替诺福韦艾拉酚胺富马酸盐化合物及其药物组合物的制作方法
本发明属于医药领域,具体涉及替诺福韦艾拉酚胺富马酸盐的新晶型及其制备方法,以及含有所述替诺福韦艾拉酚胺富马酸盐化合物的药物组合物。
背景技术:
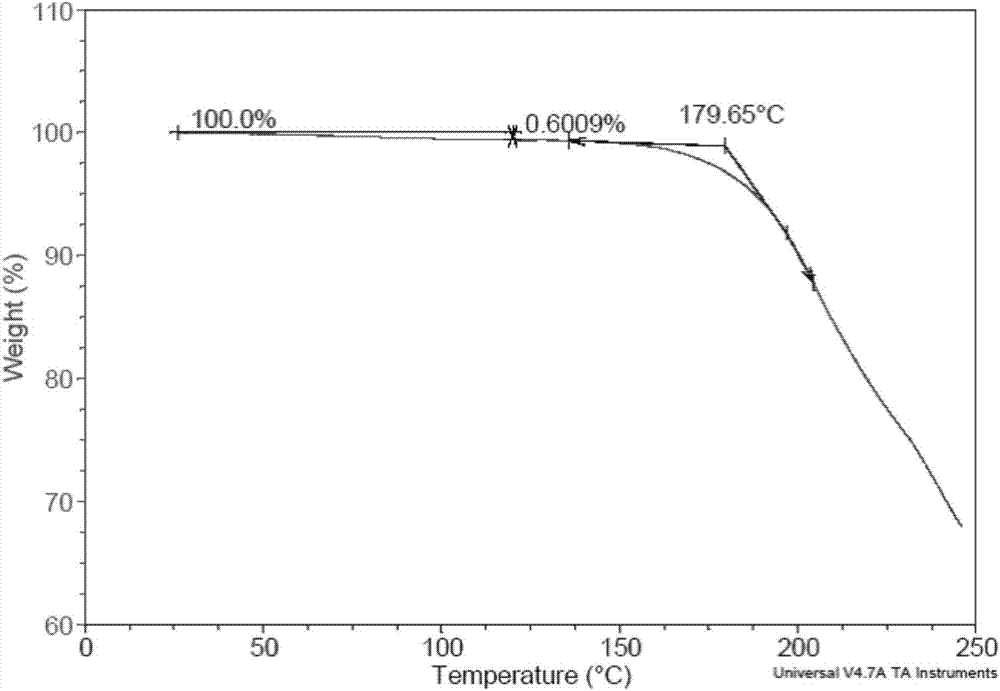
:替诺福韦艾拉酚胺(tenofoviralafenamide,结构式如下所示),化学名为9-[(r)-2-[[(s)-1-(异丙氧基羰基)乙基]氨基苯氧基氧膦基]甲氧基]丙基]腺嘌呤],是由美国gileadsciences公司研发的一种新型核苷酸类逆转录酶抑制剂。2015年在美国上市,用于治疗成人hiv感染。该药物也被用于治疗乙肝,目前处于iii期临床。本品口服后迅速转变成替诺福韦,在细胞激酶的作用下被磷酸化为替诺福韦二磷酸盐,通过竞争性地与天然脱氧核糖底物相结合抑制病毒聚合酶和插入到病毒dna后引起dna链延长终止,从而抑制hiv和hbv活性。cn1291994c公开了替诺福韦艾拉酚胺富马酸盐的制备方法,但没有公开任意相关晶型及数据。cn103732594a公开了替诺福韦艾拉酚胺半富马酸盐及其晶体形式。cn104558036a公开了替诺福韦艾拉酚胺半富马酸盐的一种晶体形式。目前尚没有针对替诺福韦艾拉酚胺富马酸盐的晶型和稳定性的相关研究。由于化合物的不同晶型可以在稳定性、物理性质等方面具有差异,直接影响到原料药和制剂的稳定性与生物利用度,因此开发具有稳定性和适用性的晶型具有重要意义。技术实现要素:本发明的目的在于提供一种式(i)所示的替诺福韦艾拉酚胺反富马酸盐的晶型,该晶型具有很好的稳定性,符合药用要求,并且制备方法简单,适合工业化生产。为了实现上述目的,本发明提供以下技术方案:本发明提供一种如式(i)所示的替诺福韦艾拉酚胺富马酸盐化合物,其特征在于,所述替诺福韦艾拉酚胺富马酸盐化合物为i型结晶形式,其使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射图谱在约6.724±0.2、8.347±0.2、10.785±0.2、16.068±0.2、17.446±0.2、18.150±0.2、20.028±0.2、20.606±0.2度处有特征峰,其中相对强度最大的峰为6.724±0.2度的特征峰。进一步,所述x-射线粉末衍射图谱在约9.584±0.2、11.829±0.2、12.010±0.2、13.627±0.2、21.192±0.2、21.469±0.2、22.930±0.2、24.855±0.2、26.356±0.2、27.598±0.2度处有特征峰。更进一步,上述i型结晶形式的替诺福韦艾拉酚胺富马酸盐的xrd图基本如图4所示,所述2θ角允许有±0.2度的误差。本发明还提供一种如式(i)所示的替诺福韦艾拉酚胺富马酸盐的i型结晶的方法,其特征在于,将替诺福韦艾拉酚胺富马酸盐化合物溶于第一溶剂,搅拌下加入第二溶剂,析晶,过滤,干燥,即得;所述第一溶剂选自甲醇、乙酸乙酯、乙腈、四氢呋喃;所述第二溶剂选自异丙醚、甲基叔丁基醚。例如,在一种示范性实施方式中,所述第一溶剂为甲醇,所述第二溶剂为异丙醚;或者所述第一溶剂为四氢呋喃,所述第二溶剂为甲基叔丁基醚;或者所述第一溶剂为乙腈,所述第二溶剂为异丙醚;或者所述第一溶剂为乙酸乙酯,所述第二溶剂为异丙醚;等等。替诺福韦艾拉酚胺富马酸盐与第一溶剂的比例为0.01~1g/ml,例如0.05~0.5g/ml,例如该范围的任意数值0.05g/ml,0.08g/ml,0.1g/ml,0.2g/ml,0.3g/ml,0.4g/ml等;第一溶剂与第二溶剂的体积比为1:(3~20),例如1:(4~15)或者1:(4~10)。所述化合物溶于第一溶剂和/或加入第二溶剂的步骤可以在室温或加热(例如50-70℃)进行,所述析晶可以是室温或低温(如-20℃~10℃)进行。所述干燥可以是室温或加热干燥,更优选室温真空干燥。