一种维帕他韦新晶型及其制备方法与流程
本发明涉及药物化学领域,尤其涉及一种维帕他韦新晶型,具体地涉及一种维帕他韦与糖精的共晶化合物。
背景技术:
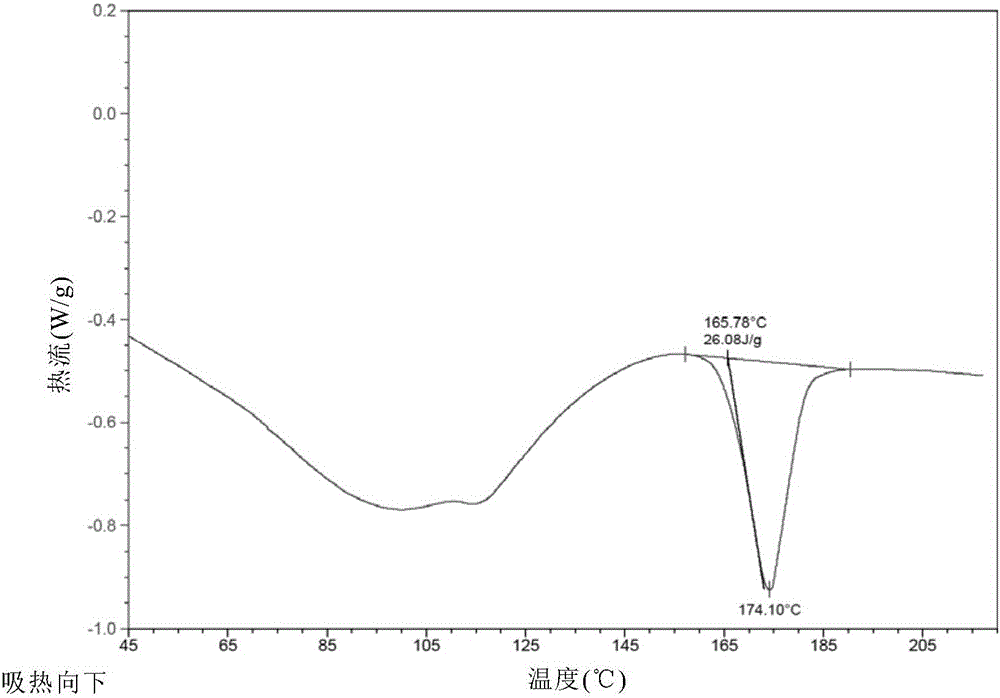
:维帕他韦(Velpatasvir,VLP,式II)是Gilead开发的丙肝治疗药物,已于2016年6月被FDA批准上市,EMA也已于2016年7月批准该药。通过和索非布韦(Sofosbuvir,SOF)联用,该组合疗法可以在短至8周的时间里治愈所有已知基因型(1-6)HCV患者,同时无需注射干扰素或联合利巴韦林(Ribavirin)。由于分子结构的特点,维帕他韦的结晶性较差。目前,Gilead公司的专利US20150361085A1申请保护了维帕他韦的通道溶剂化物、盐酸盐、磷酸盐、氢溴酸盐、和酒石酸盐。但是这些晶型的稳定性、纯化效果、吸湿性和工艺稳定性不够完善,不能很好地满足实际应用需求。因此,本领域急需开发一种新的维帕他韦晶型,以进一步改善维帕他韦的结晶性、稳定性、吸湿性和工艺稳定性,从而推进其生产和应用。技术实现要素:本发明的目的在于提供一种新的维帕他韦和糖精的共晶,以进一步改善维帕他韦的结晶性、稳定性和理化性质,从而推进其生产和应用。本发明第一方面,提供一种式I所示化合物,所述化合物由维帕他韦与糖精形成,在另一优选例中,所述式I化合物为晶型或无定形物。在另一优选例中,采用HPLC测定,所述式I化合物的纯度≥99.0%(峰面积归一化法),较佳地≥99.2%(峰面积归一化法),更佳地≥99.5%(峰面积归一化法)。本发明第二方面,提供一种制备如本发明第一方面所述的式I化合物的方法,包括步骤:(1)将式II化合物与糖精反应,生成式I化合物:在另一优选例中,所述式II化合物与糖精的摩尔比为1:1.5-1:2.5,较佳地为1:1.8-1:2.2。在另一优选例中,所述方法还包括步骤(2):在惰性溶剂中,对步骤(1)得到的式I化合物进行析晶处理,得到式I化合物的晶体,其中,所述惰性溶剂选自下组:乙醇、异丙醇、丙酮、乙腈、四氢呋喃、乙酸乙酯、乙酸异丙酯、甲苯、甲基叔丁基醚、水、或其组合。本发明第三方面,提供一种如本发明第一方面所述的式I化合物的晶型,所述晶型为晶型A,所述晶型A的X射线粉末衍射图谱包括3个或3个以上选自下组的2θ值:3.5°±0.2°、5.3°±0.2°、5.8°±0.2°、7.2°±0.2°、10.7°±0.2°或16.3°±0.2°。在另一优选例中,所述晶型A的X射线粉末衍射图谱包括3个或3个以上选自下组的2θ值:3.5°±0.2°、5.3°±0.2°、5.8°±0.2°、7.2°±0.2°、10.7°±0.2°、11.1°±0.2°、11.7°±0.2°或16.3°±0.2°。在另一优选例中,所述晶型A的X射线粉末衍射图谱包括3个或3个以上选自下组的2θ值:3.5°±0.2°、5.3°±0.2°、5.8°±0.2°、7.2°±0.2°、8.0±0.2°、9.57±0.2°、10.7°±0.2°、11.1°±0.2°、11.7°±0.2°或16.3°±0.2°。