或单环或多环C2至C60杂芳基。
根据本申请的示例性实施方式,Z选自取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的三苯基、取代或未取代的萘基、取代或未取代的蒽基、取代或未取代的咔唑基、取代或未取代的苯并咔唑基、取代或未取代的二苯并咔唑基、取代或未取代的吲哚[2,3-a]咔唑基、取代或未取代的吲哚[2,3-b]咔唑基、取代或未取代的芴基、取代或未取代的苯并呋喃基、取代或未取代的二苯并呋喃基、取代或未取代的苯硫基、取代或未取代的苯并噻吩基、取代或未取代的二苯并噻吩基、取代或未取代的苯并[9,10]菲基、取代或未取代的三嗪基、取代或未取代的芘基、-Si(Ph)3、-P(=O)(Ph)2以及取代或未取代的二苯基胺,“取代或未取代的”意指未取代的或被选自甲基、直链或支链丙基、直链或支链丁基、直链或支链戊基、苯基、联苯基、三苯基、萘基、蒽基、咔唑基、苯并咔唑基、二苯并咔唑基、吲哚[2,3-a]咔唑基、吲哚[2,3-b]咔唑基、芴基、苯并呋喃基、二苯并呋喃基、噻吩基、苯并噻吩基、二苯并噻吩基、苯并[9,10]菲基、三嗪基、芘基、-Si(Ph)3、-P(=O)(Ph)2、以及二苯基胺中的至少一个取代,并且所述取代基可以额外地被进一步取代。
根据本申请的示例性实施方式,R11至R15彼此相同或不同,并且为单环或多环C6至C60芳基。
根据本申请的示例性实施方式,R11至R15彼此相同或不同,并且为苯基、联苯基、三苯基、萘基或蒽基。
根据本申请的示例性实施方式,R4至R9为氢或氘。
根据本申请的示例性实施方式,式1由下式2或下式3表示。
[式2]
[式3]
在式2和3中,Y、X1、X2、R1、R2以及R4至R9的定义与在式1中定义的那些相同。在本申请的示例性实施方式中,式1由下式4至7中的任一个表示。
[式4]
[式5]
[式6]
[式7]
在式4至7中,Y、R1、R2以及R4至R9与在式1中定义的那些相同,并且R3与式1中的R10的定义相同。
根据本申请的示例性实施方式,在式4至7中,
R1至R3中的至少一个为–(L)m-(Z)n,并且其他取代基与在式1中定义的那些相同,并且
Y、R4至R9、L、m以及Z的定义与在式1中定义的那些相同。
根据本申请的示例性实施方式,在式4至7中,R1为–(L)m-(Z)n,R2和R3为氢、氘或苯基,并且L、m、n以及Z与以上所述的那些相同。
根据本申请的示例性实施方式,在式4至7中,R2为–(L)m-(Z)n,R1和R3为氢、氘或苯基,并且L、m、n以及Z与以上所述的那些相同。
根据本申请的示例性实施方式,在式4至7中,R3为–(L)m-(Z)n,R1和R2为氢、氘或苯基,并且L、m、n以及Z与以上所述的那些相同。
根据本申请的示例性实施方式,在式4至7中,m为0或1。
根据本申请的示例性实施方式,式1由下式8至11中的任一个表示。
[式8]
[式9]
[式10]
[式11]
在式8至11中,
A选自直接键;取代或未取代的直链或支链C2至C60亚烷基;取代或未取代的直链或支链C2至C60亚烯基;取代或未取代的直链或支链C2至C60亚炔基;取代或未取代的单环或多环C3至C60亚环烷基;取代或未取代的单环或多环C2至C60亚杂环烷基;取代或未取代的单环或多环C6至C60亚芳基;取代或未取代的单环或多环C2至C60亚杂芳基;以及未取代的或被C1至C20烷基、取代或未取代的单环或多环C6至C60芳基、或取代或未取代的单环或多环C2至C60杂芳基取代的胺基,
R16至R19彼此相同或不同,并且各自独立地选自氢;氘;卤素;取代或未取代的直链或支链C1至C60烷基;取代或未取代的直链或支链C2至C60烯基;取代或未取代的直链或支链C2至C60炔基;取代或未取代的直链或支链C1至C60烷氧基;取代或未取代的单环或多环C3至C60环烷基;取代或未取代的单环或多环C2至C60杂环烷基;取代或未取代的单环或多环C6至C60芳基;取代或未取代的单环或多环C2至C60杂芳基;以及未取代的或被C1至C20烷基、取代或未取代的单环或多环C6至C60芳基、或取代或未取代的单环或多环C2至C60杂芳基取代的胺基,
p、q、r以及s为0至4的整数,并且
Y以及R6至R9的定义与式1中定义的那些相同。
式8至11意指二聚体结构,并且A意指所述二聚体的连接基团。
根据本申请的示例性实施方式,在式8至11中,A选自取代或未取代的单环或多环C6至C60亚芳基;以及取代或未取代的单环或多环C2至C60亚杂芳基。
根据本申请的示例性实施方式,在式8至11中,A为未取代的或被烷基或芳基取代的C6至C60亚芳基;或未取代的或被烷基或芳基取代的C2至C60亚杂芳基。
根据本申请的示例性实施方式,在式8至11中,A为未取代的或被烷基或芳基取代的咔唑基团;或未取代的或被烷基或芳基取代的芴基团。
根据本申请的示例性实施方式,在式8至11中,A为未取代的或被烷基或芳基取代的咔唑基团;或未取代的或被烷基或芳基取代的芴基团,所述烷基为C1至C10直链或支链,并且所述芳基为C6至C20芳基。
根据本申请的示例性实施方式,式1至7的Y为并且X3和X4可为取代或未取代的单环或多环C6至C60芳烃环;或取代或未取代的单环或多环C2至C60芳杂环。
所述可由以下结构式中的任一个表示,但不限于此。
在所述结构式中,Z1至Z3彼此相同或不同,并且各自独立地为S或O,
Z4至Z9彼此相同或不同,并且各自独立地为CR’R”、NR’、S或O,以及
R’与R”彼此相同或不同,并且各自独立地为氢;取代或未取代的直链或支链C1至C60烷基;或取代或未取代的单环或多环C6至C60芳基。
根据本申请的示例性实施方式,式1可选自以下化合物。
上述化合物可基于将在下文描述的制备实施例来制备。将在下文描述的制备实施例中描述代表性的实例,但在必要时,可以添加或除去取代基,并且可以改变取代基的位置。此外,可基于本领域已知的技术改变起始原料、反应物、反应条件等。在必要时,本领域的普通技术人员可以通过使用本领域已知的技术改变在其他位置的取代基的种类或位置。
例如,在式4的化合物中,母核结构可按照下式1和2制备。在下式1和2中,例示式4的Y为S的情形,但是Y为氧(O)的情形也是可以的。
可通过本领域已知的方法键合取代基,并且可根据本领域已知的技术改变取代基的位置或取代基的数目。
[反应式1]
[反应式2]
反应式1是取代基键合到式4的母核结构中R2位置的反应的实例。具体地,反应式1中最后的化合物是式4中的R2为被Ar取代的苯基的情形。Ar与上述Z的定义相同。
反应式2是取代基键合到式4的母核结构中R3位置的反应的实例。具体地,反应式2中最后的化合物是式4中的R3为被Ar取代的苯基的情形。Ar与上述Z的定义相同。
例如,在式5的化合物中,可按照下反应式3制备母核结构。可通过本领域已知的方法键合取代基,并且可根据本领域已知的技术改变取代基的位置或取代基的数目。
[反应式3]
反应式3是取代基键合到式5的母核结构中R3位置的反应的实例。具体地,反应式3中最后的化合物是式5中的R3为被Ar取代的苯基的情形。Ar与上述Z的定义相同。
例如,在式6的化合物中,可按照下反应式4制备母核结构。可通过本领域已知的方法键合取代基,并且可根据本领域已知的技术改变取代基的位置或取代基的数目。
[反应式4]
反应式4是取代基键合到式6的母核结构中R3位置的反应的实例。具体地,反应式4中最后的化合物是式6中的R3为被Ar取代的苯基的情形。Ar与上述Z的定义相同。
例如,在式7的化合物中,可按照下反应式5制备母核结构。可通过本领域已知的方法键合取代基,并且可根据本领域已知的技术改变取代基的位置或取代基的数目。
[反应式5]
反应式5是取代基键合到式7的母核结构中R3位置的反应的实例。具体地,反应式5最后的化合物是式7中的R3为被Ar取代的苯基的情形。Ar与上述Z的定义相同。
本申请的另一个示例性实施方式提供包含上述式1化合物的有机发光器件。具体地,根据本申请的有机发光器件包括阳极、阴极以及设置在所述阳极和所述阴极之间的一层或更多层有机材料层,其中所述有机材料层中的一层或更多层包含式1的化合物。
图1至3图示根据本申请的示例性实施方式的有机发光器件的电极和有机材料层的堆叠次序。然而,本申请的范围不意图受到这些附图的限制,本领域已知的有机发光器件的结构也可被应用到本申请。
根据图1,图示了阳极200、有机材料层300以及阴极400顺序堆叠在基板100上的有机发光器件。然而,有机发光器件不限于该结构,并且如在图2中,也可以实施顺序堆叠阴极、有机材料层以及阳极的有机发光器件。
图3示例有机材料层为多层的情形。根据图3的有机发光器件包括空穴注入层301、空穴传输层302、发光层303、空穴阻挡层304、电子传输层305以及电子注入层306。然而,本申请的范围不限于如上所述的堆叠结构,必要时,可以省略除了发光层的其他层,并且可以进一步添加其它必要的功能层。
除了所述有机材料层中的一层或更多层包含式1的化合物以外,可通过本领域已知的材料和方法制造根据本申请的有机发光器件。
式1的化合物可单独构成有机发光器件的有机材料层的一层或更多层。然而,必要时,可将式1的化合物与其它材料混合来构成有机材料层。
可将式1的化合物用作有机发光器件中的空穴注入材料、空穴传输材料、发光材料、电子传输材料、电子注入材料等。
例如,可将根据本申请的示例性实施方式的化合物用作用于有机发光器件中的电子注入层、电子传输层、或同时注入和传输电子的层的材料。
