用于制备间三联苯化合物类型的不对称OCO钳形配体的方法与流程
本发明涉及用于制备间三联苯类不对称化合物(下文任选地也称为间三联苯化合物或不对称的OCO钳形配体)的方法,包括如下方法步骤:a) 使第一取代或未取代的酚与1,3-二取代且在2和5位同样可以取代的芳烃反应,以获得酚-芳烃-偶合产物,b) 任选地用保护基团保护所述酚-芳烃-偶合产物的OH基团,以获得受保护的酚-芳烃-偶合产物,和c) 使由a)或b)获得的酚-芳烃-偶合产物与第二取代或未取代的酚反应,以获得不对称的间三联苯,条件是,所述第一酚和所述第二酚是不同取代的,其特征在于,方法步骤a)和c)的至少一步作为电化学方法步骤实施。
背景技术:
间三联苯通常是指1,3-二苯基苯。间三联苯类型的化合物因此包含1,3-二苯基苯特征结构。在本发明中,不对称的OCO钳形配体是指具有1,3-二苯基苯单元的化合物,其中两个外环不是芳烃,而是酚。两个酚单元的OH基团分别取代在芳烃组分的邻位。
在本发明中,不对称的OCO钳形配体是指这样的化合物,其中两个酚在结构上不同,即其是不相同的。在本申请中,在两个不同的酚组分之间布置的芳烃组分不是必须对称的,即,不是必须在芳烃分子中存在一个镜面,使得一侧与另一侧互成镜像。
此外,在本发明中在芳烃组分 (B) 中采用如下的位置编号:
R9 定义为在苯环的C原子1上的取代基,R10 为在C原子2上的取代基,R11 为在C原子3上的取代基,R12 为在C原子5上的取代基。
在本发明中,取代的酚是指在一个或多个环碳原子上的氢原子被取代的酚。
目前,OCO-钳形配体的经典合成局限于对称的衍生物。已知的方法实现了CAr-CAr键合,例如通过Ullmann偶合 (B. G. Pring, Acta Chem. Scand. 1973, 27, 3873−3880),Grignard-Wurtz偶合 (R. S. Grewal, H. Hart, T. K. Vinod, J. Org. Chem.1992, 57, 2721–2726)或者钯催化的偶合反应 (S. Sarkar, A. R. Carlson, M. K. Veige, J. M. Falkowski, K. A. Abboud, A. S. Veige, J. Am. Chem. Soc. 2008, 130, 1116–1117)。在实质的偶合反应之前,必须保护羟基官能团,这通常通过甲基化而实现。在所有已知的情况下,需要在排除湿气和无氧的条件下实施所述反应。
示意图1:用于制备OCO-钳形配体的现有技术 (S. Sarkar, A. R. Carlson, M. K. Veige, J. M. Falkowski, K. A. Abboud, A. S. Veige, J. Am. Chem. Soc. 2008, 130, 1116–1117)。
用于使酚衍生物与芳烃进行交叉偶合的已知方法的很大缺点在于,在偶合反应过程中必须使用干燥的溶剂和排除空气。在传统的交叉偶合方法中,在大多数情况下必须保护羟基官能团。此外,对于区域选择性的交叉偶合,将离去官能团引入所使用的底物中,其在实质的偶合反应之后作为环境有害的反应废料出现 (参见示意图1)。由于使用的试剂而经常限制了官能团的耐受性,从而主要可以使用较少取代的衍生物。在反应过程中经常出现有毒的副产物 (例如溴化合物),其与所希望的产物很难分离并且处理费用昂贵。另外,所述反应必须经过非常多的步骤进行,这同样是非常不利的。越来越稀有的原材料使得通过钯催化的交叉偶合或者化学氧化剂制备配体体系非常昂贵。
目前为止,不对称的钳形配体的制备通过传统方法还无法实现。目前也未知用于将酚交叉偶合成不对称的间三联苯化合物的电化学方法。
因此,本发明的目的在于提供用于制备间三联苯化合物类型的不对称OCO钳形配体的简单方法,其避免了现有技术中的一项或多项缺点。
令人惊奇地发现,所述目的可以如下实现:在具有两个方法反应步骤的方法中,该方法步骤的至少之一作为电化学方法步骤实施。
技术实现要素:
本发明因此提供了用于制备间三联苯类不对称化合物(所谓的不对称的OCO钳形配体)的方法,其包括如下方法步骤:
a) 使第一取代或未取代的酚与1,3-二取代且在2和5位同样可以取代的芳烃反应,以获得酚-芳烃-偶合产物,和
b) 任选地用保护基团保护所述酚-芳烃-偶合产物的OH基团,以获得受保护的酚-芳烃-偶合产物,
c) 使由a)或b)获得的酚-芳烃-偶合产物与第二取代或未取代的酚反应,以获得不对称的间三联苯,
条件是,所述第一酚和所述第二酚是不同取代的,
其特征在于,
方法步骤a)和c)的至少一步作为电化学方法步骤实施。
本发明同样提供了如下定义的式(CDC')的化合物。
本发明还提供了本发明的化合物作为用于制备金属-配体-催化剂体系的配体的用途。
本发明的方法具有如下优点:通过借助电化学方法步骤的部分或完全反应而基本上简化了不对称的间三联苯化合物的制备,因为不必保证排除湿气或保持无氧的反应条件。这主要降低了在实施所述方法时的消耗。
此外,以电化学方法步骤实施一个或多个方法步骤具有如下优点:可以显著降低不希望的副产物的形成。
通过电化学交叉偶合而不必再使用昂贵和有毒的催化剂,以及不必在酚-芳烃-偶合反应(本发明的方法步骤a))之前引入活化的离去官能团。