在另一优选例中,所述晶型A具有选自下组的一个或多个特征:(1)所述晶型A的X射线粉末衍射图谱基本如图1所表征;(2)所述晶型A的差示扫描量热法分析图谱的起始温度(onset)为165.8±3℃,和/或峰值温度(peak)为174.1±3℃;(3)所述晶型A的差示扫描量热法分析图谱基本如图2所表征;和/或(4)所述晶型A的热重分析图谱基本如图3所表征。本发明第四方面,提供一种制备如本发明第三方面所述的晶型A的方法,包括步骤:(a)在溶剂中,将式II化合物与糖精反应,生成式I化合物,并且,所述溶剂选自下组:乙醇、异丙醇、丙酮、乙腈、四氢呋喃、乙酸乙酯、乙酸异丙酯、甲苯、甲基叔丁基醚、水、或其组合;(b)对步骤(a)生成的式I化合物进行结晶处理,从而形成所述晶型A。在另一优选例中,所述步骤(a)中,所述式II化合物为无定形物。在另一优选例中,所述步骤(a)中,生成的式I化合物为无定形物。在另一优选例中,所述步骤(b)包括步骤(b-1):加入晶种,对步骤(a)生成的式I化合物进行结晶处理,从而形成所述晶型A。在另一优选例中,所述步骤(b-1)中,所述晶种为权利要求3所述的晶型A。在另一优选例中,所述步骤(b)包括:任选地,在惰性溶剂中,对步骤(a)生成的式I化合物进行结晶处理,从而形成所述晶型A,所述惰性溶剂选自下组:乙醇、异丙醇、丙酮、乙腈、四氢呋喃、乙酸乙酯、乙酸异丙酯、甲苯、甲基叔丁基醚、水、或其组合。在另一优选例中,所述步骤(a)中,反应温度为0-50℃,较佳地为4-35℃,更佳地为10-25℃。在另一优选例中,所述步骤(a)中,反应时间为0.1-24小时,较佳地为0.2-12小时,更佳地为0.5-3小时。应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。附图说明图1显示了式I化合物晶型A的X-射线粉末衍射谱图(XRPD)。图2显示了式I化合物晶型A的差示扫描量热分析谱图(DSC)。图3显示了式I化合物晶型A的热失重分析谱图(TGA)。图4显示了式I化合物晶型A的吸湿曲线、维帕他韦无定形物的吸湿曲线、维帕他韦磷酸盐的吸湿曲线、维帕他韦磷酸盐的解吸附曲线、维帕他韦无定形物的解吸附曲线、以及式I化合物晶型A的解吸附曲线。具体实施方式本发明人经过广泛而深入的研究,对维帕他韦的制备工艺进行了大量优化研究,首次意外地获得了一种特别适合于生产的维帕他韦和糖精的共晶化合物,并且所述共晶化合物优选为晶型A。本发明的式I化合物的晶型A易于制备,有良好的理化性质和稳定性,有助于减少杂质,进而提高维帕他韦的质量。发明人在此基础上完成了本发明。术语XRPD:X-射线粉末衍射,X射线粉末衍射图谱为以下条件下测定:Cu-Ka,DSC:差示扫描量热TGA:热重分析DVS:动态水分吸附wt%:重量百分比化合物及其晶型如本文所用,术语“本发明化合物”指式I化合物,包括其无定形、晶型或其混合物。如本文所用,所述“本发明的晶体”、“本发明的晶型”、“本发明的式I化合物的晶型”、“式I化合物的晶型”、“维帕他韦和糖精共晶晶型A”、“晶型A”可互换使用,均指式I化合物的晶型A。结晶在本发明中,可以通过操作溶液,使得感兴趣化合物的达到过饱和,从而完成生产规模的结晶。