本发明的还提供一种如式(i)所示的替诺福韦艾拉酚胺富马酸盐化合物,其特征在于,所述替诺福韦艾拉酚胺富马酸盐化合物为ii型结晶形式,其使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射图谱在约4.380±0.2、4.799±0.2、13.385±0.2、17.927±0.2、22.490±0.2、24.529±0.2、26.794±0.2、27.093±0.2度处有特征峰,其中相对强度最大的峰为4.380±0.2度的特征峰。进一步,所述x-射线粉末衍射图谱在约6.591±0.2、9.258±0.2、9.647±0.2、19.530±0.2、19.808±0.2、22.271±0.2、23.629±0.2、28.153±0.2、31.735±0.22度处有特征峰。更进一步,上述ii型结晶形式的替诺福韦艾拉酚胺富马酸盐的xrd图基本如图7所示,所述2θ角允许有±0.2度的误差。本发明还提供一种替诺福韦艾拉酚胺富马酸盐的ii型结晶形式的方法,包括将替诺福韦艾拉酚胺富马酸盐加入三氯甲烷中,室温搅拌,析晶,过滤,干燥,即得。本发明的还提供一种如式(i)所示的替诺福韦艾拉酚胺富马酸盐化合物,其特征在于,所述替诺福韦艾拉酚胺富马酸盐化合物为iii型结晶形式,其使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射图谱在约4.524±0.2、13.966±0.2、16.566±0.2、19.350±0.2、21.490±0.2、22.269±0.2、23.494±0.2、28.317±0.2、33.197±0.2度处有特征峰,其中相对强度最大的峰为23.494±0.2度的特征峰。进一步,所述x-射线粉末衍射图谱在约9.231±0.2、10.646±0.2、14.228±0.2、14.804±0.2、17.083±0.2、19.911±0.2、20.550±0.2、21.169±0.2、22.069±0.2、23.991±0.2、28.734±0.2、33.595±0.2、38.139±0.2度处有特征峰。更进一步,上述iii型结晶形式的替诺福韦艾拉酚胺富马酸盐的xrd图基本如图8所示,所述2θ角允许有±0.2度的误差。本发明还提供一种替诺福韦艾拉酚胺富马酸盐的iii型结晶形式的方法,包括将替诺福韦艾拉酚胺富马酸盐室温下置于三氟乙醇溶剂气氛中扩散1天,取出室温晾干得到。本发明还提供一种如式(i)所示的替诺福韦艾拉酚胺富马酸盐化合物,其特征在于,所述替诺福韦艾拉酚胺富马酸盐化合物为iv型结晶形式,其使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射图谱在约4.438±0.2、5.081±0.2、10.863±0.2、18.488±0.2、21.029±0.2、21.392±0.2、26.413±0.2、26.913±0.2度处有特征峰,其中相对强度最大的峰为5.081±0.2度的特征峰。进一步,所述x-射线粉末衍射图谱在约9.236±0.2、9.525±0.2、10.345±0.2、15.947±0.2、16.429±0.2、19.407±0.2、19.882±0.2、20.368±0.2、25.555±0.2度处有特征峰。更进一步,上述iv型结晶形式的替诺福韦艾拉酚胺富马酸盐的xrd图基本如图9所示,所述2θ角允许有±0.2度的误差。本发明还提供一种替诺福韦艾拉酚胺富马酸盐的iv型结晶形式的方法,包括将替诺福韦艾拉酚胺富马酸盐加入异丙醇中,室温搅拌,析晶,过滤,干燥,即得。本发明的第六方面提供一种如式(i)所示的替诺福韦艾拉酚胺富马酸盐化合物,其特征在于,所述替诺福韦艾拉酚胺富马酸盐化合物为v型结晶形式,其使用cu-kα辐射,以2θ角度表示的x-射线粉末衍射图谱在约4.880±0.2、8.783±0.2、9.321±0.2、9.904±0.2、20.029±0.2、25.150±0.2、25.653±0.2、30.273±0.2度处有特征峰,其中相对强度最大的峰为4.880±0.2度的特征峰。