根据本发明方法的实施类型,所述方法首次实现了不对称化合物的直接合成以及低的合成消耗和同时对使用的基材中的官能团的高耐受性。
此外,在本发明的方法中可以部分或优选完全放弃使用化学氧化剂。以这种方式避免了由于氧化剂残余物而形成所述化合物的副产物以及杂质的风险。
此外,本发明的方法具有如下优点:可以制备大量不同的间三联苯化合物,而这些化合物根据现有技术已知的方法无法获得或仅在高消耗的情况下才能获得。
特别地,本发明的方法具有如下优点:使用该方法可以制备本发明的间三联苯化合物 (间三联苯类)。
下面示例性地描述本发明的方法、本发明的化合物以及其用途,但本发明不限于这些示例性的实施方案。如果下文给出范围、通式或化合物类型,则其不仅包括明确提到的相应范围或化合物类型,还应包括通过去除个别值(范围)或化合物而得到的所有范围子集和化合物的类型子集。如果在本说明书中引用文献,则其内容(特别是关于参考的事项)完全并入本发明的公开内容。如果下文中给出百分比数值,在没有另外说明的情况下,其是指体积%的数值。如果下文给出平均值,在没有另外说明的情况下,其是指数均平均值。如果下文给出物质特征,例如粘度等,在没有另外说明的情况下,其是指在25℃下的物质特征。
用于制备间三联苯类不对称化合物的本发明方法包括如下方法步骤:a) 使第一取代或未取代的酚与1,3-二取代且在2和5位同样可以取代的芳烃反应,以获得酚-芳烃-偶合产物,b) 任选地用保护基团保护所述酚-芳烃-偶合产物的OH基团,以获得受保护的酚-芳烃-偶合产物,和c) 使由a)或b)获得的酚-芳烃-偶合产物与第二取代或未取代的酚反应,以获得不对称的间三联苯,条件是,所述第一酚和所述第二酚是不同取代的,其特征在于,方法步骤a)和c)的至少一步,优选方法步骤c) ,优选两个方法步骤a) 和c) 都作为电化学方法步骤实施。
优选地,在本发明的方法中使用取代的酚作为第一酚以及作为第二酚。
有利地,将步骤a) 的酚-芳烃-偶合产物在输送到方法步骤c) 之前进行分离。
根据本发明优选地,在方法步骤a)、b)和c)中回收未反应的反应物,这优选通过蒸馏进行。回收的反应物可以再次作为原料输送到相应的方法步骤a)、b)和/或c)。
优选地,所述一个或多个电化学方法步骤如此实施,使得
aa) 产生至少一种溶剂和至少一种导电盐的混合物,
bb) 向该混合物中加入待反应的化合物,其中加入摩尔不足量的所述取代的酚(第一或第二取代的酚),基于其它待反应的组分(二取代的芳烃或酚-芳烃-偶合产物),
cc) 将至少两个电极引入在bb)中获得的反应溶液和在所述电极上施加电压,
dd) 使所述组分反应生成相应的偶合产物(酚-芳烃-或间三联苯-偶合产物),
ee) 关闭电压,和任选地
ff) 分离和/或纯化所述反应产物。
如果两个方法步骤a)和c)都作为电化学方法步骤实施,则本发明的方法优选如此实施,使其包括如下方法步骤:
a1) 混合至少一种溶剂和至少一种导电盐,
a2) 向a1)的混合物中加入第一取代或未取代的酚和1,3-二取代且在2和5位同样可以取代的芳烃,其中加入摩尔过量的1,3-二取代芳烃,基于第一取代或未取代的酚,
a3) 将至少两个电极引入所述反应溶液和在所述电极上施加电压,
a4) 将第一取代或未取代的酚在1,3-二取代的芳烃上偶合而得到酚-芳烃-偶合产物,
a5) 关闭电压,分离和任选地纯化所述酚-芳烃-偶合产物,
b1) 任选地用保护基团保护所述酚-芳烃-偶合产物的OH基团,以获得受保护的酚-芳烃-偶合产物,和
c1) 将步骤a5) 的酚-芳烃-偶合产物或步骤b1) 的受保护的酚-芳烃-偶合产物与由至少一种溶剂和其中所含的导电盐组成的反应溶液混合,
c2) 加入第二取代或未取代的酚(其与所述第一取代或未取代的酚是不同取代的),其中在反应溶液中存在摩尔过量的所述酚-芳烃-偶合产物,基于第二取代或未取代的酚,
c3) 将至少两个电极引入所述反应溶液和在所述电极上施加电压,
c4) 将第二取代或未取代的酚在所述酚-芳烃-偶合产物上偶合而得到不对称的间三联苯,
c5) 关闭电压,和任选地
c6) 分离和/或后处理所述不对称的间三联苯。
所述电化学方法步骤可以例如根据在如下文献中描述的用于碳-碳-键电化学偶合的方法而实施:a.) A. Kirste, B. Elsler, G. Schnakenburg, S. R. Waldvogel, J. Am. Chem. Soc. 2012, 134, 3571−3576;b.) A. Kirste, S. Hayashi, G. Schnakenburg, I. M. Malkowsky, F. Stecker, A. Fischer, T. Fuchigami, S. R. Waldvogel, Chem. Eur. J. 2011, 17, 14164–14169;c.) B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke, S. R. Waldvogel, Angew. Chem. Int. Ed. 2014, 53, 5210-5213;和d.) A. Kirste, M. Nieger, I. M. Malkowsky, F. Stecker, A. Fischer, S. R. Waldvogel, Chem. Eur. J. 2009, 15, 2273-2277。