这可以通过多种方法来完成,例如,在相对高的温度下溶解化合物,然后冷却溶液至饱和溶解度以下。或者通过沸腾、常压蒸发、真空干燥或通过其它的一些方法来减小液体体积。可通过加入反溶剂来降低感兴趣化合物的溶解度。另一种可选方法是调节pH值以降低溶解度。有关结晶方面的详细描述请见Crystallization,第三版,JWMullens,Butterworth-HeinemanLtd.,1993,ISBN0750611294。结晶的优化可包括用所需形式的晶体作为晶种接种于结晶介质中。另外,许多结晶方法使用上述策略的组合。一个实施例是在高温下将感兴趣的化合物溶解在溶剂中,随后通过受控方式加入适当体积的抗溶剂,以使体系正好在饱和水平之下。此时,可加入所需形式的晶种(并保持晶种的完整性),将体系冷却以完成结晶。共晶药物共晶是活性药物成分通过非共价键和共晶形成物结合在一个晶格中形成的。药物共晶组分中至少有一个是分子或离子型药物,同时所有组分在室温下均为固体。它是一种新的药物固体型态,可以改善药物的理化性质,比如改善溶解度、增加稳定性、提高生物利用度等。本发明的主要优点包括:(1)本发明所述的式I化合物(包括晶型或无定形物)的制备方法简单、快速,便于大规模化生产。(2)本发明所述的式I化合物在某些溶剂中的溶解度特别低,因此在短时间内形成沉淀或结晶,因此纯化和分离非常方便和快速。(3)本发明的晶型I具有非常高的纯度(如≥99%),特别适合用作生产高质量的维帕他韦。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。仪器和通用方法XRPD:BrukerD8AdvanceX-射线粉末衍射仪,Cu靶,Ka波长,管电压40KV,管电流40mA。扫描范围:3-40°2-Theta;步进:0.02°;扫描速度:1步/秒。DSC:TAQ2000差示扫描量热仪;温度范围:40-200℃;加热速率:10℃/分钟。TGA:NETZSCHTG209F3热重分析仪;温度范围:30-400℃;加热速率:10℃/分钟。实施例1制备式I化合物的无定形物称取84mg式II化合物粗品(纯度99%)至1.5ml离心管,加入1mL的乙酸乙酯,超声至溶解,加入40mg糖精并超声5分钟,有浅黄色油状沉淀形成。离心并移去上清液,置于60℃烘箱干燥12小时,得到黄色固体,经偏光显微镜确定为式I化合物的无定形物。实施例2制备式I化合物的晶型A称取84mg式II化合物粗品(纯度99%)至1.5ml离心管,加入0.4ml丙酮,超声至溶解,然后加入38mg糖精,超声至溶解。将此溶液于置于室温放置。7天后观察到有固体沉淀析出,经偏光显微镜确认为晶体。将该离心管置于离心机(Eppendorfminispin)在12000转离心5分钟,移去上清液,将分离到的固体于室温下干燥1小时,得到维帕他韦和糖精的共晶晶型A。结果对实施例2所得晶型A进行XRD、DSC、TGA和HPLC等检测。本发明实施例2的式I化合物晶型A的XRD图如图1所示,其中,晶型A的主要衍射峰以及相对强度如表1所示。表1.