一种相选择性凝胶剂及其制备方法和应用与流程
本发明属于功能材料技术领域,涉及一种相选择性凝胶剂及其制备方法和应用。
背景技术:
油类泄漏对于环境和水域生态系统有着灾难性的影响,而且对社会带来重大的经济损失。治理油类泄漏的传统手段有吸附剂、分散剂、生物降解和固化剂等,但在实际应用中的效果不大。近期,越来越多的人对发现新型治理油类泄漏方法投以关注。其中最有前途的一种方法是用相选择性有机凝胶因子来使水中的油形成凝胶。这种方法不仅有效且对环境友好。但是,通常需要大量的载体溶剂来均匀分散凝胶分子。这些溶剂,有的是溶于水的非凝胶溶剂,会扩散到水中;有的则是不溶于水的凝胶溶剂,会和油一起形成凝胶。在这两种情况下,或者会给生态系统带来影响,或者不利于油类的回收。因此,将凝胶因子以粉末的形式直接应用会更加实际且理想。
CN105686971A公开了一种聚氨基酸凝胶剂及其制备方法和应用,所述聚氨基酸凝胶剂使用以质量百分数计的1-8%的-聚谷氨酸、1-4%的聚赖氨酸、3-18%的-环糊精、70-95%的水以及余量的功能成分制备而成,其主要用于面膜或者乳液等护肤品中作为添加剂,而并没有胶凝油类的作用。
CN104803850A公开了一种酯类糖基相选择性亲油凝胶因子及其制备方法以及在油品胶凝中的应用,该发明以甘露糖醇和中长基链酰氯为原料采用O-酯化反应制备得到酯类糖基相选择性亲油凝胶因子,其可以进入油相,起到胶凝的作用。
CN105111108A公开了一种脂肪类叔胺双脲小分子凝胶剂及其触变性分子凝胶,该凝胶剂分子具有以下结构:
其中R代表H或C1-C5烷基;R1-R4各自独立的代表H或C1-C2烷基。虽然该凝胶剂具有形成剪切触变凝胶的能力,但是只有将其在有机溶剂中加热溶解,而后冷却才可以起到形成稳定的剪切触变凝胶,其可用于制备有机或无机材料的无缺陷晶体的良性介质,但却不能用于室温下的油水分离。
因此,在本领域期望得到一种能够直接以粉末的形式与水中的油类或有机溶剂形成凝胶的脲基有机凝胶因子。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种相选择性凝胶剂及其制备方法和应用。本发明的凝胶剂可以粉末的形式与水中的油类或有机溶剂形成凝胶,在室温下完成油水分离。
为达此目的,本发明采用以下技术方案:
一方面,本发明提供一种相选择性凝胶剂,所述相选择性凝胶剂具有式I所示结构:
式I中,R为C8-C18(例如C8、C9、C10、C11、C12、C13、C14、C15、C16、C17或C18)的直链烷基,优选C12直链烷基(即-(CH2)11CH3)。
本发明的式I所示凝胶剂在油水混合物中可以选择性进入油相,在油相中实现自组装,形成物理交联的超分子聚合物,可以使得油类(如汽油、煤油、柴油、机油)以及非极性的有机溶剂(例如正己烷、环己烷、苯、甲苯或二甲苯)形成凝胶,是一种具有相选择性的凝胶剂。可以粉末的形式与水中的油类或有机溶剂形成凝胶,在室温下完成油水分离。
通常在两个脲基或酰胺基之间连上平面的芳香基团,使π-π相互作用和氢键共同作用形成超分子聚合物,因此认为如果在两个脲基之间引入弯曲的桥连双萘可能会影响π-π键和氢键这两种作用的共同作用,但是事实情况却并不是这样,本发明的相选择性凝胶剂中两个脲基通过桥连的双萘骨架连接在一起后,两个脲基可以促进桥连双萘骨架之间的非共价相互作用,使得式I所示化合物具有胶凝剂性质。然而,如果在两个酰胺基之间引入弯曲的桥连双萘却不会产生这样的效果。
