金属β‑内酰胺酶抑制剂环状胺基二硫代甲酸盐衍生物及其制备方法与流程
本发明属于药物化学领域,涉及潜在应用价值的金属β-内酰胺酶抑制剂环状胺基二硫代甲酸盐衍生物及其制备方法。
背景技术:
自抗生素用于临床治疗细菌感染以来,细菌耐药性问题日益严重。导致细菌耐药性产生的原因有多种,其中可水平传播的药物水解酶是细菌获得耐药性产生的重要原因。2009年在印度新德里发现了第一株产NDM-1型金属β-内酰胺酶的超级细菌,该细菌仅对多黏菌素等少数抗菌药物敏感,耐药谱广,治疗困难(The Lancet Infectious Diseases 2010,10,597.)。产NDM-1酶菌株自发现以来,目前已在全球40多个国家出现流行,严重威胁临床抗感染治疗。因此研发新的抗菌药物迫在眉睫,但是,先导化合物的寻找和临床治疗药物的修饰并不能一蹴而就。所以,寻找耐药菌株产生水解抗生素的酶抑制剂开始成为研究热点。
金属β-内酰胺酶属于碳青霉烯酶的一种,碳青霉烯酶按其活性位点差异可分为丝氨酸类β-内酰胺酶和金属β-内酰胺酶两大类;而按照Almber分类又可以将其分为A、B、C和D四大类。其中A和D类属于丝氨酸类β-内酰胺酶,而B类属于金属β-内酰胺酶。目前进入临床研究阶段的碳青霉烯酶抑制剂的只有针对丝氨酸酶类的而金属β-内酰胺酶抑制剂尚未见报道。
2014年,Gerard D.Wright等人报道从真菌中得到的一种天然的化合物aspergillomarasmine A(AMA),它可以快速、有效的抑制NMD-1和IMP型金属β-内酰胺酶的活性(Nature 2014,510,503.)。与碳青霉烯类抗生素美罗培南联合用药的实验结果证明AMA能够使产金属酶的菌株恢复对碳青霉烯类抗生素的敏感性。2015年,Sabiha Y.Essack等人,报道了两种锌离子螯合剂NOTA和DOTA,此两种螯合剂可以使产金属酶的碳青霉烯耐药株恢复对美罗培南的敏感性(Journal of Antimicrobial Chemotherapy 2015,70,1594.)。2015年,西北大学杨科武课题组报道了一系列巯基乙酸硫酯氨基酸衍生物,其对于金属β-内酰胺酶L1的IC50最小可以达到18nM,但是其体外活性实验表明该类化合物恢复耐药株对美罗培南敏感性的效果并不佳(Acs Medicinal Chemistry Letters 2015,6,660.)。
技术实现要素:
本发明目的在于提供系列氮杂环类衍生物——金属β-内酰胺酶抑制剂环状胺基二硫代甲酸盐衍生物,该类化合物可以使产金属β-内酰胺酶耐药株恢复对美罗培南的敏感性。另一目的在于提供其制备方法。
为实现本发明目的,技术方案如下:
该化合物具有以下结构通式1,2或3:
R为钠、甲基、正己烷基、正癸烷基。具体选如下化合物:
(1)1a:R=钠
(2)1b:R=甲基
(3)1c:R=正己烷基
(4)1d:R=正癸烷基
R为钠、甲基、正己烷基、正十二烷基。具体选如下化合物:
(1)2a:R=钠
(2)2b:R=甲基
(3)2c:R=正己烷基
(4)2d:R=正十二烷基
R为钠、甲基、正己烷基、正癸烷基。具体选如下化合物:
(1)3a:R=钠
(2)3b:R=甲基
(3)3c:R=正己烷基
(4)3d:R=正癸烷基
合成本发明金属β-内酰胺酶抑制剂(1至3系列)路线如下:
(a)室温下,以醇为溶剂,哌嗪与二硫化碳在无机碱条件下反应得到化合物1a;溶剂为甲醇、乙醇、异丙醇等。无机碱可以是氢氧化钠,碳酸钠,碳酸氢钠,磷酸钠等。哌嗪与二硫化碳、无机碱摩尔比为:1:2.0-5.0:2.0-5.0。