进一步,所述x-射线粉末衍射图谱在约10.549±0.2、14.100±0.2、15.784±0.2、16.083±0.2、17.969±0.2、18.370±0.2、20.652±0.2、22.013±0.2度处有特征峰。更进一步,上述v型结晶形式的替诺福韦艾拉酚胺富马酸盐的xrd图基本如图10所示,所述2θ角允许有±0.2度的误差。本发明还提供一种替诺福韦艾拉酚胺富马酸盐的v型结晶形式的方法,包括将替诺福韦艾拉酚胺富马酸盐室温下加入乙腈和三氯甲烷的混合溶剂中,0~5℃搅拌析晶,过滤,干燥,即得。乙腈和三氯甲烷的体积比为1:1。本发明还提供一种药物组合物,其含有上述晶体形式的替诺福韦艾拉酚胺富马酸盐化合物以及药学上可接受的载体。所述药物组合物可以制成任意剂型,优选口服制剂,例如片剂、胶囊等。本发明提供了替诺福韦艾拉酚胺富马酸盐的多种晶型,尤其是晶型i,其在高温高湿及溶液环境下,稳定性很好,适合制备各种剂型,并且该晶型的制备方法简单,适合工业化生产。附图说明图1是替诺福韦艾拉酚胺富马酸盐a晶型的xrd图。图2是替诺福韦艾拉酚胺富马酸盐a晶型的tga图。图3是替诺福韦艾拉酚胺富马酸盐a晶型的dsc图。图4是替诺福韦艾拉酚胺富马酸盐a晶型的dvs图。图5是替诺福韦艾拉酚胺富马酸盐i晶型的xrd图。图6是替诺福韦艾拉酚胺富马酸盐i晶型的tga图。图7是替诺福韦艾拉酚胺富马酸盐i晶型的dsc图。图8是替诺福韦艾拉酚胺富马酸盐i晶型的dvs图。图9是替诺福韦艾拉酚胺富马酸盐ii晶型的xrd图。图10是替诺福韦艾拉酚胺富马酸盐ii晶型的tga图。图11是替诺福韦艾拉酚胺富马酸盐ii晶型的dsc图。图12是替诺福韦艾拉酚胺富马酸盐iii晶型的xrd图。图13是替诺福韦艾拉酚胺富马酸盐iii晶型的tga图。图14是替诺福韦艾拉酚胺富马酸盐iii晶型的dsc图。图15是替诺福韦艾拉酚胺富马酸盐iv晶型的xrd图。图16是替诺福韦艾拉酚胺富马酸盐iv晶型的tga图。图17是替诺福韦艾拉酚胺富马酸盐iv晶型的dsc图。图18是替诺福韦艾拉酚胺富马酸盐v晶型的xrd图。图19是替诺福韦艾拉酚胺富马酸盐v晶型的tga图。图20是替诺福韦艾拉酚胺富马酸盐v晶型的dsc图。具体实施方式以下通过具体实施例详细说明本发明,然而本领域技术人员可以理解,下述实施例仅是解释说明的目的,而不以任何方式限制本发明的范围。替诺福韦艾拉酚胺富马酸盐原料可以按现有技术方法制备,可以是任意晶型。如无特别说明,实施例中的操作步骤均为常规操作室温,析晶时间可以是3小时至3天。测试方法:xrd测试:仪器型号:brukerd8advancediffractometer,靶:cu-kα(40kv,40ma),扫描范围:3°-40°(2θ值),步长:0.02°2θ,速度:0.2s.step-1dsc测试:仪器型号:tainstrumentsq200dsc,温度范围:20-200℃,扫描速率:10℃/min,氮气流速:40ml/mintga测试:仪器型号:tainstrumentsq500tga,温度范围:5-250℃,扫描速率:10℃/min,氮气流速:40ml/mindvs测试:仪器型号:tainstrumentsq5000tga,氮气流速:40ml/min,equilibrateat25℃;humidity0%;isothermalfor180min;abortnextisoifweight(%)<0.0100for15.00min预备例:替诺福韦艾拉酚胺富马酸盐的制备(参考zl01813161.1实施例4)将替诺福韦艾拉酚胺(5.2g),富马酸(1.15g),乙腈(100g)加入反应瓶中,加热到65-70℃使固体完全溶解,趁热过滤,冷却至5℃并保持16小时。