此外,可将根据本申请的示例性实施方式的化合物用作用于有机发光器件的发光层的材料。具体地,还可以将所述化合物单独用作发光材料,以及用作发光层的主体材料或掺杂剂材料。
另外,可将根据本申请的示例性实施方式的化合物用作有机发光器件的磷光主体材料。在这种情形中,根据本申请的示例性实施方式的化合物与磷光掺杂剂一起被包含。
另外,可将根据本申请的示例性实施方式的化合物用作用于有机发光器件的空穴阻挡层的材料。
在根据本申请的有机发光器件中,将在下文中例示除了式1的化合物以外的材料,但是提供这些材料仅用于举例并且不用于限制本申请的范围,并且这些材料可以用本领域公知的材料代替。
作为阳极材料,可以使用具有相对大的功函数的材料,以及可以使用透明导电氧化物、金属或导电性聚合物等。
作为阴极材料,可以使用具有相对小的功函数的材料,以及可以使用金属、金属氧化物或导电性聚合物等。
作为空穴注入材料,还可以使用公知的空穴注入材料,以及例如,可以使用酞菁化合物,如在美国专利第4,356,429号公开的酞菁铜或在文件[Advanced Material,6,p.677(1994)]中描述的星爆型胺衍生物(starburst-type amine derivatives),例如,TCTA、m-MTDATA、m-MTDAPB、聚苯胺/十二烷基苯磺酸(Pani/DBSA)或聚(3,4-亚乙基二氧噻吩)/聚(4-苯乙烯磺酸盐)(PEDOT/PSS)(其为可溶性导电聚合物)、聚苯胺/樟脑磺酸(Pani/CSA)或聚苯胺/聚(4-苯乙烯-磺酸盐)(PANI/PSS)等。
作为空穴传输材料,可以使用吡唑啉衍生物、基于芳基胺的衍生物、二苯基乙烯衍生物、三苯基二胺衍生物等,并且还可以使用低分子量材料或聚合物材料。
作为电子传输材料,可以使用噁二唑衍生物、蒽醌二甲烷及其衍生物、苯醌及其衍生物、萘醌及其衍生物、蒽醌及其衍生物、四氰基蒽醌二甲烷及其衍生物、芴酮衍生物、二苯基二氰基乙烯及其衍生物、联苯醌衍生物、8-羟基喹啉的金属配合物及其衍生物等,并且还可以使用低分子量材料以及聚合物材料。
作为电子注入材料,例如,本领域中通常使用LiF,但本申请不限于此。
作为发光材料,可以使用红光、绿光或蓝光发光材料,而且必要时,可以混合以及使用两种或更多种发光材料。此外,作为发光材料,还可以使用荧光材料,但是也可以使用磷光材料。作为发光材料,也可以单独使用通过使从阳极和阴极分别注入的空穴和电子结合来发光的材料,但还可以使用其中主体材料和掺杂剂材料一起作用来发光的材料。
当将根据本申请的化合物用作磷光主体材料时,可以使用本领域已知的材料作为磷光掺杂剂材料来一起使用。
例如,可以使用由LL’MX、LL’L”M、LMXX’、L2MX以及L3M表示的磷光掺杂剂材料,但本申请的范围不限于这些实例。
此处,L、L’、L”、X以及X’为彼此不同的双齿配体,并且M为形成八面体配合物的金属。
M可为铱、铂、锇等。
L为通过sp2碳和杂原子在M上配位的阴离子双齿配体,并且X可执行捕获电子或空穴的功能。L的非限制性实例包括2-(1-萘基)苯并噁唑、(2-苯基苯并噁唑)、(2-苯基苯并噻唑)、(7,8-苯并喹啉)、(噻吩基吡嗪)、苯基吡啶、苯并噻吩基吡嗪、3-甲氧基-2-苯基吡啶、噻吩基吡嗪、甲苯基吡啶等。X的非限制性实例包括乙酰丙酮化物(acac)、六氟乙酰丙酮化物、亚水杨基、吡啶甲酸盐(picolinate)、8-羟基喹啉酸盐(8-hydroxyqunolinate)等。
其更多具体实例将在下文中示出,但是本申请不仅限于这些实例。
[本发明的方式]
下文中,将通过实施例更详细描述本申请,但是提供这些实施例仅用于例示本申请,并且不用于限制本申请的范围。
<实施例>
<制备实施例1>化合物1的制备
化合物1-1的制备
将30g(168.5mmol)的苯并[b]噻吩-2-基硼酸、37.8g(202.26mmol)的1-溴-4-甲氧基苯、9.7g(8.4mmol)的Pd(PPh3)4以及35.7g(336.9mmol)的Na2CO3与300mL的甲苯、120mL的乙醇(EtOH)以及120mL的H2O一起放入容器中,使所得的混合物在120℃下回流1小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到18.0g(51%)的目标化合物1-1。
化合物1-2的制备
将8g(38.0mmol)的化合物1-1与400mL的乙酸(AcOH)放入容器中,室温下搅拌所得的混合物10分钟,接着混合400mL的乙酸和20mL的HNO3并向其中缓慢添加。1小时后,反应结束,接着将生成的产物冷却至室温,并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到5.8g(53%)的目标化合物1-2。
化合物1-3的制备
将6g(21.0mmol)的化合物1-2、300mL的乙醇以及3.6g(65.1mmol)的铁(Fe)粉放入容器中,并在室温下搅拌所得的混合物10分钟。向其中缓慢滴加30mL的乙酸,接着使所得的混合物在60℃下回流1小时。反应结束后,将反应产物冷却至室温,且接着过滤通过向其中添加H2O而产生的固体并接着用H2O和己烷洗涤得到5.3g(99%)的目标化合物1-3。
化合物1-4的制备
将3.9mL的HCOH以及9.77mL的乙酸放入容器中,在60℃下搅拌2小时,接着冷却至室温。之后,向其中添加360mL的乙醚以及12g(46.9mmol)的化合物1-3,且在室温下搅拌所得的混合物。1小时后,过滤所得的固体并用乙醚洗涤得到6.5g(49%)的目标化合物1-4。
化合物1-5的制备
将6.5g(22.94mmol)的化合物1-4、0.43mL(4.59mmol)的POCl3以及30mL的硝基苯放入容器中,将所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加3.76mL(32.12mmol)的SnCl4。在将所得的混合物在回流下搅拌2小时后,结束该反应,接着将所得的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇洗涤所得的产物接着纯化得到2.74g(45%)的目标化合物1-5。
化合物1-6的制备
使4.0g(15.08mmol)的化合物1-5以及3.65g(45.22mmol)HBr与50mL的H2O一起回流1小时,并将所得的混合物冷却至室温,接着用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇洗涤所得的产物接着纯化得到3.49g(92%)的目标化合物1-6。
化合物1-7的制备
使5.0g(19.89mmol)的化合物1-6以及2.0g(19.89mmol)的三乙胺放入容器中,在室温下搅拌所得的混合物约1小时,接着向其中逐滴缓慢滴加4.18g(19.89mmol)的Tf2O。在将该所得的混合物回流下搅拌2小时后,结束反应,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇洗涤生成的产物接着纯化得到6.71g(88%)的目标化合物1-7。
化合物1的制备
使5.0g(13.04mmol)的化合物1-7、4.77g(14.35mmol)的11-苯基-11,12-二氢吲哚[2,3-a]咔唑、0.60g(0.65mmol)的Pd2(dba)3、0.75g(1.30mmol)的4,5-双二苯基膦-9,9-二甲基氧杂蒽以及5.28g(26.08mmol)的NaOtBu与80mL的甲苯一起在130℃下回流3小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着将所得的混合物完全溶解在甲苯中,并用硅胶过滤所得的溶液。之后,用热甲苯过滤产物并纯化得到5.9g(80%)的化合物1。
<制备实施例2>化合物17的制备
化合物2-1的制备
使10g(26.09mmol)的化合物1-7、6.29g(31.31mmol)的(3-溴苯基)硼酸、1.5g(1.3mmol)的Pd(PPh3)4以及5.53g(52.18mmol)的Na2CO3与200mL的甲苯、40mL的乙醇以及40mL的H2O一起在120℃下回流6小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到6.82g(67%)的目标化合物3-1。
化合物17的制备
使5.0g(12.81mmol)的化合物2-1、4.18g(15.37mmol)的苯并菲-2-基硼酸、0.74g(0.64mmol)的Pd(PPh3)4以及2.71g(25.62mmol)的Na2CO3与100mL的甲苯、20mL的乙醇以及20mL的H2O一起在120℃下回流4小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到6.06g(88%)的目标化合物17。
<制备实施例3>化合物29的制备
化合物A-1的制备
在氮气下在单颈圆底烧瓶中,将1.4mL(0.0374mol)的硫酸逐滴缓慢滴加到30.0g(0.374mol)的1,2-二环己酮和77.37g(0.749mol)的盐酸苯肼在1,000mL的乙醇中的混合物中,接着在60℃下搅拌所得的混合物4小时。过滤冷却至室温的溶液得到黄棕色固体(69g,93%)。在单颈圆底烧瓶中将46.5mL(0.6mol)的三氟乙酸放入68.9g(0.25mol)的该固体和700ml的乙酸的混合物中,在100℃下搅拌所得的混合物12小时。用乙酸和己烷洗涤并过滤冷却至室温的溶液得到乳白色的固体A-1(27.3g,42%)。