所述电化学方法步骤可以在所有合适的现有技术中已知的电解池中实施。在本发明的方法中优选使用法兰池(Flanschzelle)、烧杯玻璃池(Becherglaszelle)或筛选池(Screeningzelle)。这样的电解池在文献中描述过和所述前两个玻璃池可以例如以该名称在HWS Labortechnik Mainz获取。
所述电解池具有至少两个电极。作为电极,可以使用市售常见的电极。作为阳极,优选可以使用例如BDD (0.015 mm的在硅上的硼掺杂金刚石或者0.05 mm的在铌上的硼掺杂金刚石)、铂电极、均衡石墨电极或玻璃碳电极。作为阴极,优选使用BDD电极、镍网电极或玻璃碳电极。这些BDD电极 (在例如铌或硅的载体上的硼掺杂金刚石) 例如以名称DIACHEM从Condias GmbH Itzehoe可以获得。其余的电极可以由常见的化学品和材料供应商(例如Goodfellow或Aldrich)获得。
所述一个或多个电化学方法步骤优选在溶剂的存在下实施。使用的溶剂优选是乙腈、碳酸亚丙酯、碳酸甲酯、硝基甲烷、乙二醇二甲醚、甲磺酸、苯、甲苯、水、甲醇、乙醇、丙醇、异丙醇、卤化的溶剂或卤化的或未卤化的酸或者它们的混合物。
作为溶剂优选添加羧酸,优选甲酸,使用氟化的羧酸或氟化的醇,优选三氟乙酸或1,1,1,3,3,3-六氟-2-丙醇(HFIP),优选1,1,1,3,3,3-六氟-2-丙醇。
特别优选地,使用的溶剂是甲醇、甲酸、三氟乙酸、六氟异丙醇或者它们的混合物,优选甲醇、六氟异丙醇或者它们的混合物,和特别优选六氟异丙醇(1,1,1,3,3,3-六氟-2-丙醇)。
所述一个或多个电化学方法步骤优选在至少一种导电盐的存在下实施,其中使用的导电盐优选选自四(C1-C6-烷基)铵和1,3-二(C1-C6-烷基)咪唑鎓的盐,条件是,所述烷基可以是卤素取代的,特别是氟取代的。优选地,使用的导电盐的抗衡离子选自砷酸根离子、硫酸根离子、硫酸氢根离子、烷基硫酸根离子、烷基磷酸根离子、高氯酸根离子、氟离子、芳基硫酸根离子、六氟磷酸根离子和四氟硼酸根离子。优选的导电盐选自硼酸季铵、氟烷基磷酸季铵、氟烷基砷酸季铵、三氟甲磺酸季铵、双(氟甲磺酰基)亚氨基季铵、三(氟甲磺酰基)甲基化季铵、甲基硫酸甲基三丁基铵、甲基硫酸甲基三乙基铵、六氟磷酸四丁基铵、四氟硼酸四乙基铵、六氟磷酸锂或四氟硼酸四乙基铵。特别优选甲基硫酸甲基三乙基铵或Bu3NMe+MeOSO3-(甲基硫酸甲基三丁基铵),非常特别优选甲基硫酸甲基三丁基铵作为导电盐使用。
优选地,所述导电盐的浓度为0.001至10 mol/l,优选0.01至1 mol/l,特别优选0.075至0.125 mol/l和非常特别优选0.09 mol/l,基于所述反应混合物。
优选地,在本发明的方法中分别使用浓度为0.001至5 mol/l,优选0.05至0.5 mol/l,优选0.1至0.3 mol/l和特别优选0.15 mol/l的第一和/或第二取代的酚。
所述一个或多个电化学方法步骤优选在室温或提高的温度下实施。优选地,所述一个或多个电化学方法步骤在电解池中的温度为25至80℃,优选35至70℃和优选45至55℃的情况下实施。
所述一个或多个电化学方法步骤优选在恒电流下实施。
在实施所述一个或多个电化学方法步骤时,电流强度优选如此选择,使得电流密度为1至10 mA/cm2,优选2至5.5 mA/cm2,优选2.5至3 mA/cm2和优选2.8 mA/cm2。为了实施所述一个或多个电化学方法步骤,优选在电极之间施加2至10 V,优选2.5至7.5 V和优选3至6 V的(端)电压。
有利地,使用如此多的1,3-二取代的芳烃和第一取代或未取代的酚,使得1,3-二取代的芳烃与第一取代或未取代的酚的摩尔比为1.5:1至6:1,优选2:1至3:1。
酚-芳烃-偶合产物与第二取代或未取代的酚的摩尔比优选1.5:1至6:1,优选2:1至3:1。
特别优选地,如此选择量的比例,使得在步骤a) 中1,3-二取代的芳烃与第一取代或未取代的酚的摩尔比为1.5:1至6:1,优选2:1至3:1,和在步骤b) 中酚-芳烃-偶合产物与第二取代或未取代的酚的摩尔比为1.5:1至6:1,优选2:1至3:1。
在根据本发明的方法中,使用的第一取代或未取代的酚优选是式(A)的那些,
使用的第二取代或未取代的酚优选是式(A')的那些,
和使用的1,3-二取代且在2和5位同样可以取代的芳烃优选是式(B)的那些,
其中所述基团
R1、R2、R3、R4、R5、R6、R7、R8、R10和R12彼此独立地选自氢、(C1-C12)-烷基、O-(C1-C12)-烷基、(C6-C20)-芳基、O-(C6-C20)-芳基和卤素,优选氢、(C1-C12)-烷基、O-(C1-C12)-烷基和卤素,特别优选氢、(C1-C12)-烷基、O-(C1-C12)-烷基,
其中R9和R11彼此独立地选自(C1-C12)-烷基、O-(C1-C12)-烷基、(C6-C20)-芳基、O-(C6-C20)-芳基和卤素,优选(C1-C12)-烷基、O-(C1-C12)-烷基和卤素,特别优选(C1-C12)-烷基、O-(C1-C12)-烷基,
和其中在对R1/R5、R2/R6、R3/R7和R4/R8 中的至少一对由不同的基团组成。