维帕他韦和糖精的共晶晶型A的XRD数据2θ位置[°]相对强度[%]3.519.45.346.45.8100.07.245.610.733.411.111.711.712.516.349.8实施例2的式I化合物晶型A的DSC图如图2所示。从图2中可知,在45-200℃之间,晶型A的热流曲线包含有一个熔融吸热峰:起始温度和峰值温度分别为165.8℃和174.1℃。实施例2的式I化合物晶型A的TGA图如图3所示。从图3中可知,在30-190℃之间,晶型A的热重曲线有3.52%的缓慢失重,是由于残留溶剂缓慢挥发造成。200℃之后,化合物开始分解,造成进一步的失重。HPLC分析结果表明,晶型A中糖精的含量为29.8wt%,证明晶型A中维帕他韦和糖精的摩尔比例为1:2(理论糖精含量为30.3wt%)。HPLC纯度为96%的维帕他韦粗品在经实施例2所述方法结晶为晶型A后,其HPLC纯度可以大幅度提高至98.6%。进一步地,将HPLC纯度已经为99.0%的维帕他韦无定形粉进行实施例2所述操作结晶为晶型A后,所得晶型A的HPLC纯度还可以得到进一步的提高,达到99.4%。这表明,经实施例2所述结晶方法将维帕他韦粗品结晶为晶型A可高效去除维帕他韦中的杂质,显著提高产品的纯度。实施例3制备式I化合物的晶型A称取100mg式I化合物无定形(纯度99%)至1.5ml离心管,加入1ml丙酮,超声至溶解,然后加入晶型A的晶种,将此溶液于置于室温放置。4小时后观察到有固体沉淀析出。将该离心管置于离心机(Eppendorfminispin)在12000转离心5分钟,移去上清液,将分离到的固体于室温下干燥1小时,经XRPD确认为维帕他韦和糖精的共晶晶型A。实施例4晶型A吸湿性评估吸湿性通过动态水分吸附(DVS)进行了评估,具体测试方法如下:仪器:SMSDVSAdvantage方法:相对湿度0-90%,step:10%药物吸湿性是关键理化性质之一,会对药物稳定性、粉体学性质和后继加工工艺有显著影响。晶型A的吸湿性结果如图4所示,同时对比了无定形的维帕他韦和维帕他韦磷酸盐(晶型XIV,参见专利US20150361085A1)的吸湿性。结果表明,晶型A的吸湿性明显优于无定形和维帕他韦磷酸盐。实施例5光照稳定性评估(1)对维帕他韦和糖精的共晶晶型A、维帕他韦磷酸盐(晶型XIV,参见专利US20150361085A1)和维帕他韦无定形的光照稳定性进行了考察。将下述样品:维帕他韦和糖精的共晶晶型A、维帕他韦磷酸盐和维帕他韦无定形物分别置于表面皿中,摊成厚度不超过1mm的薄层,放置于光照强度为4400lux的LS-3000光照试验箱中,持续12天光照。(2)应用HPLC对光照后样品纯度进行检查并与初始纯度进行对比。结果如表2所示,经过120万lux·小时的光照后,维帕他韦无定形的HPLC纯度下降了6.5%,维帕他韦磷酸盐的HPLC纯度下降了28.5%,维帕他韦和糖精的共晶晶型A的HPLC纯度下降了3.9%。维帕他韦和糖精的共晶晶型A的光稳定性明显优于维帕他韦无定形物和维帕他韦磷酸盐。表2.维帕他韦不同晶型光照稳定性在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1