化合物A-2的制备
在氮气下在二颈圆底烧瓶中回流下搅拌2.1g(0.0082mol)的化合物A-1、2.5g(0.013mol)的碘苯、0.312g(0.0049mol)的Cu、0.433g(0.0016mol)的18-冠-6-醚以及3.397g(0.0246mol)的K2CO3在20ml的邻-二氯苯(o-DCB)中的混合物16小时。用二氯甲烷/H2O萃取并浓缩,通过柱层析(SiO2,己烷:乙酸乙酯=10:1)分离冷却至室温的溶液得到白色固体化合物A-2(1.76g,64%)。
化合物3-1的制备
使30g(168.5mmol)的苯并[b]噻吩-2-基硼酸、41.2g(202.26mmol)碘苯、19.0g(16.9mmol)的Pd(PPh3)4以及35.7g(336.9mmol)的Na2CO3与300mL的甲苯、120mL的乙醇(EtOH)以及120mL的H2O一起在120℃下回流1小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到18.0g(51%)的目标化合物3-1。
化合物3-2的制备
将10g(47.55mmol)的化合物3-1和400mL的乙酸放入容器中,室温下搅拌所得的混合物10分钟,接着混合400mL的乙酸和20mL的HNO3并向其中缓慢添加。1小时后,反应结束,接着将生成的产物冷却至室温,并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到5.9g(50%)的目标化合物3-2。
化合物3-3的制备
将6g(23.5mmol)化合物3-2、300mL乙醇以及4.06g(72.8mmol)铁(Fe)放入容器中,并在室温下搅拌所得的混合物10分钟。向其中缓慢滴加30mL的乙酸,接着使所得的混合物回流1小时。反应结束后,将反应产物冷却至室温,且接着过滤通过向其中添加H2O而产生的固体并接着用H2O和己烷洗涤得到5.5g(99%)的目标化合物3-3。
化合物3-4的制备
使2.93mL(22.19mmol)的4-溴苯酰氯完全溶解在30mL的二氯甲烷(MC)中,接着向其中添加3.12mL(22.19mmol)的三乙胺(TEA),在室温下搅拌所得的混合物15分钟后接着将其保持在0℃,向其中缓慢添加2.93mL(22.19mmol)的化合物3-3。约1小时后,产生白色固体并过滤,且接着用己烷洗涤生成的产物并干燥得到8.0g(87%)的目标化合物3-4。
化合物3-5的制备
将6.5g(22.94mmol)的化合物3-4、0.43mL(4.59mmol)的POCl3以及30mL的硝基苯放入容器中,使所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加3.76mL(32.12mmol)的SnCl4。在回流下搅拌所得的混合物2小时后,反应结束,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到2.74g(45%)的目标化合物3-5。
化合物29的制备
使8g(20.5mmol)的化合物3-5、6.1g(18.44mmol)的化合物A-2、0.38g(0.41mmol)的Pd2(dba)3、0.47g(0.82mmol)的4,5-双二苯基膦-9,9-二甲基氧杂蒽以及8.3g(41.0mmol)的NaOtBu与80mL的甲苯一起在130℃下回流3小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着将所得的混合物完全溶解在甲苯中,并用硅胶过滤所得的溶液。之后,用热甲苯过滤并纯化产物得到8.8g(67%)的化合物29。
<制备实施例4>化合物42的制备
化合物4-1的制备
使2.93mL(22.19mmol)的3-溴苯酰氯完全溶解在30mL的二氯甲烷(MC)中,接着向其中添加3.12mL(22.19mmol)的三乙胺(TEA),在室温下搅拌所得的混合物15分钟后接着将其保持在0℃,向其中缓慢添加2.93mL(22.19mmol)的化合物3-3。约1小时后,产生白色固体并过滤,且接着用己烷洗涤生成的产物并干燥得到8.0g(87%)的目标化合物4-1。
化合物4-2的制备
将8.0g(19.59mmol)的化合物4-1、1.8mL(19.59mmol)的POCl3以及80mL的硝基苯放入容器中,使所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加4.5mL(54.85mmol)的SnCl4。在回流下搅拌所得的混合物2小时后,反应结束,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着用甲醇洗涤并接着纯化生成的产物得到5.03g(66%)的目标化合物4-2。
化合物42的制备
使5g(12.81mmol)的化合物4-2、3.5g(15.37mmol)二苯并[b,d]噻吩-4-基硼酸、0.74g(0.64mmol)的Pd(PPh3)4以及3.5g(25.62mmol)的K2CO3与50mL的甲苯、5mL的乙醇以及5mL的H2O一起在120℃下回流5小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到5.56g(88%)的化合物42。
<制备实施例5>化合物48的制备
化合物5-1的制备
将化合物4-2(10g,25.62mmol)溶解在100mL的THF中,接着将所得的溶液冷却至-78℃。向其中缓慢滴加正丁基锂(2.5M,在己烷中)(13.3ml,33.31mmol),接着搅拌所得的混合物1小时。向溶液中逐滴添加二苯基氯化膦(5.65ml,25.62mol),并在室温下搅拌所得的溶液12小时。用MC/H2O萃取反应混合物,接着减压蒸馏。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到4.19g(33%)的化合物5-1。
化合物48的制备
使5g(10.09mmol)的化合物5-1完全溶解在50mL的二氯甲烷(MC)中,接着在室温下将所得的混合物与10mL的H2O2水溶液(30重量%)一起搅拌1小时。用MC/H2O萃取反应混合物,接着减压蒸馏。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到1.14g(22%)的化合物48。
<制备实施例6>化合物74的制备
化合物6-1的制备
使20g(112.34mmol)的苯并[b]噻吩-2-基硼酸、20.4g(101.11mmol)的2-溴苯胺、6.5g(5.12mmol)的Pd(PPh3)4、以及31.05g(224.68mmol)的K2CO3与200mL的甲苯、40mL的乙醇以及40mL的H2O一起在120℃下回流16小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到16.9g(67%)的化合物6-1。
化合物6-2的制备
将10g(44.38mmol)的化合物6-1完全溶解在二氯甲烷(MC)中,接着在室温下将所得的溶液与6.2mL(44.38mmol)的三乙胺(TEA)一起搅拌15分钟。之后,将所述溶液保持在0℃,接着向其中缓慢添加9.7g(44.38mmol)的4-溴苯酰氯。约1小时后,生成白色固体并过滤,接着用EA和己烷洗涤生成的产物得到17.2g(95%)的目标化合物6-2。
化合物6-3的制备
将15g(36.74mmol)的化合物6-2、3.4mL(36.74mmol)的POCl3以及150mL的硝基苯放入容器中,将所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加12.04mL(102.87mmol)的SnCl4。在将所得的混合物在回流下搅拌2小时后,结束该反应,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇(MeOH)和己烷洗涤生成的产物得到6.17g(43%)的目标化合物6-3。
化合物74的制备
使5g(12.81mmol)的化合物6-3、2.38g(14.09mmol)的二苯胺、0.74g(0.64mmol)的Pd(PPh3)4、以及3.5g(25.62mmol)的K2CO3与50mL的甲苯、5mL的乙醇以及5mL的H2O一起在120℃下回流6小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到4.72g(77%)的化合物74。
<制备实施例7>化合物85的制备
使5g(12.81mmol)的化合物6-3、6.37g(14.09mmol)的(3,5-二(9H-咔唑-9-基)苯基)硼酸、0.74g(0.64mmol)的Pd(PPh3)4、以及3.5g(25.62mmol)的K2CO3与50mL的甲苯、5mL的乙醇以及5mL的H2O一起在120℃下回流6小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到6.4g(69%)的化合物85。
<制备实施例8>化合物86的制备