在本发明中,卤素是指氟、氯、溴或碘,尤其是氟、氯和溴。术语"烷基"在本发明中是指直链或支化的烃基,其中任选一个或多个氢原子可以优选被卤素原子或羟基或烷(基)氧基取代。芳基在本发明中是指芳族的烃基,例如苯基- (C6H5-)、萘基- (C10H7-)、蒽基- (C14H9-),优选苯基,其中任选一个或多个氢原子可以优选被烷基或烷(基)氧基取代。
在本发明中,表述-(C1-C12)-烷基包含直链和支化的烷基基团。优选地,这些基团是未取代的直链或者支化的-(C1-C8)-烷基基团和最优选-(C1-C6)-烷基基团。例如,(C1-C12)-烷基基团尤其是甲基、乙基、丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、2-戊基、2-甲基丁基-、3-甲基丁基-、1,2-二甲基丙基-、1,1-二甲基丙基、2,2-二甲基丙基-、1-乙基丙基-、正己基-、2-己基-、2-甲基戊基-、3-甲基戊基-、4-甲基戊基-、1,1-二甲基丁基-、1,2-二甲基丁基-、2,2-二甲基丁基-、1,3-二甲基丁基-、2,3-二甲基丁基-、3,3-二甲基丁基-、1,1,2-三甲基丙基-、1,2,2-三甲基丙基-、1-乙基丁基-、1-乙基-2-甲基丙基-、正庚基-、2-庚基-、3-庚基-、2-乙基戊基-、1-丙基丁基-、正辛基-、2-乙基己基-、2-丙基庚基-、壬基-、癸基-和十二烷基-。
基团“(C1-C12)-烷基和O-(C1-C12)-烷基”可以各自是未取代的或者被一个或多个相同或不同的选自如下的基团取代的:(C3-C12)-环烷基、(C3-C12)-杂环烷基、(C6-C20)-芳基、氟、氯、氰基、甲酰基、酰基或烷氧基羰基。
关于表述-(C1-C12)-烷基的说明也适用于在-O-(C1-C12)-烷基,即-(C1-C12)-烷氧基中的烷基。优选地,这些基团是未取代的直链或者支化的-(C1-C6)-烷氧基基团。
在本发明中,表述-(C6-C20)-芳基包括单环或者多环的芳族的烃基。它们具有6-20个环原子,更优选6-14个环原子,尤其6-10个环原子。芳基是优选-(C6-C10)-芳基和-(C6-C10)-芳基-(C6-C10)-芳基-。芳基尤其是苯基、萘基、茚基、芴基、蒽基、菲基、并四苯基、苯并菲基、芘基、蒄基。更具体地,芳基是苯基、萘基和蒽基。
取代的-(C6-C20)-芳基基团可以具有一个或者多个(例如1、2、3、4或5个)取代基,这取决于它们的环大小。这些取代基优选各自独立地选自-H、-(C1-C12)-烷基、-O-(C1-C12)-烷基、-O-(C6-C20)-芳基、-(C6-C20)-芳基、-卤素 (例如 Cl、F、Br、I)、-COO-(C1-C12)-烷基、-CONH-(C1-C12)-烷基、-(C6-C20)-芳基-CON[(C1-C12)-烷基]2、-CO-(C1-C12)-烷基、-CO-(C6-C20)-芳基、-COOH、-OH、-SO3H、-SO3Na、-NO2、-CN、-NH2、-N[(C1-C12)-烷基]2。
下面,通过两个反应示意图示例性地展示本发明的方法的实施,其中在此两个方法步骤a) 和c) 作为电化学方法步骤实施。在示意图中使用的基团R1至R12 如上定义。在第一方法步骤a)中 (示意图2),取代或未取代的酚在1,3-二取代且在2和5位同样可以取代的芳烃上的偶合生成酚-芳烃-偶合产物 (AB)。
示意图2:方法步骤a)。
在第二方法步骤c) 中,使酚-芳烃-偶合产物 (AB) 与另一个取代或未取代的酚 (A') 反应而生成化合物ABA' 和由此转化为不对称的OCO钳形配体 (示意图3)。
示意图3:方法步骤c)。
这些化合物以传统的方法目前未被描述过或仅以高的合成消耗才能获得。
有利地,在方法步骤a) 和方法步骤c) 之间实施方法步骤b) ,其中酚-芳烃-偶合产物的羟基被合适的保护基团保护。当两个方法步骤a) 和c) 都以电化学方法步骤实施时,这样的方法步骤是特别有利的。
因此,在本发明方法的一个变型方案中,在方法步骤c) 中使用受保护的酚-芳烃-偶合产物,优选式(A''B)的那些
以及第二取代或未取代的酚,优选式(A')之一,
其中X = -OR'和R' = 保护基团,优选甲硅烷基-保护基团,优选三甲基甲硅烷基-、三乙基甲硅烷基-、三异丙基甲硅烷基-、叔丁基二苯基甲硅烷基-或(1,1-二甲基乙基)二甲基甲硅烷基-保护基团,特别优选三异丙基甲硅烷基-或(1,1-二甲基乙基)二甲基甲硅烷基-保护基团,和
R1、R2、R3、R4、R5、R6、R7、R8、R10和R12彼此独立地选自氢、(C1-C12)-烷基、O-(C1-C12)-烷基、(C6-C20)-芳基、O-(C6-C20)-芳基和卤素,优选氢、(C1-C12)-烷基、O-(C1-C12)-烷基和卤素,特别优选氢、(C1-C12)-烷基、O-(C1-C12)-烷基,
其中所述基团R9和R11彼此独立地选自(C1-C12)-烷基、O-(C1-C12)-烷基、(C6-C20)-芳基、O-(C6-C20)-芳基和卤素,优选(C1-C12)-烷基、O-(C1-C12)-烷基和卤素,特别优选(C1-C12)-烷基、O-(C1-C12)-烷基,
和
其中在对R1/R5、R2/R6、R3/R7和R4/R8 中的至少一对由不同的基团组成。