过滤分离出产物,用乙腈洗涤,干燥得5.6g白色固体。经检测该固体为无水晶型,下称晶型a。此外,将100mg替诺福韦艾拉酚胺富马酸盐加入3ml乙醇中,搅拌下缓慢加入50ml异丙醚,析晶,离心过滤,真空干燥得到固体,仍为晶型a。使用cu-kα辐射测量的xrd图谱如图1所示,图谱数据列于下表1。tga检测显示,样品在100℃之前失重0.6%,为无水物,分解温度约为175℃(图2)。dsc检测显示样品的熔点约为122℃(图3)。dvs/等温吸附图显示,0%rh至80%rh范围内重量变化约为0.8%(图4)。表1.替诺福韦艾拉酚胺富马酸盐的a晶型的xrd图谱数据实施例1:替诺福韦艾拉酚胺富马酸盐的晶型i的制备方法1:取500mg替诺福韦艾拉酚胺富马酸盐,室温下加入10ml甲醇,搅拌下滴加200ml异丙醚,析出大量固体,离心过滤,室温真空干燥过夜得到。方法2:取500mg替诺福韦艾拉酚胺富马酸盐,室温下加入8ml甲基叔丁基醚,搅拌析晶,离心过滤,室温真空干燥得到。方法3:取500mg替诺福韦艾拉酚胺富马酸盐,加入15ml四氢呋喃中得到溶清液,将溶液加入到200ml甲基异丙醚中,析晶,离心过滤,室温真空干燥过夜得到。使用cu-kα辐射测量的xrd图谱如图5所示,图谱数据列于下表2。tga检测显示在120℃之前失重0.6%,样品为无水物,分解温度约为180℃(图6)。dsc检测显示样品熔点约为132℃(图7)。dvs/等温吸附图显示,0%rh至80%rh范围内重量变化约为0.4%(图8)。表2.替诺福韦艾拉酚胺富马酸盐的晶型i的xrd图谱数据序号2θd值相对强度序号2θd值相对强度16.72412.1353100.01720.0284.429727.528.34710.584329.91820.6064.306755.539.5849.220911.01921.1924.188911.9410.7858.196746.02021.4694.13565.2511.8297.47549.82122.3103.98155.1612.0107.36284.92222.9303.875312.6713.6276.49255.92323.0713.85185.8814.5106.09973.52423.9523.71215.3914.6666.03512.02524.3493.65257.31015.3045.78493.22624.6083.61475.11115.6715.65017.12724.8553.579412.21216.0685.511631.02825.3363.51245.81316.9255.23415.32926.3563.378713.91417.4465.079024.53026.7753.32693.91518.1504.883818.33127.5983.229411.11619.4034.57116.43228.4783.13176.3实施例2:替诺福韦艾拉酚胺富马酸盐的晶型ii的制备取500mg替诺福韦艾拉酚胺富马酸盐,室温下加入5ml三氯甲烷,室温磁力搅拌3天,离心,真空干燥过夜得到。使用cu-kα辐射测量的xrd图谱如图9所示,图谱数据列于下表3。tag检测显示在100℃之前失重3.2%,约合一个水分子(理论重量比例为3.0%),分解温度约为174℃,该晶型为一水合物(图10)。dsc检测显示样品在40℃~85℃之间有一宽吸收峰,90℃~105℃之间有一转晶放热峰,发生转晶(图11)。表3.