化合物7-1的制备
将10g(44.38mmol)的化合物6-1完全溶解在二氯甲烷(MC)中,接着在室温下将所得的溶液与6.2mL(44.38mmol)的三乙胺(TEA)一起搅拌15分钟。之后,将所述溶液保持在0℃,接着向其中缓慢添加9.7g(44.38mmol)的4-溴苯酰氯。约1小时后,生成白色固体并过滤,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到17.2g(95%)的目标化合物7-1。
化合物7-2的制备
将15g(36.74mmol)的化合物7-1、3.4mL(36.74mmol)的POCl3以及150mL的硝基苯放入容器中,将所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加12.04mL(102.87mmol)的SnCl4。在将所得的混合物在回流下搅拌2小时后,结束该反应,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇(MeOH)和己烷洗涤生成的产物得到7.89g(55%)的目标化合物7-2。
化合物86的制备
使8g(20.5mmol)的化合物7-2、6.1g(18.44mmol)的11-苯基-11,12-二氢吲哚[2,3-a]咔唑、0.38g(0.41mmol)的Pd2(dba)3、0.47g(0.82mmol)的4,5-双二苯基膦-9,9-二甲基氧杂蒽(XantPhos)以及8.3g(41.0mmol)的NaOtBu与80mL的甲苯一起在130℃下回流3小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着将所得的混合物完全溶解在甲苯中,并用硅胶过滤所得的溶液。之后,用热甲苯过滤并纯化产物得到7.3g(56%)的化合物86。
<制备实施例9>化合物87的制备
使5g(12.81mmol)的化合物7-2、3.5g(15.37mmol)的二苯并[b,d]噻吩-4-基硼酸、0.74g(0.64mmol)的Pd(PPh3)4以及3.5g(25.62mmol)的K2CO3与50mL的甲苯、5mL的乙醇以及5mL的H2O一起在120℃下回流5小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到4.80g(76%)的化合物87。
<制备实施例10>化合物88的制备
使5g(12.81mmol)的化合物7-2、5.7g(12.81mmol)的2-(9,9-二苯基-9H-芴-2-基)-4,4,5,5-四甲基-1,3,2-二氧硼戊烷、0.74g(0.64mmol)的Pd(PPh3)4以及3.5g(25.62mmol)的K2CO3与100mL的甲苯、20mL的乙醇以及20mL的H2O一起在120℃下回流24小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和MC萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到6.0g(74%)的化合物88。
<制备实施例11>化合物90的制备
使5g(12.81mmol)的化合物7-2、5.9g(12.81mmol)的三苯基(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)硅烷、0.74g(0.64mmol)的Pd(PPh3)4以及3.5g(25.62mmol)的K2CO3与100mL的甲苯、20mL的乙醇以及20mL的H2O一起在120℃下回流24小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和MC萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到6.5g(78%)的化合物90。
<制备实施例12>化合物91的制备
使5g(12.81mmol)的化合物7-2、5.6g(12.81mmol)的9-(3-(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)苯基)-9H-咔唑、0.74g(0.64mmol)的Pd(PPh3)4以及3.5g(25.62mmol)的K2CO3与100mL的甲苯、20mL的乙醇以及20mL的H2O一起在120℃下回流24小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和MC萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到5.0g(70%)的化合物91。
<制备实施例13>化合物92的制备
化合物7-3的制备
使10g(25.6mmol)的化合物7-2、13.0g(51.2mmol)的双(频那醇)二硼、0.93g(1.3mmol)的PdCl2(dppf)以及7.5g(51.2mmol)的KOAc与200mL的DMF一起在130℃下回流4小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和MC萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到7.5g(67%)的化合物7-3。
化合物92的制备
使7.5g(17.1mmol)的化合物7-3、8.35g(17.1mmol)的9,9'-(5-溴-1,3-亚苯基)双(9H-咔唑)、1.0g(0.85mmol)的Pd(PPh3)4以及4.7g(34.0mmol)的K2CO3与200mL的甲苯、40mL的乙醇以及40mL的H2O一起在100℃下回流24小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和MC萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到9.0g(73%)的化合物92。
<制备实施例14>化合物93的制备
使6.15g(14.1mmol)的化合物7-3、5.3g(16.9mmol)的2-溴-4,6-二苯基嘧啶、0.81g(0.70mmol)的Pd(PPh3)4以及3.9g(28.1mmol)的K2CO3与100mL的甲苯、20mL的乙醇以及20mL的H2O一起在120℃下回流24小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和MC萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到7.3g(96%)的化合物93。
<制备实施例15>化合物110的制备
化合物8-1的制备
将10g(46.93mmol)的化合物3-溴苯并[b]噻吩以及400mL的乙酸放入容器中,室温下搅拌所得的混合物10分钟,接着混合400mL的乙酸和20mL的HNO3并向其中缓慢添加,1小时后,反应结束,接着将生成的产物冷却至室温,并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到9.34g(77%)的目标化合物8-1。
化合物8-2的制备
将9g(34.87mmol)的化合物8-1、300mL的乙醇以及6.03g(108.1mmol)的铁(Fe)粉放入容器中,并在室温下搅拌所得的混合物10分钟。向其中缓慢滴加30mL的乙酸,接着使所得的混合物在60℃下回流1小时。反应结束后,将反应产物冷却至室温,且接着过滤并接着用H2O和己烷洗涤通过向反应产物中添加H2O而产生的固体得到7.9g(99%)的目标化合物8-2。
化合物8-3的制备
将10g(43.84mmol)的化合物8-2、5.87g(48.22mmol)的苯基硼酸、4.27g(3.69mmol)的Pd(PPh3)4以及9.29g(87.66mmol)的Na2CO3与100mL的甲苯、20mL的乙醇以及20mL的H2O一起在120℃下回流1小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到9.08g(92%)的目标化合物8-3。
化合物8-4的制备
将9g(39.94mmol)的化合物8-3完全溶解在二氯甲烷(MC)中,接着在室温下将所得的溶液与5.6mL(39.94mmol)的TEA一起搅拌15分钟。之后,将所述溶液保持在0℃,接着向其中缓慢添加8.77g(39.94mmol)的4-溴苯酰氯。约1小时后,生成白色固体并过滤,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到15.0g(92%)的目标化合物8-4。
化合物8-5的制备
将15g(36.74mmol)的化合物8-4、3.4mL(36.74mmol)的POCl3以及150mL的硝基苯放入容器中,将所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加12.04mL(102.87mmol)的SnCl4。在将所得的混合物在回流下搅拌2小时后,结束该反应,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇(MeOH)和己烷洗涤生成的产物得到11.1g(77%)的目标化合物8-5。