方法步骤b) 可以如此实施,使得方法步骤a) 的酚-芳烃-偶合产物在后处理之前或之后与合适的化合物反应。优选地,添上甲硅烷基保护基团。特别优选地,使用化合物(1,1-二甲基乙基)二甲基甲硅烷基氯和三(甲基乙基)甲硅烷基氯。
甲硅烷基保护基团及其引入是本领域技术人员已知的并且可以从常见的专业文献中获取(为此参见:P. G. M. Wuts, T. W. Greene "Greene's Protective Groups in Organic Synthesis", 第四版, 2007, John Wiley and Sons; Hoboken, New Jersey)。
令人惊奇地发现,当在方法步骤c) 中使用具有受保护的羟基(特别是通过与甲硅烷基反应而受保护的羟基)的酚-芳烃-偶合产物时,当其作为电化学方法步骤实施时,保护基团再次消除,从而获得又具有两个OH基团的间三联苯化合物。
令人惊奇地还发现,通过在酚-芳烃-偶合产物中引入保护基团(特别是甲硅烷基),在接下来的在电化学实施的方法步骤c) 的偶合中可以增加又具有两个OH基团的间三联苯化合物的产率。因此,总方法的效率可以再次明显改善。
本发明的方法优选如此实施,使得在电化学方法步骤中的反应,优选整个反应在不使用有机氧化剂的情况下实施。
借助本发明的方法可以制备大量不同的间三联苯化合物,尤其是式(ABA')的那些
其中X、Y和R1至R12如上定义。使用本发明的方法特别可以制备本发明的式(CDC')化合物。本发明的式(CDC')化合物
其中所述基团R13、R14、R15、R16、R19、R20、R21和R22彼此独立地选自氢、(C1-C12)-烷基、O-(C1-C12)-烷基、(C6-C20)-芳基、O-(C6-C20)-芳基、卤素,优选氢、(C1-C12)-烷基、O-(C1-C12)-烷基、卤素,特别优选氢、(C1-C12)-烷基和O-(C1-C12)-烷基,
其中R17和R18彼此独立地选自(C1-C12)-烷基、O-(C1-C12)-烷基、(C6-C20)-芳基、O-(C6-C20)-芳基和卤素,优选(C1-C12)-烷基、O-(C1-C12)-烷基和卤素,特别优选(C1-C12)-烷基和O-(C1-C12)-烷基,和
其中在对R13/R22、R14/R21、R15/R20和R16/R19中的至少一对由不同的基团组成。
优选地,基团R13、R14、R15、R16、R19、R20、R21和R22彼此独立地选自氢、(C1-C12)-烷基、O-(C1-C12)-烷基、卤素,
和基团R17和R18彼此独立地选自(C1-C12)-烷基、O-(C1-C12)-烷基或卤素。
优选地,基团R13、R14、R15、R16、R19、R20、R21和R22彼此独立地选自氢、(C1-C12)-烷基、O-(C1-C12)-烷基,
和基团R17和R18彼此独立地选自(C1-C12)-烷基、O-(C1-C12)-烷基。
非常特别优选的本发明化合物是满足式(1)、(2)或(3)的那些
。
本发明的化合物可以作为配体用于制备金属-配体-催化剂体系。
附图说明
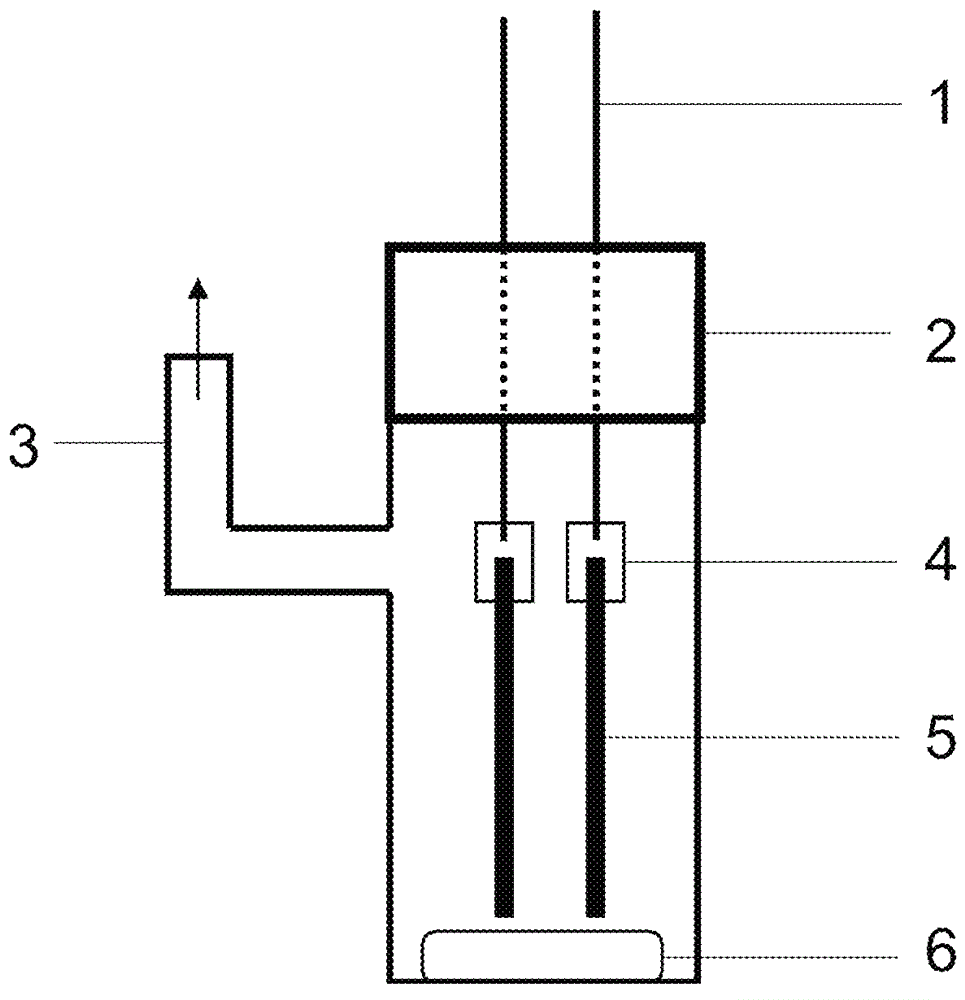
本发明借助图1至4示例性描述,而不应将本发明限于在图中显示的实施方案;本发明的应用范围由整个说明书和权利要求书而获得。在图1至4中显示了如在本发明的方法中可以使用的和特别是在下面的实施例中使用的电解池的示意图。
图1示意性地显示了筛选池。在图1中用1标记BDD电极,用2标记特氟隆覆盖体,用3标记特氟隆杯体和用4标记搅拌磁体。
图2示意性地显示法兰池 (玻璃)。在图2中,用1标记具有固定的镍网阴极的不锈钢棒,用2标记特氟隆塞子,用3标记冷却罩(玻璃),用4标记螺旋夹钳,用5标记BDD电极,用6标记EPDM密封件和用7标记搅拌磁体。
图3示意性地显示小型的烧杯玻璃池(玻璃)。在图3中,用1标记用于接触的不锈钢棒,用2标记特氟隆塞子,用3标记电解池出口/连接,例如用于Dimroth冷却器,用4标记电极支撑体(不锈钢),用5标记玻璃碳电极和用6标记搅拌磁体。
图4示意性地显示大型的烧杯玻璃池(玻璃)。在图4中,用1标记BDD电极,用2标记用于接触的连接体,用3标记玻璃罩,用4标记搅拌磁体,用5标记电解池出口/连接,例如用于Dimroth冷却器和用6标记导电触点(不锈钢箔膜)。
在下面给出的实施例中,示例性地描述了本发明,但不应将本发明限于实施例中提及的实施方案;本发明的应用范围由整个说明书和权利要求书而获得。
具体实施方式
实验部分
通用的方法
色谱法 (GC/GCMS)
用于分离物质混合物的制备型液相色谱法使用60 M硅胶 (0.040-0.063 mm)(Machery-Nagel GmbH & Co.KG, Düren)在2 bar的最大压力下进行。所有使用的洗脱液 (乙酸乙酯 (工业纯度)、环己烷 (工业纯度)) 预先在旋转蒸发仪上蒸馏提纯。
薄膜色谱 (TLC)在现成的PSC 硅胶板 60 F254(Merck KGaA, Darmstadt)上进行。不同物质的检测首先在UV光下和然后借助铈/钼磷酸试剂(5.6 g的钼磷酸、2.2 g的四水合硫酸铈(IV)和13.3 g的浓缩硫酸在200 ml的水中)的染色和然后通过热吹风机加热而进行。
气相色谱 (GC/GCMS)