(b)室温下,以醇为溶剂,1,4,7-三氮杂环壬烷与二硫化碳在无机碱条件下反应得到化合物2a;溶剂为甲醇、乙醇、异丙醇等。无机碱可以是氢氧化钠,碳酸钠,碳酸氢钠,磷酸钠等。1,4,7-三氮杂环壬烷与二硫化碳、无机碱摩尔比为:1:3.0-6.0:3.0-6.0。
(c)室温下,以醇为溶剂,1,4,7,10-四氮杂环十二烷与二硫化碳在无机碱条件下反应得到化合物3a;溶剂为甲醇、乙醇、异丙醇等。无机碱可以是氢氧化钠,碳酸钠,碳酸氢钠,磷酸钠等。1,4,7,10-四氮杂环十二烷与二硫化碳、无机碱摩尔比为:1:4.0-8.0:4.0-8.0。
R为甲基、正己烷基、正癸烷基或正十二烷基。
(d)室温下,溶剂中,哌嗪与二硫化碳,卤代烷在无机碱条件下反应得到化合物1b-1d;溶剂为丙酮,甲醇,乙醇等,卤代烷为与R对应的溴代烷、碘代烷和氯代烷,无机碱可以是磷酸钠,氢氧化钠,碳酸钠,碳酸氢钠等。哌嗪、二硫化碳、卤代烷、无机碱摩尔比为:1:2.0-5.0:2.0-5.0:2.0-5.0。
(e)室温下,溶剂中,1,4,7-三氮杂环壬烷与二硫化碳,卤代烷在无机碱条件下反应得到化合物2b-2d;溶剂为丙酮,甲醇,乙醇等,卤代烷为与R对应的的溴代烷、碘代烷和氯代烷,无机碱可以是磷酸钠,氢氧化钠,碳酸钠,碳酸氢钠等。1,4,7-三氮杂环壬烷、二硫化碳、卤代烷、无机碱摩尔比为:1:3.0-6.0:3.0-6.0:3.0-6.0。
(f)室温下,溶剂中,1,4,7,10-四氮杂环十二烷与二硫化碳,卤代烷在无机碱条件下反应得到化合物3b-4d;溶剂为丙酮,甲醇,乙醇等,卤代烷为与R对应的溴代烷、碘代烷和氯代烷,无机碱可以是磷酸钠,氢氧化钠,碳酸钠,碳酸氢钠等。1,4,7,10-四氮杂环十二烷、二硫化碳、卤代烷、无机碱摩尔比为:1:4.0-8.0:4.0-8.0:4.0-8.0。
本发明成功合成了一系氮杂环类衍生物,该类化合物可以使产金属β-内酰胺酶耐药株恢复对美罗培南的敏感性,能将美罗培南的抑菌活性提高128倍。且红细胞溶血试验和细胞毒实验证实该类化合物毒性较低,对于目前耐药情况愈发严重的情况具有临床潜在应用价值,开发前景良好。
附图说明
图1为本发明化合物3a在体外对于红细胞溶血性的实验结果。
图2为本发明化合物3a在体外对于Hela细胞毒性的实验结果。
图3为本发明化合物3a在不同浓度下作用于Hela细胞24h后于荧光显微镜下的成像结果.(a–c)为阴性对照(不加药组);(d–f)为化合物3a(1000μg/mL);(g–i)为化合物3a(500μg/mL);和(j–l)为阳性对照组0.1%Triton X-100,图中比例尺为100nm。
具体实施方式
下面结合具体实施例,进一步阐述本发明。这些实施例仅用于说明本发明而不用于限制本发明要求保护的范围。
合成化合物表征使用的仪器:NMR谱使用瑞典Bruker DPX-400型超导核磁共振仪测定,TMS为内标;高分辨质谱使用Waters-Micromass公司Q-Tof质谱仪测定。
实施例1化合物1a的制备
室温下,取化合物无水哌啶(100mg,1.16mmol)和氢氧化钠(93mg,2.32mmol)置于10mL单口瓶内,加入2mL乙醇,室温搅拌,体系未全溶;加入二硫化碳(169μL,2.79mmol),体系变为金白色浑浊,约半个小时停止反应。减压蒸馏除去溶剂,加入适量乙酸乙酯,勺子刮下黏附在瓶壁上的固体,抽滤,再次用乙酸乙酯冲洗滤饼,真空干燥得到白色固体325mg,收率99%。
m.p.:>300℃;1H NMR(400MHz,D2O)δ4.38(s,1H).13C NMR(100MHz,D2O)δ209.13,50.21.