化合物110的制备
将5g(12.81mmol)的化合物8-5、6.37g(14.09mmol)的(3,5-二(9H-咔唑-9-基)苯基)硼酸、0.74g(0.64mmol)的Pd(PPh3)4以及3.5g(25.62mmol)的K2CO3与50mL的甲苯、5mL的乙醇以及5mL的H2O一起在120℃下回流6小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到7.1g(77%)的化合物110。
<制备实施例16>化合物119的制备
化合物9-1的制备
将10g(44.38mmol)的化合物8-3完全溶解在二氯甲烷(MC)中,接着在室温下将所得的溶液与6.2mL(44.38mmol)的TEA一起搅拌15分钟。之后,将所述溶液保持在0℃,接着向其中缓慢添加9.7g(44.38mmol)的3-溴苯酰氯。约1小时后,生成白色固体并过滤,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到17.7g(98%)的目标化合物9-1。
化合物9-2的制备
将15g(36.74mmol)的化合物9-1、3.4mL(36.74mmol)的POCl3以及150mL的硝基苯放入容器中,使所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加12.04mL(102.87mmol)的SnCl4。在回流下搅拌所得的混合物2小时后,反应结束,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着用甲醇(MeOH)和己烷洗涤生成的产物得到9.18g(64%)的目标化合物9-2。
化合物119的制备
使8g(20.5mmol)的化合物9-2、6.1g(18.44mmol)的化合物A-2、0.38g(0.41mmol)的Pd2(dba)3、0.47g(0.82mmol)的4,5-双二苯基膦-9,9-二甲基氧杂蒽以及8.3g(41.0mmol)的NaOtBu与80mL的甲苯一起在130℃下回流3小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到6.8g(52%)的目标化合物119。
<制备实施例17>化合物156的制备
化合物10-1的制备
使20g(112.34mmol)的苯并[b]噻吩-3-基硼酸、20.4g(101.11mmol)的2-溴苯胺、6.5g(5.12mmol)的Pd(PPh3)4以及31.05g(224.68mmol)的K2CO3与200mL的甲苯、40mL的乙醇以及40mL的H2O一起在120℃下回流16小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,且接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到15.1g(60%)的目标化合物10-1。
化合物10-2的制备
使10g(44.38mmol)的化合物10-1完全溶解在二氯甲烷(MC)中,接着在室温下将所得的溶液与6.2mL(44.38mmol)的三乙胺(TEA)一起搅拌15分钟。之后,将所述溶液保持在0℃,接着向其中缓慢添加9.7g(44.38mmol)的4-溴苯酰氯。约1小时后,生成白色固体并过滤,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到15.9g(88%)的目标化合物10-2。
化合物10-3的制备
将15g(36.74mmol)的化合物10-2、3.4mL(36.74mmol)的POCl3以及150mL的硝基苯放入容器中,将所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加12.04mL(102.87mmol)的SnCl4。在将所得的混合物在回流下搅拌2小时后,结束该反应,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇(MeOH)和己烷洗涤生成的产物得到9.47g(66%)的目标化合物10-3。
化合物156的制备
使5g(12.81mmol)的化合物10-3、5.1g(15.37mmol)的9H-3,9'-二咔唑、0.74g(0.64mmol)的Pd(PPh3)4、以及3.5g(25.62mmol)的K2CO3与50mL的甲苯、5mL的乙醇以及5mL的H2O一起在120℃下回流5小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到6.3g(77%)的化合物156。
<制备实施例18>化合物185的制备
化合物11-1的制备
使10g(44.38mmol)的化合物10-1完全溶解在二氯甲烷(MC)中,接着在室温下将所得的溶液与6.2mL(44.38mmol)的三乙胺(TEA)一起搅拌15分钟。之后,将所述溶液保持在0℃,接着向其中缓慢添加9.7g(44.38mmol)的3-溴苯酰氯。约1小时后,生成白色固体并过滤,接着用乙酸乙酯(EA)和己烷洗涤生成的产物得到17.3g(96%)的目标化合物11-1。
化合物11-2的制备
将15g(36.74mmol)的化合物11-1、3.4mL(36.74mmol)的POCl3以及150mL的硝基苯放入容器中,将所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加12.04mL(102.87mmol)的SnCl4。在将所得的混合物在回流下搅拌2小时后,结束该反应,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇(MeOH)和己烷洗涤生成的产物得到7.89g(55%)的目标化合物11-2。
化合物185的制备
使5g(12.81mmol)的化合物11-2、3.5g(15.37mmol)的(4,6-二苯基-1,3,5-三嗪-2-基)硼酸、0.74g(0.64mmol)的Pd(PPh3)4、以及3.5g(25.62mmol)的K2CO3与50mL的甲苯、5mL的乙醇以及5mL的H2O一起在120℃下回流5小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到4.80g(69%)的化合物185。
<制备实施例19>化合物243的制备
化合物12-1的制备
使20g(123.49mmol)的化合物苯并呋喃-3-基硼酸、36.4g(123.49mmol)的4-溴-2-碘苯胺、14.2g(12.35mmol)的Pd(PPh3)4、以及51.2g(370.47mmol)的K2CO3与400mL的甲苯、80mL的乙醇以及80mL的H2O一起在120℃下回流24小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到28.8g(81%)的化合物12-1。
化合物12-2的制备
将3.9mL的HCOH以及9.77mL的乙酸放入容器中,并在60℃下搅拌该所得的混合物2小时,接着冷却至室温。之后,向其中添加360mL的乙醚和13.5g(46.9mmol)的化合物12-1,并在室温下搅拌所得的混合物。1小时后,过滤并用乙醚洗涤生成的固体得到8.45g(57%)的目标化合物12-2。
化合物12-3的制备
将8.45g(26.7mmol)的化合物12-2、3.4mL(36.74mmol)的POCl3以及150mL的硝基苯放入容器中,将所得的混合物回流1小时并冷却至室温,接着向其中缓慢滴加12.04mL(102.87mmol)的SnCl4。在将所得的混合物在回流下搅拌2小时后,结束该反应,接着将生成的产物冷却至室温并用蒸馏水和二氯甲烷(MC)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着用甲醇(MeOH)和己烷洗涤生成的产物得到7.00g(88%)的目标化合物12-3。
化合物243的制备
使7g(23.48mmol)的化合物12-3、7.1g(23.48mmol)的(3-(二苯并[b,d]噻吩-4-基)苯基)硼酸、2.7g(2.35mmol)的Pd(PPh3)4、以及9.73g(70.44mmol)的K2CO3与150mL的甲苯、30mL的乙醇以及30mL的H2O一起在120℃下回流7小时。反应结束后,将反应产物冷却至室温,接着用蒸馏水和乙酸乙酯(EA)萃取。在用无水MgSO4干燥有机层后,通过旋转蒸发仪除去溶剂,接着使用二氯甲烷和己烷作为洗脱溶剂通过柱层析纯化生成的产物得到8.75g(78%)的化合物243。
<制备实施例20>化合物245的制备
化合物6-1的制备
使2-溴苯胺(50g,290mmol)、四(三苯基膦)钯(0)(16.75g,14.5mmol)以及碳酸氢钠(58.4g,696mmol)在甲苯/乙醇/水(1,000ml/200ml/200ml)中的混合物在单颈圆底烧瓶中在100℃下回流1小时。
将温度降低至80℃,向其中添加固体状态的苯并[b]噻吩-2-基硼酸(62g,348mmol),接着搅拌所得的混合物2小时。用MC萃取混合物,接着用MgSO4干燥有机层。浓缩后,通过柱层析(SiO2,己烷:二氯甲烷=1:1)分离该混合物(50g,76%)。