产物混合物和纯物质的气相色谱分析 (GC) 借助气相色谱仪GC-2010(Shimadzu, Japan)进行。测量在HP-5 石英毛细管柱(Agilent Technologies, USA)(长度: 30 m; 内径: 0.25 mm; 共价结合的固定相的膜厚度: 0.25 μm; 载气: 氢气; 注射器温度: 250℃; 检测器温度: 310℃; 程序: “硬”方法: 起始温度 50℃ 1 min, 加热速率: 15℃/min, 结束温度 290℃ 8 min)上进行。产物混合物和纯物质的气相色谱-质谱分析 (GC-MS) 借助GC-2010气相色谱仪结合GCMS-QP2010质量检测器(Shimadzu, Japan)进行。测量在HP-1石英毛细管柱(Agilent Technologies, USA)(长度: 30 m; 内径: 0.25 mm; 共价结合的固定相的膜厚度: 0.25 μm; 载气: 氢气; 注射器温度: 250℃; 检测器温度: 310℃; 程序: “硬”方法: 起始温度 50℃ 1 min, 加热速率: 15℃/min, 结束温度 290℃ 8 min; GC-MS: 离子源温度: 200℃)上进行。
质谱
所有的电喷雾电离测量 (ESI+) 在QTof Ultima 3(Waters Micromasses, Milford, Massachusetts)上进行。EI质谱以及高分辨率的El质谱在型号MAT 95 XL扇区仪器(Thermo Finnigan, Bremen)上测量。
核磁共振谱
核磁共振谱分析在多核共振波谱仪AC 300或AV II 400型号(Bruker, Analytische Messtechnik, Karlsruhe)上进行。使用的溶剂是CDCl3。1H和13C波谱根据残余量的未氘化溶剂按照NMR溶剂数据表(Cambridge Isotopes Laboratories, USA)校对。1H和13C信号的归属部分借助H,H-COSY、H,H-NOESY、H,C-HSQC和H,C-HMBC波谱。化学位移作为δ值以ppm为单位给出。对于NMR信号的多重性使用下列缩写:s (单峰)、bs (宽单峰)、d (双峰)、t (三重峰)、q (四重峰)、m (多重峰)、dd (双重-双峰)、dt (双重-三重峰)、tq (三重-四重峰)。所有的耦合常数J借助其中包含的键的数目以赫兹 (Hz)为单位给出。在信号归属时给出的编号相应于结构式中给出的标记,其不是必须与IUPAC命名法相一致。
晶体结构分析
晶体结构分析在美因茨Johannes Gutenberg大学的有机化学研究所在型号IPDS 2T的仪器(出自公司STOE & Cie GmbH, Darmstadt)上进行。
熔点
相对熔点借助SG 2000熔点测定仪(HW5, Mainz)测量,并且是未校对的。
通用操作规范:
AAV1:在L-电解池中的电化学交叉偶合
将5 mmol的待氧化的物质A或A'(不足量的组分)与2至3倍过量(10-15 mmol)的偶合对应物B或AB在33 ml的1,1,1,3,3,3-六氟异丙醇(HFIP) 或33 ml的HFIP和甲醇混合溶剂(具有18体积%的甲醇(MeOH),基于HFIP和MeOH的总和)中在未分割的法兰池中使用BDD阳极和镍网阴极而反应。作为导电盐使用浓度为0.09 M的Bu3NMe+MeOSO3- (MTBS)。进行恒电流的电解。电解池的外壳通过恒温器控温至大约10℃,而搅拌反应混合物并借助油浴将其加热到50℃。在电解结束之后,将池内的物质转移到50 ml的圆颈烧瓶中,并且在减压下在50℃和200-70 mbar的旋转蒸发仪上除去溶剂。矿物质化产物以及所含有的导电盐通过乙酸乙酯 (300 ml) 作为洗脱液经过50 g的硅胶60而分离。借助短程蒸馏在球形管蒸馏器上 (100℃和10-3 mbar) 回收未反应的反应物。所得到的反应产物如分别说明地通过柱色谱法而分离。
电极材料:
阳极: 在硅载体上的BDD(15μm的金刚石层)
阴极: 镍网
电解条件:
温度: 50℃
电流密度: 2.8 mA/cm2
电荷量: 2-4 F,基于不足量的组分
端电压: 3-6 V。
AAV2:在烧杯玻璃池中采用叠放的电极排列的电化学交叉偶合
将120 mmol的待氧化的物质A(不足量的组分)与2至3倍过量(240-360 mmol)的偶合对应物B在750 ml的1,1,1,3,3,3-六氟异丙醇或750 ml的HFIP和甲醇混合溶剂(具有18体积%的MeOH,基于HFIP和MeOH的总和)中在烧杯玻璃池中采用10个BDD电极的叠放排列(5个阳极和5个阴极)而反应。作为导电盐使用浓度为0.09 M的MTBS。进行恒电流的电解。蒸发的HFIP借助Dimroth冷却器再蒸馏并且输送到电解中,而搅拌反应混合物并借助油浴将其加热到50℃。在电解结束之后,将池内的物质转移到1 l的圆颈烧瓶中,并且在减压下在50℃和200-70 mbar的旋转蒸发仪上除去溶剂。矿物质化产物以及所含有的导电盐通过乙酸乙酯 (2 l) 作为洗脱液经过300 g的硅胶60而分离。借助短程蒸馏在球形管蒸馏器上 (100℃和10-3 mbar) 回收未反应的反应物。
电极材料:
阳极: 5个在铌上的BDD
阴极: 5个在铌上的BDD
电解条件:
温度: 50℃