在两个脲基通过桥连的双萘骨架连接在一起形成式I所示凝胶剂,桥连的双萘骨架在固体状态下,以反向平行的方式形成“从背后拥抱”的类似于“超分子聚合物”的结构,相邻分子间存在的C-H…π相互作用和偏离中心的π…π相互作用得到最大化,并且满足偶极作用,这种完美的组织方式能够使所有的相互作用得到最大化,桥连双萘之间的堆积能够与脲基的氢键相互促进,在这些作用下能够形成非常稳定的超分子聚合物。在非极性溶液中,这种效果得到放大,从而形成聚集度很高的超分子聚合物,这些聚合物进一步缠绕从而使这些溶剂形成凝胶。
另一方面,本发明提供了如上所述的相选择性凝胶剂的制备方法,所述制备方法为:采用式II所示的异氰酸酯与式III所示的双胺化合物反应得到所述相选择性凝胶剂,反应式如下:
其中R为C8-C18(例如C8、C9、C10、C11、C12、C13、C14、C15、C16、C17、C18、)的直链烷基,优选C12直链烷基(即-(CH2)11CH3)。
优选地,所述式II所示的异氰酸酯与式III所示的双胺化合物的摩尔比≥2:1,例如2:1、2.2:1、2.4:1、2.5:1、2.8:1、3:1、3.5:1、4:1等,优选(2-2.5):1。
优选地,所述反应在催化剂存在下进行,所述催化剂为N,N-二异丙基乙胺。在本发明中所述催化剂N,N-二异丙基乙胺除了可以催化反应外,还可以提高式III所示化合物在反应介质中的溶解性。
优选地,所述催化剂的用量为式II所示的异氰酸酯质量的3-8%,例如3%、3.5%、4%、4.5%、5%、5.5%、6%、6.5%、7%、7.5%或8%。
优选地,所述反应的温度为20-30℃,例如20℃、21℃、23℃、25℃、27℃、29℃或30℃。
优选地,所述反应的时间为3-20h,例如3h、5h、8h、10h、12h、14h、16h、18h或20h。
优选地,所述反应的介质为二氯甲烷和/或三氯甲烷;
优选地,所述反应的介质为干燥的二氯甲烷和/或三氯甲烷,即进行过除水干燥处理的二氯甲烷和/或三氯甲烷。
第三方面,本发明提供了所述相选择性凝胶剂在胶凝油类或有机溶剂中的应用。
优选地,所述油类为汽油、煤油、柴油或机油中的任意一种或至少两种的组合。
优选地,所述有机溶剂为正己烷、环己烷、苯、甲苯或二甲苯中的任意一种或至少两种的组合。
本发明的相选择性凝胶剂可使油类或发生有机溶剂发生胶凝,具有油水分离能力,可用于治理油类以及有机溶剂泄漏。
相对于现有技术,本发明具有以下有益效果:
本发明通过将两个脲基连接在桥连双萘骨架上得到式I所示的凝胶剂,该凝胶剂可以选择性地进入油相,两个脲基可以促进桥连双萘骨架之间的非共价相互作用,以发生聚集,使得油相发生胶凝,形成稳定的凝胶,可以使得油水分离,可用于油类以及有机溶剂泄漏的治理,并且该凝胶剂不溶于水,可以回收再利用,安全无污染,在回收海上溢油和快速分离工业含油或有机溶剂废水等领域具有很好的应用前景。
附图说明
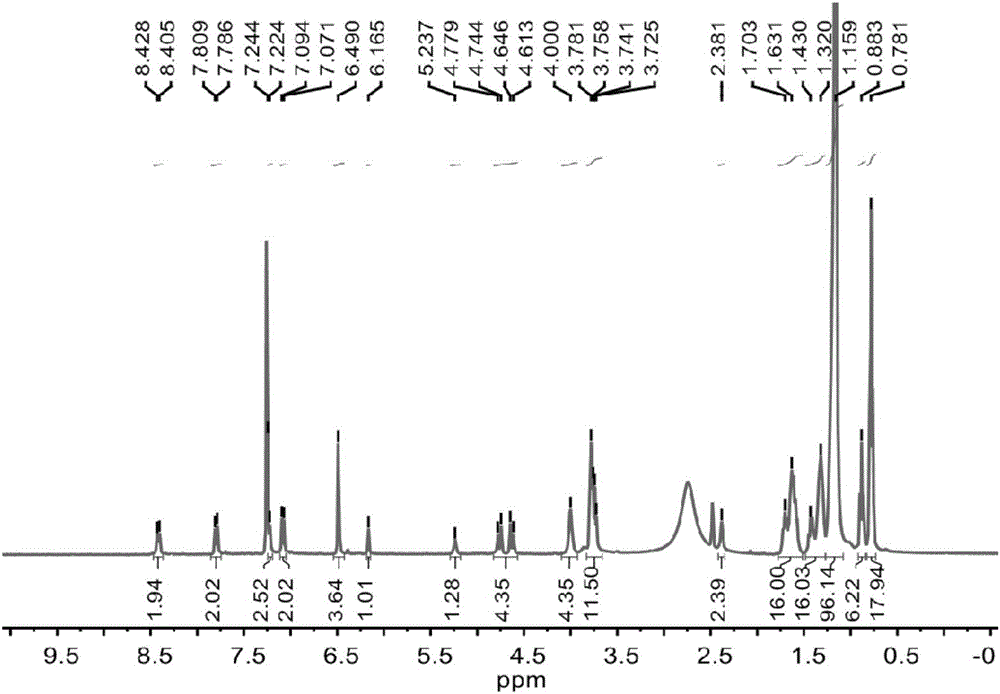
图1为实施例1制备得到的凝胶剂的核磁氢谱图。
图2为实施例1制备得到的凝胶剂的核磁碳谱图。
图3为实施例1制备得到的凝胶剂的质谱图。
图4为本发明凝胶剂使汽油形成凝胶的过程示意图。
图5A为实施例1制备得到的凝胶剂的加入量为汽油质量的5.0%的情况下形成的凝胶在旋转模式下,储能模量(G')和损耗模量(G”)随剪切速率的变化图。