化合物6-2的制备
氮气下在单颈圆底烧瓶中将三乙胺(15.5ml,110mmol)添加到6-1(22.6g,100mmol)和四氢呋喃(400ml)的混合物中,接着搅拌所得的混合物10分钟。将温度降低至0℃,向其中添加4-溴苯酰氯(26.4g,120mmol)在四氢呋喃(100ml)中的混合物,接着搅拌所得的混合物30分钟。在用MC萃取该混合物后,浓缩有机层,接着向其中添加甲醇,超声处理并接着过滤所得的混合物(33g,81%)。
化合物6-3的制备
在充满氮气的单颈圆底烧瓶中将三氯氧磷(7.2ml,77.8mmol)添加到6-2(31.8g,77.8mmol)在硝基苯(320ml)中的混合物中,接着在150℃下搅拌所得的混合物2小时。在用0℃饱和碳酸氢钠水溶液终止反应物的反应,接着用二氯甲烷萃取生成的产物。浓缩后,除去硝基苯,接着向其中添加MeOH,并搅拌并接着过滤所得的混合物(26.6g,87%)。
化合物245-4的制备
在氮气下在单颈圆底烧瓶中使6-3(26.6g,68.15mmol)、频那醇二硼(pinacol diboron)(34.6g,136.3mmol)、PdCl2(dppf)(2.5g,3.4mmol)以及KOAc(20g,204mmol)在1,4-二噁烷(70ml)中的混合物在120℃下回流3小时。用二氯甲烷萃取生成的产物,接着用硫酸镁干燥有机层。浓缩后,通过柱层析(SiO2,己烷:二氯甲烷=1:4)分离该混合物。
化合物245的制备
在单颈圆底烧瓶中120℃下搅拌245-4(6g,13.7mmol)、2-氯-4,6-二苯基-1,3,5-三嗪(4g,15.09mmol)、Pd(PPh3)4(1.58g,1.37mmol)以及K2CO3(3.78g,27.4mmol)在1,4-二噁烷(120ml)/H2O(30ml)中的混合物3小时。在110℃的状态下过滤反应物,接着用1,4-二噁烷以及用H2O和MeOH洗涤生成的产物(6.4g,87%)。
<制备实施例21>化合物246的制备
除了使用化合物2-(4-溴苯基)-1-苯基-1H-苯并[d]咪唑代替化合物2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物246(10.1g,76%)。
<制备实施例22>化合物250的制备
除了使用化合物2-氯-4,6-二苯基嘧啶代替化合物2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物250(9.7g,78%)。
<制备实施例23>化合物248的制备
在氮气下在单颈圆底烧瓶中将10g(25.6mmol)的化合物6-3溶解在20ml的无水THF中,接着将所得的溶液冷却至-78℃。向其中缓慢滴加正丁基锂(2.5M,在己烷中)(10.2ml,25.6mmol),接着搅拌所得的混合物1小时。向该溶液中滴加二苯基氯化膦(4.7ml,25.6mmol),并在室温下搅拌所得的溶液12小时。用MC/H2O萃取反应混合物,接着减压蒸馏。将反应混合物溶解在MC(200ml)中,接着在室温下将所得的溶液与30%H2O2水溶液(10ml)一起搅拌1小时。用MC/H2O萃取反应混合物,接着通过柱层析(SiO2,MC:甲醇=25:1)分离浓缩的混合物得到固体化合物248(7.2g,54%)。
<制备实施例24>化合物253的制备
除了使用化合物4-([1,1'-联苯基]-4-基)-6-氯-2-苯基嘧啶代替化合物2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物253(11.5g,82%)。
<制备实施例25>化合物256的制备
除了使用化合物2-(4-溴苯基)-4,6-二苯基-1,3,5-三嗪代替化合物2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物256(10.2g,72%)。
<制备实施例26>化合物259的制备
除了使用化合物4-溴-2-苯基喹唑啉代替化合物2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物259(7.3g,62%)。
<制备实施例27>化合物260的制备
除了使用化合物2-溴-1,10-邻二氮杂菲代替化合物2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物260(7.7g,69%)。
<制备实施例28>化合物261的制备
化合物7-1的制备
在氮气下在单颈圆底烧瓶中将三乙胺(26ml,186mmol)添加到6-1(35g,155mmol)和四氢呋喃(600ml)的混合物中,接着搅拌所得的混合物10分钟。将温度降低至0℃,向其中添加3-溴苯酰氯(40.8g,186mmol)在四氢呋喃(150ml)中的混合物,接着搅拌所得的混合物30分钟。在用MC萃取该混合物后,浓缩有机层,接着向其中添加甲醇,超声处理并接着过滤所得的混合物(58g,91.7%)。
化合物7-2的制备
在充满氮气的单颈圆底烧瓶中将三氯氧磷(V)(12ml,127mmol)添加到7-1(52g,127mmol)在硝基苯(1,000ml)中的混合物中,接着在150℃下搅拌所得的混合物3小时。在0℃下用饱和碳酸氢钠水溶液终止反应物的反应,接着用二氯甲烷萃取生成的产物。浓缩后,除去硝基苯,接着向其中添加MeOH,并且搅拌并接着过滤所得的混合物(43g,86%)。
化合物7-3的制备
在氮气下在单颈圆底烧瓶中使7-2(43g,110mmol)、频那醇二硼(pinacol diboron)(56g,220mmol)、PdCl2(dppf)(4g,5.5mmol)以及KOAc(32.3g,330mmol)在1,4-二噁烷(400ml)中的混合物在120℃下回流3小时。用二氯甲烷萃取生成的产物,接着用硫酸镁干燥有机层。浓缩后,进行硅胶过滤,使混合物与MeOH搅拌接着过滤得到题述化合物(36g,74%)。
化合物261的制备
除了使用化合物7-3代替245-4以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物261(9.4g,76%)。
<制备实施例29>化合物274的制备
除了使用化合物4-([1,1'-联苯基]-4-基)-2-氯-6-苯基嘧啶代替化合物2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例28中化合物261的制备方式相同的方式进行制备得到目标化合物274(11.1g,79%)。
<制备实施例30>化合物278的制备
化合物10-1的制备
在单颈圆底烧瓶中使2-溴苯胺(100g,580mmol)、四(三苯基膦)钯(0)(33.5g,29mmol)以及碳酸氢钠(116.8g,1,392mmol)在甲苯/乙醇/水(2,000ml/400ml/400ml)中的混合物在100℃下回流1小时。
将温度降低至80℃,向其中添加固体状态的苯并[b]噻吩-3-基硼酸(124g,696mmol),接着搅拌所得的混合物3小时。用MC萃取混合物,接着用MgSO4干燥有机层。浓缩后,通过柱层析(SiO2,己烷:二氯甲烷=1:1)分离该混合物(100g,76%)。
化合物10-2的制备
氮气下在单颈圆底烧瓶中将三乙胺(100ml,707mmol)添加到10-1(145g,643mmol)和四氢呋喃(2,000ml)的混合物中,接着搅拌所得的混合物10分钟。将温度降低至0℃,向其中添加4-溴苯酰氯(155.3g,707.9mmol)在四氢呋喃(1,000ml)中的混合物,接着搅拌所得的混合物30分钟。在用MC萃取该混合物后,浓缩有机层,接着向其中添加甲醇,超声处理并接着过滤所得的混合物(220g,83%)。
化合物10-3的制备
在充满氮气的单颈圆底烧瓶中将三氯氧磷(55ml,592.6mmol)添加到10-2(220g,538.8mmol)在硝基苯(2,000ml)中的混合物中,接着在150℃下搅拌所得的混合物3小时。在0℃下用饱和碳酸氢钠水溶液终止反应物的反应,接着用二氯甲烷萃取生成的产物。浓缩后,除去硝基苯,接着向其中添加MeOH,并且搅拌并接着过滤产生的混合物(167g,80%)。
化合物278的制备
除了使用化合物10-3代替6-3以外,以与在制备实施例23中化合物248的制备方式相同的方式进行制备得到目标化合物278(8.6g,69%)。
<制备实施例31>化合物294的制备
化合物11-1的制备
氮气下在单颈圆底烧瓶中将三乙胺(26ml,186mmol)添加到10-1(35g,155mmol)和四氢呋喃(600ml)的混合物中,接着搅拌所得的混合物10分钟。将温度降低至0℃,向其中添加4-溴苯酰氯(40.8g,186mmol)在四氢呋喃(100ml)中的混合物,接着搅拌所得的混合物30分钟。在用MC萃取该混合物后,浓缩有机层,接着向其中添加甲醇,超声处理并接着过滤所得的混合物(58g,91%)。
化合物11-2的制备
在充满氮气的单颈圆底烧瓶中将三氯氧磷(V)(12ml,127mmol)添加到11-1(52g,127mmol)在硝基苯(1,000ml)中的混合物中,接着在150℃下搅拌所得的混合物2小时。在0℃下用饱和碳酸氢钠水溶液终止反应物的反应,接着用二氯甲烷萃取生成的产物。浓缩后,除去硝基苯,接着向其中添加MeOH,并且搅拌并接着过滤所得的混合物(43g,86%)。