实施例2化合物2a的制备
室温下,取化合物1,4,7-三氮杂环壬烷于溶剂中,同实施例1化合物1a的制备,得到白色晶状无36mg,收率15%。
m.p.:174-176℃;1H NMR(400MHz,CDCl3)δ4.78–3.85(m,12H),2.64(s,9H).13C NMR(100MHz,CDCl3)δ201.42,29.71,20.49.HR-MS(ESI)Calcd for C12H21N3S6[M+Na]+:421.9957,found:421.9957.
实施例3化合物3a的制备
室温下,取化合物1,4,7,10-四氮杂环十二烷于溶剂中,同实施例1化合物1a的制备,得到白色固体327mg,收率99%
m.p.:278-280℃ 1H NMR(400MHz,D2O)δ4.70.13C NMR(100MHz,D2O)δ55.01.
实施例4化合物1b的制备
室温下,取无水哌啶(50mg,0.58mmol)和十二水磷酸钠(265mg,0.696mmol)置于25mL单口瓶内,加入2mL丙酮,二硫化碳(210μL,3.48mmol),该混合体系于室温下搅拌,体系白色浑浊;30min后,加入碘甲烷(83μL,1.34mmol),继续室温下搅拌,约5h后反应结束。加入少量水淬灭反应。抽滤,适量乙酸乙酯冲洗,静止分层,收集有机相,水反洗(10mL×1),饱和食盐水萃洗(10mL×1),收集有机相,无水硫酸钠静置干燥后浓缩,柱层析纯化(PE:EA=10:1),最终得到白色固体约50mg,收率30%。
m.p.:188-190℃;1H NMR(400MHz,CDCl3)δ4.30(s,8H),2.70(s,6H),1.27(s,1H).13C NMR(100MHz,CDCl3)δ199.14,29.71,20.05.HR-MS(ESI)Calcd for C8H14N2S4[M+Na]+:288.9938,found:288.9936.
实施例5化合物1c的制备
同实施例4化合物1b的制备,卤代烷为溴代正己烷。得到白色晶状固体16mg,收率6.7%。
m.p.:75-77℃;1H NMR(400MHz,CDCl3)δ4.66–3.77(m,8H),3.37–3.30(m,4H),1.72(dd,J=15.1,7.6Hz,4H),1.45(dd,J=10.2,4.6Hz,4H),1.38–1.30(m,8H),0.92(dd,J=9.0,4.7Hz,6H).13C NMR(100MHz,CDCl3)δ198.46,48.45,37.36,31.36,29.71,28.68,28.47,22.54,14.04.HR-MS(ESI)Calcd for C18H34N2S4[M+Na]+:429.1503,found:429.1505.
实施例6化合物1d的制备
同实施例4化合物1b的制备,卤代烷为溴代正十二烷。得到白色晶状固体218mg,收率65.3%。
m.p.:56-57℃;1H NMR(400MHz,CDCl3)δ4.63–3.89(m,8H),3.31(t,J=7.4Hz,4H),1.77–1.65(m,4H),1.40(dd,J=14.5,6.8Hz,4H),1.26(s,24H),0.88(t,J=6.8Hz,6H).13C NMR(101MHz,CDCl3)δ197.44,47.36,36.34,30.87,28.53,28.48,28.29,28.17,27.99,27.48,21.67,13.11.HR-MS(ESI)Calcd for C26H50N2S4[M+Na]+:541.2755,found:541.2756.
实施例7化合物2b的制备
同实施例4化合物1b的制备,原料为1,4,7-三氮杂环壬烷,卤代烷为碘甲烷。得到白色晶状无36mg,收率15%。
m.p.:51-52℃;1H NMR(400MHz,CDCl3)δ4.31(d,J=76.4Hz,12H),3.25(t,J=7.5Hz,6H),1.75–1.61(m,6H),1.49–1.24(m,18H),0.90(t,J=6.9Hz,9H).13C NMR(101MHz,CDCl3)δ200.75,54.83,37.64,31.41,28.77,28.37,22.55,14.05.HR-MS(ESI)Calcd for C27H51N3S6[M+Na]+:632.2305,found:632.2303.