替诺福韦艾拉酚胺富马酸盐的晶型ii的xrd图谱数据序号2θd值相对强度序号2θd值相对强度14.38020.1580100.01419.5304.541412.624.79918.399022.21519.8084.478511.636.59113.40013.01622.2713.988431.549.2589.54433.11722.4903.950160.159.6479.16047.21823.6293.76229.3610.7538.22053.21924.5293.626119.6712.2267.23332.42026.7943.324617.6813.3856.609417.02127.0933.288562.4914.1476.25532.32228.1533.167015.61015.2185.81732.72328.7703.10056.41116.6615.31642.52429.4933.02625.91217.9274.943825.82531.7352.817316.21318.4714.79952.52632.7142.73522.4实施例3:替诺福韦艾拉酚胺富马酸盐的晶型iii的制备取500mg替诺福韦艾拉酚胺富马酸盐,室温下置于三氟乙醇溶剂气氛中扩散1天,取出室温晾干2分钟得到。使用cu-kα辐射测量的xrd图谱如图12所示,图谱数据列于下表4。tga图谱显示在120℃之前约有14.0%的失重,约合一个三氟乙醇分子,分解温度约为171℃(图13)。dsc图谱显示,在50~105℃之间有一个脱溶剂吸热峰,熔点约为120℃(图14)。表4.替诺福韦艾拉酚胺富马酸盐的晶型iii的xrd图谱数据实施例4:替诺福韦艾拉酚胺富马酸盐的晶型iv的制备取500mg替诺福韦艾拉酚胺富马酸盐,室温下加入4ml异丙醇,室温搅拌3天,离心,室温真空干燥过夜,即得。使用cu-kα辐射测量的xrd图谱如图15所示,图谱数据列于下表5。tga图谱显示在120℃之前失重0.6%,样品为无水物,分解温度约为173℃(图16)。dsc检测显示样品的熔点约为122℃(图17)。表5.替诺福韦艾拉酚胺富马酸盐的晶型iv的xrd图谱数据序号2θd值相对强度序号2θd值相对强度14.43819.895220.01418.4884.795215.325.08117.3765100.01519.4074.57009.139.2369.56723.21619.8824.46185.049.5259.27794.31720.3684.35657.0510.3458.543911.21821.0294.221028.0610.8638.137720.51921.3924.150358.0712.1457.28155.02022.6503.92257.3814.2236.22184.92125.5553.48299.9915.2425.80844.72226.4133.371630.21015.6835.64585.82326.9133.310134.31115.9475.55299.92430.1162.96492.91216.4295.391110.12531.8542.80705.41317.9494.937911.42632.4352.75809.5实施例5:替诺福韦艾拉酚胺富马酸盐的晶型v的制备取500mg替诺福韦艾拉酚胺富马酸盐,室温下加入10ml乙腈和10ml三氯甲烷,4℃搅拌3天,离心过滤,室温真空干燥过夜,即得。使用cu-kα辐射测量的xrd图谱如图18所示,图谱数据列于下表6。tga图谱显示在120℃之前失重0.2%,样品为无水物,分解温度约为175℃(图19)。dsc图谱显示样品在90℃~112℃之间有一个熔融转晶峰,转晶后熔点约为121℃(图20)。表6.