化合物294-3的制备
在氮气下在单颈圆底烧瓶中使11-2(43g,110mmol)、频那醇二硼(pinacol diboron)(56g,220mmol)、PdCl2(dppf)(4g,5.5mmol)以及KOAc(32.3g,330mmol)在1,4-二噁烷(400ml)中的混合物在120℃下回流3小时。用二氯甲烷萃取生成的产物,接着用硫酸镁干燥有机层。浓缩后,进行硅胶过滤,浓缩后,使混合物与MeOH搅拌接着过滤得到题述化合物(36g,74%)。
化合物294的制备
除了使用化合物294-3以及4-溴-2,6-二苯基嘧啶代替245-4以及2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物294(9.8g,79%)。
<制备实施例32>化合物309的制备
化合物309-4的制备
除了使用化合物苯并呋喃-2-基硼酸代替苯并[b]噻吩-2-基硼酸以外,以与在制备实施例20中化合物245-4的制备方式相同的方式进行制备得到目标化合物309-4。
化合物309的制备
除了使用化合物309-4以及4-溴-2,6-二苯基嘧啶代替245-4以及2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物309(9.2g,73%)。
<制备实施例33>化合物321的制备
化合物321-3的制备
除了使用化合物309-1代替6-1以外,以与在制备实施例28中化合物270-3的制备方式相同的方式进行制备得到目标化合物321-3。
化合物321的制备
除了使用化合物321-3以及2-氯-4,6-二苯基-1,3,5-三嗪代替245-4以及2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物321(8.9g,71%)。
<制备实施例34>化合物343的制备
化合物343-3的制备
除了使用化合物343-1代替10-1以外,以与在制备实施例30中化合物10-3的制备方式相同的方式进行制备得到目标化合物343-3。
化合物343-4的制备
在氮气下在单颈圆底烧瓶中使343-3(21.5g,55mmol)、频那醇二硼(pinacol diboron)(28g,110mmol)、PdCl2(dppf)(2g,2.7mmol)以及KOAc(14g,165mmol)在1,4-二噁烷(200ml)中的混合物在120℃下回流4小时。用二氯甲烷萃取生成的产物,接着用硫酸镁干燥有机层。浓缩后,进行硅胶过滤,并在浓缩后,使生成的产物与MeOH搅拌接着过滤得到题述化合物(18g,73%)。
化合物343的制备
除了使用化合物343-4以及4-([1,1'-联苯基]-4-基)-2-氯喹唑啉代替245-4以及2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物343(9.3g,68%)。
<制备实施例35>化合物354的制备
化合物354-3的制备
除了使用化合物343-1代替10-1以外,以与在制备实施例31中化合物294-3的制备方式相同的方式进行制备得到目标化合物354-3。
化合物354的制备
除了使用化合物354-3以及2-(4-溴苯基)-1-苯基-1H-苯并[d]咪唑代替化合物245-4以及2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物354(10.2g,76%)。
<制备实施例36>化合物370的制备
化合物370-3的制备
除了使用化合物5-溴吡啶酰氯代替4-溴苯酰氯以外,以与在制备实施例32中化合物309-4的制备方式相同的方式进行制备得到目标化合物370-3。
化合物370的制备
除了使用化合物370-3以及2-氯-4,6-二苯基嘧啶代替化合物245-4以及2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物370(9.6g,77%)。
<制备实施例37>化合物373的制备
化合物373-3的制备
除了使用化合物6-1代替309-1以外,以与在制备实施例36中化合物370-3的制备方式相同的方式进行制备得到目标化合物373-3。
化合物373的制备
除了使用化合物373-3以及2-氯-4,6-二苯基嘧啶代替化合物245-4以及2-氯-4,6-二苯基-1,3,5-三嗪以外,以与在制备实施例20中化合物245的制备方式相同的方式进行制备得到目标化合物373(12.4g,87%)。
<制备实施例38>化合物419的制备
化合物419-1的制备
氮气下在单颈圆底烧瓶中将三乙胺(34ml,244mmol)添加到6-1(50g,221.9mmol)和四氢呋喃(800ml)的混合物中,接着搅拌所得的混合物10分钟。将温度降低至0℃,向其中添加3,5-二溴苯酰氯(100g,332.8mmol)在四氢呋喃(200ml)中的混合物,接着搅拌所得的混合物30分钟。在用MC萃取该混合物后,浓缩有机层,接着向其中添加甲醇,超声处理并接着过滤所得的混合物(107g,99%)。
化合物419-2的制备
在充满氮气的单颈圆底烧瓶中将三氯氧磷(V)(20ml,216mmol)添加到419-1(96g,197mmol)在硝基苯(2,000ml)中的混合物中,接着在150℃下搅拌所得的混合物3小时。在0℃下用饱和碳酸氢钠水溶液终止反应物的反应,接着向其中轻轻地添加二氯甲烷并接着向其中添加过量甲醇以固化产物,接着过滤该产物(60g,65%)。
化合物419的制备
在单颈圆底烧瓶中在120℃下搅拌419-2(7g,14.91mmol)、二苯并[b,d]噻吩-4-基硼酸(12.12g,37.29mmol)、Pd(PPh3)4(1.72g,1.49mmol)以及K2CO3(8.2g,59.64mmol)在1,4-二噁烷(100ml)/H2O(20ml)中的混合物并持续30小时。在110℃的状态下过滤反应物,接着在110℃下用1,4-二噁烷洗涤生成的产物(7.5g,74%)。
<制备实施例39>化合物426的制备
化合物426-1的制备
在单颈圆底烧瓶中在110℃下搅拌419-2(60g,127.8mmol)、2-苯基-1-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)苯基)-1H-苯并[d]咪唑(55.7g,140.6mmol)、Pd(PPh3)4(14.6g,12.7mmol)以及NaHCO3(21.4g,255.6mmol)在甲苯(1,000ml)/EtOH(200ml)/H2O(200ml)中的混合物并持续3小时。用MC萃取,接着浓缩并通过柱层析(SiO2,乙酸乙酯:二氯甲烷=1:20)分离反应物(47g,55%)。
化合物426的制备
在单颈圆底烧瓶中在110℃下搅拌426-1(10.7g,15.16mmol)、二苯并[b,d]呋喃-2-基硼酸(3.5g,16.67mmol)、Pd(PPh3)4(1.7g,1.5mmol)以及K2CO3(4.19g,30.32mmol)在1,4-二噁烷(100ml)/H2O(20ml)中的混合物并持续6小时。用MC萃取,接着浓缩并通过柱层析(SiO2,乙酸乙酯:二氯甲烷=1:20)分离反应物(8.3g,73%)。
<制备实施例40>化合物437的制备
化合物437-2的制备
除了使用化合物10-1代替6-1以外,以与在制备实施例38中化合物419-2的制备方式相同的方式进行制备得到目标化合物437-2。
化合物437的制备
除了使用化合物437-2以及二苯并[b,d]呋喃-4-基硼酸代替419-2以及二苯并[b,d]呋喃-2-基硼酸以外,以与在制备实施例38中化合物419的制备方式相同的方式进行制备得到目标化合物437(10.5g,76%)。
<制备实施例41>化合物454的制备
化合物454-1的制备
除了使用化合物437-2以及1-苯基-2-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)苯基)-1H-苯并[d]咪唑代替419-2以及2-苯基-1-(4-(4,4,5,5-四甲基-1,3,2-二氧硼戊烷-2-基)苯基)-1H-苯并[d]咪唑以外,以与在制备实施例39中化合物426-1的制备方式相同的方式进行制备得到目标化合物454-1。
化合物454的制备
在单颈圆底烧瓶中在110℃下搅拌454-1(11.9g,18mmol)、9H-咔唑(3.62g,21.6mmol)、Pd2(dba)3(1.6g,1.8mmol)、P(t-Bu)3(6ml,5.4mmol)以及NaOt-Bu(3.45g,36mmol)在甲苯(100ml)中的混合物并持续8小时。用MC萃取,接着浓缩并通过柱层析(SiO2,乙酸乙酯:二氯甲烷=1:20)分离反应物(11.5g,85%)。
<制备实施例42>化合物379的制备
除了使用化合物309-1代替6-1以外,以与在制备实施例38中化合物419的制备方式相同的方式进行制备得到目标化合物379。
<制备实施例43>化合物386的制备
除了使用化合物379-2代替419-2以外,以与在制备实施例39中化合物426的制备方式相同的方式进行制备得到目标化合物386。
<制备实施例44>化合物397的制备
除了使用化合物343-1代替10-1以外,以与在制备实施例40中化合物437的制备方式相同的方式进行制备得到目标化合物397。