实施例8化合物2c的制备
同实施例7化合物2b的制备,卤代烷为溴代正己烷。得到白色晶状无36mg,收率15%。得到白色晶状无50mg,收率10%。
m.p.:63-64℃;1H NMR(400MHz,CDCl3)δ4.31(d,J=74.9Hz,12H),3.25(t,J=7.4Hz,6H),1.78–1.61(m,6H),1.43–1.23(m,54H),0.88(t,J=6.8Hz,9H).13C NMR(101MHz,CDCl3)δ200.73,54.87,37.66,31.94,29.70,29.67,29.64,29.56,29.38,29.26,29.12,28.42,26.22,22.71,14.15.
实施例9化合物2d的制备
同实施例7化合物2b的制备,卤代烷为溴代正十二烷。得到白色固体46mg,收率9.1%。
m.p.:56-57℃;1H NMR(400MHz,CDCl3)δ4.63–3.89(m,8H),3.31(t,J=7.4Hz,4H),1.77–1.65(m,4H),1.40(dd,J=14.5,6.8Hz,4H),1.26(s,24H),0.88(t,J=6.8Hz,6H).13C NMR(101MHz,CDCl3)δ197.44,47.36,36.34,30.87,28.53,28.48,28.29,28.17,27.99,27.48,21.67,13.11.HR-MS(ESI)Calcd for C26H50N2S4[M+Na]+:541.2755,found:541.2756.
实施例10化合物3b的制备
同实施例4化合物1b的制备,原料为1,4,7,10-四氮杂环十二烷,卤代烷为碘甲烷。得到白色固体105mg,收率36%。
1H NMR(400MHz,CDCl3)δ4.61–3.84(m,16H),2.68(s,12H).13C NMR(101MHz,CDCl3)δ199.18,29.71,20.04.HR-MS(ESI)Calcd for C27H51N3S6[M+Na]+:632.2305,found:632.2303.
实施例11化合物3c的制备
同实施例10化合物3b的制备,卤代烷为溴代正己烷。得到白色固体55mg,收率38%。
m.p.:128-130℃;1H NMR(400MHz,CDCl3)δ5.81–3.40(m,16H),3.26(t,J=7.4Hz,8H),1.72–1.62(m,8H),1.56(s,3H),1.46–1.35(m,8H),1.36–1.22(m,20H),0.88(t,J=6.9Hz,12H).13C NMR(101MHz,CDCl3)δ36.54,30.36,27.65,27.44,21.53,13.02.
实施例12化合物3d的制备
同实施例10化合物3b的制备,卤代烷为溴代正葵烷。得到白色固体65mg,收率20%。
1H NMR(400MHz,CDCl3)δ4.49(s,16H),3.39(t,J=6.7Hz,3H),3.25(t,J=7.4Hz,8H),1.66(dd,J=14.8,7.5Hz,8H),1.40(dd,J=12.8,6.2Hz,8H),1.34–1.20(m,76H),0.88(t,J=6.7Hz,18H).13C NMR(101MHz,CDCl3)δ69.96,36.54,30.90,28.77,28.58,28.55,28.49,28.32,28.22,28.01,27.49,25.19,21.67,13.11.