电流密度: 2.8 mA/cm2
电荷量: 2-4 F,基于不足量的组分
端电压: 3-6 V。
AAV3:在烧杯玻璃池中的电化学交叉偶合
将3.8 mmol的待氧化的物质A(不足量的组分)与2至3倍过量(7.6-11 mmol)的偶合对应物B在25 ml的1,1,1,3,3,3-六氟异丙醇或25 ml的HFIP和甲醇混合溶剂(具有18体积%的MeOH,基于HFIP和MeOH的总和)中在烧杯玻璃池中在玻璃碳电极上而反应。作为导电盐使用浓度为0.09 M的MTBS。进行恒电流的电解。蒸发的HFIP借助Dimroth冷却器再蒸馏并且输送到电解中,而搅拌反应混合物并借助油浴将其加热到50℃。在电解结束之后,将池内的物质转移到100 ml的圆颈烧瓶中,并且在减压下在50℃和200-70 mbar的旋转蒸发仪上除去溶剂。矿物质化产物以及所含有的导电盐通过乙酸乙酯 (300 ml) 作为洗脱液经过50 g的硅胶60而分离。借助短程蒸馏在球形管蒸馏器上 (100℃和10-3 mbar) 回收未反应的反应物。
电极材料:
阳极: 玻璃碳
阴极: 玻璃碳
电解条件:
温度: 50℃
电流密度: 2.8 mA/cm2
电荷量: 2-4 F,基于不足量的组分
端电压: 3-6 V。
AAV4:在筛选池中的电化学交叉偶合
将0.76 mmol的待氧化的物质A(不足量的组分)与2至3倍过量(1.52-2.28 mmol)的偶合对应物B在5 ml的1,1,1,3,3,3-六氟异丙醇或5 ml的HFIP和甲醇混合溶剂(具有18体积%的MeOH,基于HFIP和MeOH的总和)中在烧杯玻璃池中在玻璃碳电极上而反应。作为导电盐使用浓度为0.09 M的MTBS。进行恒电流的电解。将所述反应混合物置于不锈钢模块中,借助加热搅拌器搅拌和保持在50℃。在电解结束之后,将池内的物质转移到50 ml的圆颈烧瓶中,并且在减压下在50℃和200-70 mbar的旋转蒸发仪上除去溶剂。矿物质化产物以及所含有的导电盐通过乙酸乙酯 (150 ml) 作为洗脱液经过20 g的硅胶60而分离。借助短程蒸馏在球形管蒸馏器上 (100℃和10-3 mbar) 回收未反应的反应物。
电极材料:
阳极: 在铌上的BDD(50μm)
阴极: 在铌上的BDD(50μm)
电解条件:
温度: 50℃
电流密度: 2.8 mA/cm2
电荷量: 2-4 F,基于不足量的组分
端电压: 3-6 V。
两步骤的钳形配体合成
实施例1:
3,4'-二甲氧基-2',5-二甲基-2-羟基联苯和2',3-二甲氧基-4',5-二甲基-2-羟基联苯:
该电解根据AAV1实施,区别在于:使用1.45 g (10.5 mmol,2.0当量)的4-甲基愈创木酚和639 mg (5.23 mmol,1.0当量)的3-甲基苯甲醚。电流密度为2.8 mA/cm2,电荷量为4 F每3-甲基苯甲醚 (Q = 2018 C)。在分离出溶剂之后,将产物混合物通过柱色谱法在硅胶60上纯化,洗脱液为9:1体积比 (CH (环己烷):EE (乙酸乙酯))。获得的产物为浅黄色的油状物质。
3,4'-二甲氧基-2',5-二甲基-2-羟基联苯
HRMS (ESI, 正极模式): m/z, C16H19O3 [M+H+]: 计算值: 259.1334 测量值: 259.1332。
2',3-二甲氧基-4',5-二甲基-2-羟基联苯
HRMS (ESI, 正极模式): m/z, C16H18O3Na [M+Na+]:计算值: 281.1154; 测量值: 281.1165。
实施例2:
2-羟基-5-甲基-2',3,4'-三甲氧基联苯
该电解根据AAV2实施,使用16.58 g (120 mmol,1.0当量)的4-甲基愈创木酚和49.74 mg (360 mmol,3.0当量)的1,3-二甲氧基苯。电流密度为2.8 mA/cm2,电荷量为2 F每4-甲基愈创木酚 (Q = 23156 C)。在分离出溶剂和导电盐之后,将产物通过短程蒸馏在球形管蒸馏器上得到黄色油状物 (140℃,10-3 mbar)。
HRMS (ESI, 正极模式): m/z,C16H18O4 (M+Na+): 计算值:297.1103; 测量值:297.1096。
实施例3:
2,2''-二羟基-5'',2'-二甲基-3-(1,1-二甲基乙基)-3'',4',5-三甲氧基[1,1';5',1'']三联苯
该电解根据AAV1实施,使用325 mg (1.81 mmol,1.0当量)的2-(1,1-二甲基乙基)-4-甲氧基苯酚和1.40 g (5.42 mmol,3.0当量)的2',3-二甲氧基-4',5-二甲基-2-羟基联苯。电流密度为2.8 mA/cm2,电荷量为2 F每2-(1,1-二甲基乙基)-4-甲氧基苯酚 (Q = 350 C)。在分离出溶剂之后,将反应产物通过柱色谱法在硅胶60上纯化,使用洗脱液混合物9:1 (CH:EE)。获得的产物为黄色油状物。
HRMS (ESI, 正极模式): m/z,C27H32O5 (M+Na+): 计算值:459.2147; 测量值:459.2147。
实施例4:
2,2''-二羟基-4'',5-二甲基-2',3,4',5''-四甲氧基[1,1';5',1'']三联苯