<制备实施例45>化合物414的制备
除了使用化合物397-2代替437-2以外,以与在制备实施例41中化合物454的制备方式相同的方式进行制备得到目标化合物414。
以在制备实施例中相同的方式制备化合物,其合成确认结果示于表1至3中。
[表1]
[表2]
[表3]
<实验实施例1>化合物CV、UV以及PL的测量
通过采用NPB(HOMO=-5.5eV)作为参比材料并使用循环伏安(CV)测量装置(制造商:Princeton Applied Research,型号名称:Parstat2273)测量CV。
在常温下通过使用UV-可见光分光光度计(制造商:Perkin Elmer,型号名称:LS35)测量并通过使用四氢呋喃(THF)分析UV。
在常温下通过使用光谱仪(设备:Perkin Elmer,型号名称:LS55)测量并通过使用四氢呋喃(THF)分析PL。
图4图示化合物29的UV和PL的测量图。
图5和图6图示由测量化合物29的CV的结果导出的Eox值。
图7图示化合物42的UV和PL的测量图。
图8和图9图示由测量化合物42的CV的结果导出的Eox值。
图10图示化合物87的UV和PL的测量图。
图11和图12图示由测量化合物87的CV的结果导出的Eox值。
图13图示化合物88的UV和PL的测量图。
图14和图15图示由测量化合物88的CV的结果导出的Eox值。
图16图示化合物90的UV和PL的测量图。
图17和图18图示由测量化合物90的CV的结果导出的Eox值。
图19图示化合物91的UV和PL的测量图。
图20和图21图示由测量化合物91的CV的结果导出的Eox值。
图22图示化合物92的UV和PL的测量图。
图23和图24图示由测量化合物92的CV的结果导出的Eox值。
图25图示化合物93的UV和PL的测量图。
图26和图27图示由测量化合物93的CV的结果导出的Eox值。
图28图示由测量化合物73的CV的结果导出的Eox值。
图29图示化合物73的UVPL的测量图。
图30图示化合物85的LTPL的测量图。
图31图示化合物85的UVPL的测量图。
图32图示化合物86的LTPL的测量图。
图33图示化合物86的UVPL的测量图。
图34图示化合物87的LTPL的测量图。
图35图示化合物87的UVPL的测量图。
图36图示化合物88的LTPL的测量图。
图37图示化合物88的UVPL的测量图。
图38图示化合物90的LTPL的测量图。
图39图示化合物90的UVPL的测量图。
图40图示化合物91的LTPL的测量图。
图41图示化合物91的UVPL的测量图。
图42图示化合物92的LTPL的测量图。
图43图示化合物92的UVPL的测量图。
图44图示化合物93的LTPL的测量图。
图45图示化合物93的UVPL的测量图。
图46图示化合物245的LTPL的测量图。
图47图示化合物245的UVPL的测量图。
图48图示化合物246的LTPL的测量图。
图49图示化合物246的UVPL的测量图。
图50图示化合物250的LTPL的测量图。
图51图示化合物250的UVPL的测量图。
图52图示化合物253的LTPL的测量图。
图53图示化合物253的UVPL的测量图。
图54图示化合物259的LTPL的测量图。
图55图示化合物259的UVPL的测量图。
图56图示化合物260的LTPL的测量图。
图57图示化合物260的UVPL的测量图。
图58图示化合物409的LTPL的测量图。
图59图示化合物409的UVPL的测量图。
图60图示化合物420的LTPL的测量图。
图61图示化合物420的UVPL的测量图。
图62图示化合物425的LTPL的测量图。
图63图示化合物425的UVPL的测量图。
图64图示化合物427的LTPL的测量图。
图65图示化合物427的UVPL的测量图。
图66图示化合物434的LTPL的测量图。
图67图示化合物434的UVPL的测量图。
在图5、6、8、9、11、12、14、15、17、18、20、21、23、24以及26至28的曲线图中,y-轴表示电流(单位:A),以及x-轴表示电势(单位:V)。
在图4、7、10、13、16、19、22以及25的曲线图中,左曲线图(蓝色)表示UV吸收,而右曲线图(红色)表示PL发光值。在图4、7、10、13、16、19、22、25以及29至67的曲线图中,y-轴和x-轴各自表示强度和波长(单位:nm)。
此外,可通过以下等式确定化合物的最高占据分子轨道(HOMO)、最低未占分子轨道(LUMO)、以及带隙。
<等式>
Homo=-5.5-(Eox(待测的化合物)-Eox(NPB))eV
带隙(Homo-Lumo)=1240/UV吸收限
<实验实施例2>OLED器件的制造
<对比实施例1>
相继使用三氯乙烯、丙酮、乙醇以及蒸馏水超声洗涤由用于OLED的玻璃获得的透明电极ITO薄膜(康宁三星有限公司制造),每种洗涤5分钟,接着将所述ITO薄膜置于异丙醇中,保存,接着使用。
然后,将ITO基板布置在真空沉积设备的基板夹中,将以下4,4',4"-三(N,N-(2-萘基)-苯基氨基)三苯基胺(2-TNATA)放置在所述真空沉积设备的小格(cell)中。
随后,排空小室中的空气直到小室中的真空度达到10-6托,接着通过将电流施加到所述小格以使2-TNATA蒸发而使得具有厚度的空穴注入层沉积到所述ITO基板上。
通过将以下N,N'-双(α-萘基)-N,N'-二苯基-4,4'-二胺(NPB)放置在所述真空沉积设备的另一个小格(cell)中并向该小格施加电流使NPB蒸发而使得具有厚度的空穴传输层沉积到空穴注入层上。
如上所述形成空穴注入层和空穴传输层,接着在其上沉积具有以下结构的蓝光发光材料作为发光层。具体地,使所述蓝光发光主体材料H1真空沉积在真空沉积设备的一个小格上具有的厚度,并使蓝光发光掺杂剂材料D1以相对于主体材料5%的量在其上真空沉积。
随后,使作为电子传输层的具有以下结构式E1的化合物真空沉积为具有的厚度。
通过沉积氟化锂(LiF)(作为电子注入层)使其具有的厚度并且使得A1阴极具有的厚度来制造OLED器件。
同时,使用于制造OLED器件所需的所有有机化合物在10-6至10-8托下经历真空升华纯化以用于每种材料,并用于制造OLED。
<对比实施例2>
制造如对比实施例1中的器件结构,除了使用E2材料代替E1材料。
<对比实施例3>
通过以下方法制造有机电致发光器件。
用蒸馏水超声洗涤玻璃基板,其中在所述玻璃基板中薄薄地涂覆有具有厚度的ITO。当用蒸馏水洗涤结束时,用溶剂(如丙酮、甲醇以及异丙醇)超声洗涤该玻璃基板,干燥并接着在UV洗涤机中使用UV对其进行UVO处理5分钟。之后,将该基板转移至等离子洗涤机(PT)中,接着真空状态下对其进行针对ITO功函数的等离子体处理并除去残留膜,并且因此,将其转移至用于有机沉积的热沉积设备。
在如上制备的ITO透明电极(阳极)上形成空穴注入层4,4′,4″-三[2-萘基(苯基)氨基]三苯基胺(2-TNATA)以及空穴传输层N,N′-二(1-萘基)-N,N′-二苯基-(1,1′-联苯基)-4,4′-二胺(NPB),作为公用层(common layers)。
如下在所述公用层上热真空沉积发光层。通过使用4,4’-N,N’-二咔唑-联苯基(CBP)作为主体以及三(2-苯基吡啶)铱(Ir(ppy)3)作为绿色磷光掺杂剂使CBP掺杂有7%的量的Ir(ppy)3)而使得发光层沉积为具有的厚度。之后,使作为空穴阻挡层的BCP沉积为具有的厚度,并在其上使作为电子传输层的Alq3沉积为具有的厚度。最后,有机电致发光器件通过以下步骤制造:在电子传输层上沉积氟化锂(LiF)以具有的厚度来形成电子注入层,接着在该电子注入层上沉积铝(Al)阴极以具有的厚度来形成阴极。
同时,使用于制造OLED器件所需的所有有机化合物在10-6至10-8托下经历真空升华纯化以用于每种材料,并用于制造OLED。
<实施例1至98>
除了使用制备实施例20至45合成的化合物代替在形成对比实施例1和2中的电子传输层时使用的E1和E2以外,以对比实施例1和2中相同的方式制造有机电致发光器件。
对在对比实施例1和2以及实施例1至98中制造的每个有机电致发光器件,在700cd/m2的发光亮度下测定并评价驱动电压、效率、色坐标以及耐久性(使用寿命),结果示于以下表4中。
[表4]
从表4可知,与通过使用对比实施例1和2中使用的电子传输层材料E1和E2制造的器件相比,当通过使用本发明的实施例1中所使用的电子传输层材料制造器件时,器件的使用寿命延长并且其驱动电压和效率得到改善。
<实施例99至118>
除了使用制备实施例1至19合成的化合物代替在对比实施例3中发光层形成期间使用的主体CBP以外,以对比实施例3中相同的方式制造有机电致发光器件。
对于对比实施例3和实施例99至118中制造的每个有机电致发光器件,通过McScience公司制造的M7000测量电致发光(EL)性质,并基于其测量结果,当参比亮度为6,000cd/m2时,通过McScience公司制造的使用寿命测量设备(M6000)测量T90。结果示于以下表5中。
[表5]
从表5的结果可见,可知晓将本发明的化合物应用到发光层的有机发光器件具有比对比实施例3中的有机发光器件更低的驱动电压和更加改善的发光效率,并且其使用寿命也得到显著改善。
- 还没有人留言评论。精彩留言会获得点赞!