应用例1本发明化合物体外抗菌活性测试:
实验方法:微量肉汤稀释法:
(1)抗菌药物贮存液制备:制备抗菌药物贮备液的浓度为5120μg/mL,溶解度低的抗菌药物可稍低于上述浓度。所需抗菌药物溶液量或粉剂量可公式进行计算。配制好的抗菌药物贮存液应贮存于-20℃以下环境,保存期不超过6个月。
(2)待测菌的制备:用接种环挑取过夜培养的MH(A)培养皿上的单菌落于MH(B)培养基中,校准为0.5麦氏比浊标准,约含菌数1×108CFU/mL,然后稀释100倍,即得到约含菌数1.0×106CFU/mL的菌液,备用。
(3)分别将抗菌药物(美罗培南)贮备液母液(5120μg/mL)稀释80倍,得到浓度为64μg/mL的抗菌药物溶液。取无菌的96孔板,第一孔加入200μL的抗菌药物,第二至十孔分别加入100μL的MH肉汤培养基,从第一孔吸取100μL加入第二孔,混匀,再吸取100μL至第三孔,依次类推,第十孔吸取100μL弃去。此时各孔药物浓度依次为:32、16、8、4、2、1、0.5、0.25、0.125、0.0625、0.03125μg/mL,第十二孔加入200μLMH(B)培养基(阴性对照)。
(4)然后将稀释好的菌液分装于EP管中,计算需要固定待测化合物的浓度,将其加入菌液中,使待测化合物的终浓度为128或64μg/mL。自稀释好美罗培南的96孔板的第1孔至第10孔分别加入菌液和本发明待测化合物的混合溶液。将加完药的96孔板放置37℃培养箱进行培养,16-18h观察菌液生长情况。同时用标准株做质控。实验结果如下表:
表一:目标化合物3a和NOTA对含有NDM-1和IMP-4的菌株的MIC(μg/mL)及其与美罗培南(MEM)联合用药的结果
由表一可见,化合物3a和对照化合物NOTA一样,对于大部分临床分离得到携带NDM-1和IMP-4型金属内酰胺酶的细菌均能恢复美罗培南的敏感,MIC值显著下降。在含NDM-1基因的大肠埃希菌SMX5中化合物3a可以将美罗培南的MIC提高128倍。同样的在含有IMP-4基因的大肠埃希菌2013091407中,同样可以将美罗培南的活性提高128倍。应用例2本发明化合物体外红细胞溶血性实验
(1)实验材料:10mL EP管,96孔板,新鲜脱脂羊血。
(2)PBS缓冲液:500mL规格,氯化钠4g,氯化钾100mg,二水合磷酸二氢钠1.49g,无水磷酸二氢钾100mg,去离子水定容至490mL,调节PH7.2-7.4之间,灭菌,用10mL灭过菌的超纯水溶解900mg葡萄糖后加入PBS溶液中。
(3)5%红细胞悬浮液的制备:新鲜的脱纤维羊血冷冻与冰箱里,配置好的PBS缓冲液放置与37℃水浴锅中,即用即取。
(4)取两支10mL EP管置于试管架,将37℃PBS取出水浴锅和冷藏的新鲜羊血一起喷酒精,放入超净台。用移液枪分别吸取5700微升PBS加入两支EP管中,再分别吸取300微升羊血,缓慢加入到PBS溶液中,盖盖子,上下缓慢颠倒混匀,放入离心机1500转离心10min,取出EP管,小心吸取上清,移除上清。再重新分别加入5~7mL PBS溶液,上下缓慢颠倒混匀,放入离心1500转离心10min。如此反复操作,直至离心后上清液不再浑浊。最后一次离心过后,撇去上清液,红细胞沉积物留置待用。
(5)取几支10mL EP管,放置试管架上,于每支EP管中加入5700μL的PBS(37℃),然后依次加入300μL的红细胞沉积物。上下缓慢颠倒混匀,如此,便配置好5%的红细胞悬液。