图5B为实施例1制备得到的凝胶剂的加入量为汽油质量的5.0%的情况下形成的凝胶在震荡模式下,储能模量(G')和损耗模量(G”)随剪切应力的变化图。
图6为在混合溶剂氘代氯仿和氘代DMSO(500:1)中不同浓度下的实施例1制备的凝胶剂的核磁氢谱图;
图7为本发明的凝胶剂在汽油中形成的凝胶制备成的干凝胶的扫描电镜图,其标尺为5μm。
图8为利用本发明的凝胶剂进行油水分离的过程示意图。
具体实施方式
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
实施例1
在本实施例中,通过以下反应式制备如下式I所示相选择性凝胶剂:
其中R为-(CH2)11CH3。
制备过程为:
(1)参照文献[Hong Yang,et al.Switchable Fluorescent Organogels and Mesomorphic Superstructure Based on Naphthalene Derivatives,Langmuir 2007,23,8224-8230]合成式II所示化合物,合成流程如下:
中间体S1的合成:在没食子酸甲酯(18g,97.8mmol)与K2CO3(54g,390mmol)的DMF(90mL)悬浮液中加入溴代十二烷(75mL,300mmol),搅拌并加热至70℃,在氩气保护下反应过夜。然后将反应液倒入蒸馏水中(500mL)。过滤沉淀并用丙酮重结晶得到白色固体S1 60.6g,收率约为90%。
中间体S2的合成:将化合物S1(4g,6.8mmol)的乙醇溶液(30mL)与过量NaOH水溶液(10mL)混合,并在常温下搅拌6小时。反应完后用1M的盐酸调反应液pH至1。用二氯甲烷萃取有机相,并用无水Na2SO4干燥,旋蒸得到白色固体S2 3.5g,收率约为90%。
式II化合物的合成:取中间体化合物S2(2.8g,4.1mmol)溶于过量的SOCl2(14mL)中,搅拌并加热至回流,反应4小时。反应完后,旋蒸掉剩余的SOCl2,得到酰氯。取过量NaN3(2g,30mmol)溶于H2O(30mL)。在冰浴下,将上述酰氯的THF(10mL)溶液逐滴加入NaN3水溶液中,常温下搅拌并反应过夜。反应完后用二氯甲烷萃取有机相,并用饱和食盐水洗涤,无水Na2SO4干燥,旋蒸得到黄色固体。将此黄色固体在油泵下减压干燥后,溶于超干的甲苯(40mL),氩气保护,搅拌并加热至回流,反应6小时。之后将溶剂旋干,得到式II化合物。
(2)参照文献[Zhenfeng He,et al.Imine Macrocycle with a Deep Cavity:Guest-Selected Formation of syn/anti Configuration and Guest-Controlled Reconfiguration(Supporting Information),Chemistry A European Journal,2015,21,3005-3012.]合成式III所示化合物,合成流程如下:
中间体化合物C1的合成:无水K2CO3(4.5g,32.5mmol)加入到溶有2,6-二羟基萘(4.0g,25.0mmol)的DMF(200mL)溶液中加热搅拌1h后,通过恒压滴液漏斗将正溴丁烷(3.2mL,30.0mmol)缓慢加入到反应液中。加热80℃反应7h。冷却至室温,向反应液中加入大量水并加入少量稀盐酸(pH=7)搅拌,大量不溶固体析出。过滤并用大量水洗涤滤渣,将滤渣溶于200mL甲醇中,搅拌过滤,得到的滤液加入无水硫酸钠干燥过夜,过滤将得到的滤液蒸干。得到的残留物通过柱层析分离(石油醚/二氯甲烷=1/1,v/v),得到白色固体(1.6g,30%)。
中间体化合物C2的合成:化合物C1(4.0g,18.5mmol)溶于到三氟乙酸(TFA,25mL)和四氢呋喃(THF,100mL)的混合溶液中,通过恒压滴液漏斗将丙二缩醛(1.6mL,9.7mmol)缓慢加入到反应液中,室温反应3h。向其中加入1mol/L NaOH溶液、饱和Na2HCO3溶液,调节pH=8。用二氯甲烷萃取有机相后,加入无水Na2SO4干燥,旋蒸干溶剂,残留物通过柱层析分离(PE/DCM=5/1,v/v)得到白色固体C2(3.4g,80%)。
中间体化合物C3的合成:冰浴下,将化合物C2(1.5g,3.2mmol)溶于干燥的二氯甲烷(30mL)中,边搅拌边缓慢加入1,1-二氯甲醚(7.5mL,13mmol)和四氯化钛(2.2g,11.6mmol)。冰浴下搅拌反应1小时后升至室温再继续反应2.5h。反应完成后向反应液中加入冰水混合物,并加入大量饱和NaHCO3搅拌使其pH=8。用二氯甲烷萃取有机相,加入无水Na2SO4干燥。过滤后旋蒸干溶剂并通过柱层析分离(PE/DCM=1/1,v/v)得到黄色固体C3(1.4g,86%)。
中间体化合物C4的合成:化合物C3(0.6g,1.1mmol)溶于MeCN(20mL)与DCM(40mL)的混合溶液中,向其中逐步加入氨基甲酸叔丁酯(0.8g,6.9mmol)、三乙基硅烷(1.1mL,7.1mmol)和三氟乙酸(1mL,5.0mmol),室温搅拌反应过夜。加入饱和NaHCO3搅拌使其pH=8,用二氯甲烷萃取有机相,加入无水Na2SO4干燥。过滤后旋蒸干溶剂并通过柱层析分离(PE/DCM=2/1,v/v)得到白色固体(0.6g,76%)。
式III化合物的合成:化合物C4(1.0g,1.9mmol)溶于三氟乙酸(15mL)与DCM(30mL)的混合溶液中室温搅拌4小时。旋蒸干溶剂后将剩余固体进一步抽干得到褐色固体粉末。将其溶于二氯甲烷中搅拌后过滤得到白色固体,加入二氯甲烷(30mL)和1M NaOH溶液(20mL)搅拌,固体逐渐溶解。萃取有机相,加入无水Na2SO4干燥,过滤后旋蒸干溶剂得到淡黄色固体(0.6g,71%)。
1H NMR(400MHz,CDCl3,300K):ppm=8.47(d,J=9.2,2H),7.81(d,J=9.2,2H),7.32(d,J=9.2,2H),7.19(d,J=9.2,2H),6.25(s,1H),5.29(s,1H),4.18(s,4H),4.15-4.02(m,4H),2.47-2.35(m,2H),1.88-1.79(m,4H),1.62-1.47(m,4H),1.00(t,J=7.4Hz,6H);13C NMR(100MHz,CDCl3,300K):ppm=152.26,148.45,128.65,126.77,125.62,123.11,123.01,119.38,119.05,114.44,91.38,69.01,36.37,31.73,27.06,23.03,19.43,13.92;ESI-TOF-HRMS:m/z calcd for[M+Na]+C33H38N2O4Na 549.2724;found 549.2725。