该电解根据AAV1实施,使用250 mg (1.8 mmol,1.0当量)的4-甲氧基-3-甲基苯酚和1.49 g (5.4 mmol,3.0当量)的2-羟基-5-甲基-2',3,4'-三甲氧基联苯。电流密度为2.8 mA/cm2,电荷量为2 F每4-甲氧基-3-甲基苯酚 (Q = 350 C)。第一纯化通过在球形管蒸馏器上分离出反应物进行 (120 ℃,10-3 mbar)。将产物通过柱色谱法在硅胶60上使用梯度4:1,然后7:3,然后2:1 (CH:EE) 纯化。获得的三联苯化合物为黄色的固体。
MS (ESI, 正极模式): m/z,C24H26O6 (M+Na+): 计算值: 433.16, 测量值: 433.16。
本发明的实施例显示了,根据本发明的方法可以以令人满意的产率制备所述不对称的OCO钳形配体。
实施例5:
通过保护基团而衍生化来自实施例2的酚-芳烃-组分
使用(1,1-二甲基乙基)二甲基甲硅烷基氯 (TBDMS氯化物)保护
示意图4:根据实施例5的反应
在具有干燥管的圆底烧瓶中,预先装入2.26 g的咪唑 (33.2 mmol,1.1 当量)。将8.29 g (30.2 mmol,1.0 当量)的实施例2的AB化合物 (2-羟基-5-甲基-2',3,4'-三甲氧基联苯)溶解在大约 50 ml的干燥二氯甲烷中和转移到所述烧瓶中。然后,将5.92 g (39.3 mmol,1.3 当量)的TBDMS氯化物同样溶解在大约50 ml的干燥二氯甲烷中和转移到所述烧瓶中。另外加入100 ml的二氯甲烷。然后将该混合物在室温下搅拌24 h。在反应过程中,产生了无色的片状沉淀物。通过三次萃取分别使用100 ml的水而分离产物,从而分离出生成的盐。在通过硫酸钠干燥和在旋转蒸发仪上除去二氯甲烷之后,蒸馏除去过量的氯化甲硅烷 (50℃,10-3 mbar),其中沉淀的产物为无色的固体。
实施例6:
通过保护基团而衍生化来自实施例2的酚-芳烃-组分
使用三(甲基乙基)甲硅烷基氯 (TIPS氯化物)保护
示意图5:根据实施例6的反应
在具有干燥管的圆底烧瓶中,预先装入5.27 g的咪唑 (27.3 mmol,2.5 当量)。将8.50 g (31.0 mmol,1.0 当量)的实施例2的所述AB化合物 (2-羟基-5-甲基-2',3,4'-三甲氧基联苯)溶解在大约60 ml的干燥二氯甲烷和转移到所述烧瓶中。然后,将11.57 g (62.0 mmol,2.0 当量)的TIPS氯化物同样加入到所述混合物,其中生成橙色/浅黄色的溶液。将该混合物在室温下搅拌24 h。然后,再加入0.5 当量的TIPS氯化物并且将该混合物在室温下继续搅拌48 h。在反应过程中,产生了无色的片状沉淀物。通过三次萃取分别使用100 ml的水而分离产物,从而分离出生成的盐。在通过硫酸钠干燥和在旋转蒸发仪上除去二氯甲烷之后,蒸馏除去过量的氯化甲硅烷 (50℃,10-3 mbar),其中沉淀的产物为无色的固体。
实施例7:
2,2''-二羟基-3''-(1,1-二甲基乙基) -5-甲基-2',3,4',5''-四甲氧基- [1,1',5',1'']-三联苯的制备
该电解根据通用规范AAV1在法兰池中实施。为此,使用33 ml电解质体积的HFIP和1.03 g的MTBS作为导电盐 (0.09 M)。针对电化学交叉偶合,使用0.90 g (5.0 mmol,1.0 当量)的2-(1,1-二甲基乙基)-4-甲氧基苯酚和6.45 g (15 mmol,3.0 当量)的实施例6的经保护的酚-芳烃-偶合产物。进行恒电流的电解,其中生成深色的溶液。在电解结束之后,将溶剂在减压下在旋转蒸发仪上除去和将棕色的残余物使用大约300 ml的乙酸乙酯通过硅胶60过滤。最后通过柱色谱法两次分离产物混合物 (柱尺寸:50 x 3 cm):在硅胶60上采用快速色谱法,使用下列梯度的洗脱液混合物:95:5至9:1到5:1至2:1 (CH:EE,每根柱使用大约3 l的CH和2 l的EE)。由此获得除去了保护基团的相应产物,其为深红色的固体。
实施例8:
2, 2''-二羟基-4'',5-二甲基-2',3,4',5''-四甲氧基- [1,1',5',1'']-三联苯的制备
该电解根据通用规范AAV1在法兰池中实施。为此,使用33 ml电解质体积的HFIP和1.03 g的MTBS作为导电盐 (0.09 M)。针对电化学交叉偶合,使用0.69 g (5.0 mmol,1.0 当量)的4-甲氧基-3-甲基苯酚和6.45 g (15 mmol,3.0 当量)的实施例6的经保护的酚-芳烃-偶合产物。进行恒电流的电解,其中生成黑色的溶液。在电解结束之后,将溶剂在减压下在旋转蒸发仪上除去和将剩余的棕色溶液使用大约300 ml的乙酸乙酯通过硅胶60过滤。最后通过柱色谱法两次分离产物混合物 (柱尺寸:50 x 3 cm):在硅胶60上采用快速色谱法,使用下列梯度的洗脱液混合物:95:5至9:1到5:1至2:1 (CH:EE,每根柱使用大约3 l的CH和2 l的EE)。由此获得除去了保护基团的相应产物,其为深红色的固体。
由实施例7和8表明,通过在酚-芳烃-偶合产物中引入保护基团(甲硅烷基)和然后在电化学实施的方法步骤c) 中进行偶合而进一步提高了间三联苯化合物的产率(提高2至3倍的产率)。因此,以这种方式可以显著改进整个方法的效率。
- 还没有人留言评论。精彩留言会获得点赞!