(6)样品溶液的配置:用少量的DMSO溶解(DMSO终浓度不能大于0.5%),并且用相同体积的DMSO做阴性对照。溶解后的DMSO用PBS稀释(例如,第一孔浓度定为1000μg/mL,那么第一孔加入的50μL中药物的含量就是2mg,配置成2mg/50μL的溶液),此时这支EP管内的药物为初始药物。然后平行取九支1.5mL EP管置于试管架中,分别加入200μL的PBS(编号2号、3号、4号……10号)。所有药物都如此平行操作。最后,由初始药物EP管中吸取200μL的药品溶液加入2号EP管中,反复吹洗后吸取200μL到3号EP管中,反复吹洗重复操作,直到10号EP管。如此,稀释好药物。
(7)铺板:取96孔板,写好实验编号,药品代码,日期。将移液枪调至150μL,将配置好的5%红细胞悬液上下轻缓颠倒混匀,依次吸取铺入96孔板中(6×10)。然后将配置好的药物对应加入96孔板中,一个药物三个复孔。加完后放置37℃恒温箱内孵育1h。
(8)后处理:将96孔板从恒温箱内取出,置于-4℃离心机内离心(3500rpm,5min)。离心完毕,每块板对应都取一块新的96孔板。标注和离心后的板子对照。然后对应地吸取100μL上清液(孔孔对应)。吸取完毕后,与酶标仪中测取OD值,分析数据,得到HC50。实验结果图1。
由图1可知,化合物3a在1000μg/mL时依旧对红细胞没有太大的损伤,由此可以证明其对于哺乳动物的红细胞损伤很小。
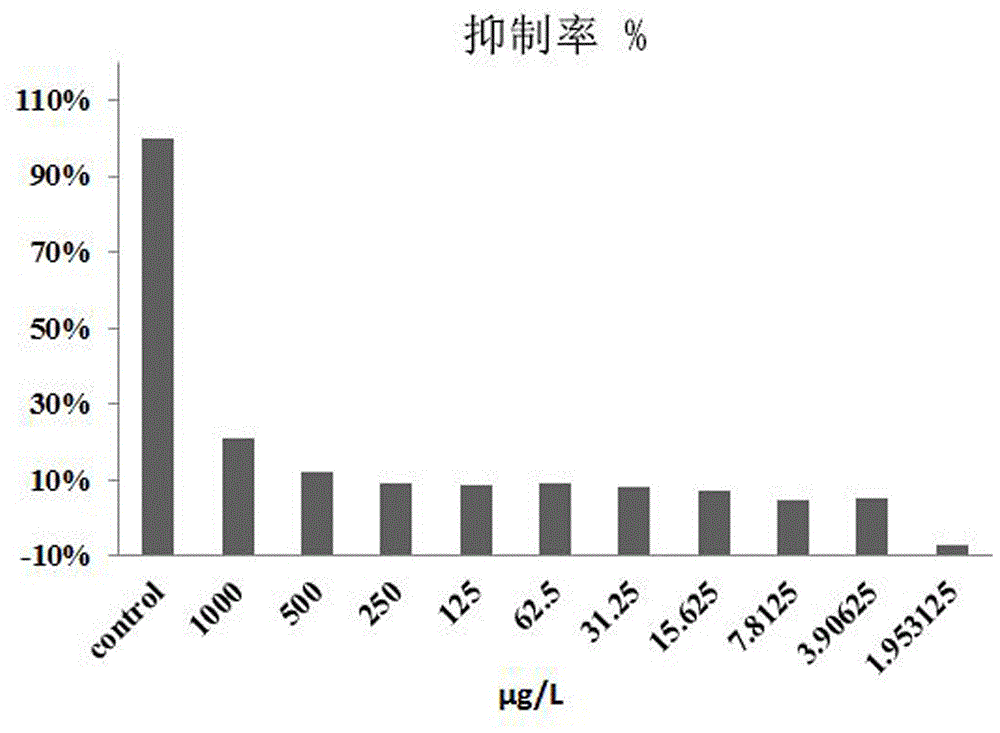
应用例3本发明化合物体外细胞毒性测试:
(1)实验材料:DMEM培养基,细胞计数板,96孔板,CCK-8,酶标仪。
(2)培养基的配置:取无菌分装瓶,FBS和DMEM培养基1:10的比例配置,于4℃冰箱内储存待用。
(3)铺板:将培养中状态良好的Hela细胞自培养箱中取出,于生物安全柜中操作。倒掉上清培养基,用2mL PBS洗细胞一次,除去残留的培养基。取1mL胰蛋白酶沿壁打入培养皿中,于37℃培养箱,体积百分比0.5%的CO2中孵育1-2min。取出消化后的培养皿,加入1mL培养基终止消化。将终止消化后的细胞混悬液转移至10mL EP管中,800rpm 3min离心。取出EP管,缓缓倒掉上清,加入2-4mL培养基,反复吹打50次。从中取10μL细胞混悬液打入细胞计数板中,于20×显微镜下计数。计算好需要细胞的数目,配置为5000个/孔的细胞混悬液。没孔100μL依次加入96孔板中,于恒温培养箱中孵育24h。
(4)加药:将待测药物在4mL EP管中梯度稀释好后,取出已贴壁生长的96孔板,弃掉上清,将稀释好的待测药物每个浓度三个副孔,每孔200μL加入板中,操作完成后将96孔板放入恒温培养箱中。24h后,CCK-8每孔7μL依次加入板中,放入恒温培养箱中培养4h后于酶标仪中,450nm波长处读取OD值,计算抑制率,回归其IC50。实验结果如图2。
由图2可知,化合物3a在1000μg/mL时对Hela细胞的抑制率在20%左右,远高于其有效剂量,由此可以初步证明,在体外实验过程中,发现化合物3a具有较小的毒性。
应用例4本发明化合物体外活死细胞双染测试:
(1)实验材料:DMEM培养基,细胞计数板,96孔板,CCK-8,钙黄绿素-AM,碘化丙啶(PI),PBS缓冲液,荧光显微镜。
(2)培养基的配置:取无菌分装瓶,FBS和DMEM培养基1:10的重量比配置,于4℃冰箱内储存待用。