替诺福韦艾拉酚胺富马酸盐的晶型v的xrd图谱数据序号2θd值相对强度序号2θd值相对强度14.88018.0928100.01316.0835.50624.026.79013.00721.51417.9694.93253.537.36711.98983.51518.3704.82574.347.83311.27802.01620.0294.429544.258.78310.05996.51720.6524.29735.669.3219.479710.71822.0134.03464.579.9048.923522.11922.4473.95753.6810.5498.37944.42022.6313.92583.2913.3886.60792.92125.1503.537922.41014.1006.27594.02225.6533.46987.21114.8655.95462.12328.6503.11332.01215.7845.60987.92430.2732.94997.2实施例6:替诺福韦艾拉酚胺富马酸盐多晶型的稳定性考察1、高温干燥考察将上述实施例制备的替诺福韦艾拉酚胺富马酸盐的不同晶型放置于密封洁净的玻璃瓶中,置于60℃恒温干燥箱中,分别于0、5、10天取样检测晶型变化。结果见表7。表7.替诺福韦艾拉酚胺富马酸盐多晶型的高温试验晶型检测结果a60℃干燥放置10天晶型不变i60℃干燥放置10天晶型不变ii60℃干燥放置5天部分转为forma+formi的混晶,10天基本转为forma+formiiii60℃干燥放置5天即转为forma+formi,后发现放置不到1天即出现转晶iv60℃干燥放置5天转为晶型a+晶型iv60℃干燥放置5天部分转为forma+formi,10天基本转为forma+formi实验结果表明,在高温干燥条件下,晶型a和晶型i的稳定性很好,晶型ii、iii、iv和v的稳定性较差,容易转变为晶型a和晶型i的混晶;晶型iii的稳定性最差,不到1天即发生转晶。2、室温高湿考察将上述实施例制备的替诺福韦艾拉酚胺富马酸盐的不同晶型均匀分摊至敞口培养皿中,厚度≤5mm,置于室温(25℃左右),相对湿度rh为75±5%的恒温培养箱中,分别于0、5、10天取样检测晶型变化。结果见表8。表8.替诺福韦艾拉酚胺富马酸盐多晶型的高湿试验晶型检测结果a室温75%rh放置10天晶型不变i室温75%rh放置10天晶型不变ii室温75%rh放置5天晶型不变但峰强有变化,10天基本转为forma+formiiii室温75%rh放置5天晶型有明显变化,转为forma+formiiv室温75%rh放置5天转为forma+formiv室温75%rh放置10天晶型不变实验结果表明,在室温高湿条件下,晶型a、晶型i、晶型v的稳定性较好,晶型ii、iii和iv的稳定性较差,容易转变为晶型a和晶型i的混晶;晶型iii的稳定性最差,很短时间内即发生转晶。3、混合溶剂竞争实验考察将替诺福韦艾拉酚胺富马酸盐的晶型a、i和v样品分别取约200mg,在25℃条件下与20ml溶剂搅拌,对所得固体产物进行xrd表征。实验条件及结果见下表9。表9.晶型稳定性比较(室温竞争)实验结果表明,晶型a、晶型i稳定性较好,二者可以共存,晶型v的稳定性较差,容易转变为晶型a和晶型i的混晶。4、单一溶剂晶浆稳定性考察将替诺福韦艾拉酚胺富马酸盐的晶型a、晶型i样品分别取约200mg,加入相应溶剂得到混悬液,在25℃和40℃下搅拌3天,对所得固体产物进行xrd表征。实验条件及结果见下表10。表10.晶型稳定性比较(40℃考察)实验结果表明,在25℃条件下置于溶剂中,晶型a、晶型i的稳定性较好;然而在40℃条件下,晶型a会发生转晶,部分转为晶型i;综合常温和高温条件,晶型i的稳定性>晶型a的稳定性。综合本发明的上述实验结果,替诺福韦艾拉酚胺富马酸盐的晶型a和晶型i均为无水晶型,在室温干燥条件下都比较稳定,然而,晶型a在40℃在部分溶剂中会转为晶型a和晶型i的混晶,在高温条件下晶型i的稳定性大于晶型a。此外dvs考察结果表明,晶型i的吸湿性很小,要明显小于晶型a,尤其是对于固体制剂,吸湿后制剂更容易氧化、水解,霉变等,有关物质增加,影响产品的稳定性和安全性。因此综合考虑,晶型i的稳定性更高,尤其适合制备替诺福韦艾拉酚胺富马酸盐的药物组合物和制剂。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1