(3)荧光染料的配置:将分装好的钙黄绿素-AM和碘化丙啶(PI)用PBS稀释,即用即稀释。
(4)铺板:将培养中状态良好的Hela细胞自培养箱中取出,于生物安全柜中操作。倒掉上清培养基,用2mL PBS洗细胞一次,除去残留的培养基。取1mL胰蛋白酶沿壁打入培养皿中,于37℃培养箱,体积百分比0.5%的CO2中孵育1-2min。取出消化后的培养皿,加入1mL培养基终止消化。将终止消化后的细胞混悬液转移至10mL EP管中,800rpm 3min离心。取出EP管,缓缓倒掉上清,加入2-4mL培养基,反复吹打50次。从中取10μL细胞混悬液打入细胞计数板中,于20×显微镜下计数。计算好需要细胞的数目,配置为30000个/孔的细胞混悬液。每孔1mL依次加入12孔板中,于恒温培养箱中孵育24h。
(5)加药与染色:将待测药物在4mL EP管中梯度稀释好后,取出已贴壁生长的12孔板,弃掉上清,将稀释好的待测药物每孔700μL加入板中,操作完成后将12孔板放入恒温培养箱中。24h后,取出,分别收集每个孔的上清,做好标记,3500rpm 5min离心上清,弃掉上清收集死细胞,接着用PBS洗一次,弃掉PBS。将12孔板用PBS每孔各洗一次。用配置好的染料每孔700μL先吹打收集该孔的死细胞,然后将其混合液加入到12孔板的孔中。操作完成后,于恒温培养箱孵育15min。接着,荧光显微镜下拍照。化合物3a在不同浓度下作用于Hela细胞24h后于荧光显微镜下的成像结果如图3。
由图3可以看出,化合物3a于1000μg/mL作用于Hela细胞后,对于细胞维持正常形态的生长几乎没有影响。
应用例5本发明化合物体外红NDM-1酶活性的测定
反应在96孔板中进行,终体积为200μL。分别设置不含美罗培南和不含NDM-1酶的实验对照组。
(1)反应体系中各物质的终浓度分别为:NDM-1酶和美罗培南(MEM)都用10mM HEPES(PH=7.5)溶液稀释;NDM-1酶终活力约3.5U,美罗培南终浓度为125mM。待测药物起始浓度为10mg/mL,待测药物用DMSO梯度稀释使浓度从10mg/mL逐步稀释到0.15625mg/mL
(2)其中96孔板中,每孔加入NDM-1酶溶液98μL,待测药物溶液2μL,每组待测药物平行做三个副孔。用作为阴性对照组,每组四个平行。在30℃条件下孵育15min,再加入美罗培南100μL,使化合物终浓度为100μg/mL到1.5625μg/mL,捶打均匀启动反应。
(3)将96孔板置入酶标仪中,测定其相应孔中于300nm处的吸光值;每隔60s测定一次,连续测定60个循环。分别计算各抑制浓度下的酶抑制率,用Graphpad Prism5分析各抑制剂的IC50。
运用上述模型监测待测样品酶活性的抑制率,其中阴性对照组为不含抑制剂,可以反应体系中酶的最大活力。空白对照组为不含抑制剂也不含酶,所测得的结果为体系本底值。因此待测样品抑制率的计算公式如下:
IR(%)=(1-VS/VN)×100%
VS代表待测样品孔的酶反应速率
VN代表阴性对照孔的平均酶反应速率
本发明化合物3a对金属beta内酰胺酶NDM-1的抑制率实验结果如下:
a表明化合物在10μg/mL条件下对于NDM-1酶的抑制率。
从结果可以看出化合物3a对于NDM-1酶的IC50结果较好,在10μg/mL的条件下对于NDM-1的抑制率就可以达到52%,IC50为17.3μM。
- 还没有人留言评论。精彩留言会获得点赞!