(3)取式III化合物(800mg,1.5mmol)和N,N-二异丙基乙胺(0.2mL)溶于干燥的二氯甲烷(20mL)中。常温下,通过恒压滴液漏斗将式II化合物(约2.4g,3.6mmol)的干燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加时间30分钟,将得到的混合物再在常温下搅拌5个小时。反应完成后,过滤沉淀并依次用二氯甲烷和甲醇洗涤,得到白色固体2.5g,收率约为89%。
对本实施例制备得到的白色固体产物进行核磁氢谱、核磁碳谱和质谱表征,结果如图1-3所示,其数据结果如下:
1H NMR(400MHz,CDCl3:DMSO-d6=99:1,25℃):δ[ppm]=8.42(d,J=9.3Hz,2H),7.80(d,J=9.3Hz,2H),7.23(d,J=8.1Hz,2H),7.08(d,J=9.2Hz,2H),6.49(s,4H),6.16(s,1H),5.24(s,1H),4.70(dd,J=52.7,13.4Hz,4H),4.07-3.90(m,4H),3.87-3.65(m,12H),2.38(s,2H),1.81-1.52(m,16H),1.50–1.26(m,16H),1.16(s,96H),0.93-0.84(m,6H),0.82-0.70(m,18H).13C NMR(126MHz,CDCl3:DMSO-d6=10:1,25℃):δ[ppm]=155.34,152.61,152.46,148.22,135.40,132.20,128.67,126.01,123.62,120.55,119.03,118.65,96.85,90.81,72.86,69.09,68.30,33.48,31.39,31.13,29.78,29.22,29.20,29.17,29.12,28.91,28.85,28.82,25.65,25.61,22.21,22.16,18.80,13.67,13.47.ESI-TOF-HRMS:m/z calcd for[M+H]+C119H193O12N4+,1870.4592;found 1870.4610(error=-0.9623ppm)。
该结果证明制备得到的白色固体产物具有本实施例式I所示结构。
实施例2
在本实施例中,通过与实施例1相同的方法制备得到式II化合物和式III化合物;通过以下方法制备得到式I化合物:
取式III化合物3(800mg,1.5mmol)和N,N-二异丙基乙胺(0.07mL)溶于干燥的二氯甲烷(20mL)中。常温下,通过恒压滴液漏斗将式II化合物(约2g,3.0mmol)的干燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加时间30分钟,将得到的混合物再在30℃下搅拌12个小时。反应完成后,过滤沉淀并依次用二氯甲烷和甲醇洗涤,得到白色固体2.3g,收率约为82%。
1H NMR(400MHz,CDCl3:DMSO-d6=99:1,25℃):δ[ppm]=8.42(d,J=9.3Hz,2H),7.80(d,J=9.3Hz,2H),7.23(d,J=8.1Hz,2H),7.08(d,J=9.2Hz,2H),6.49(s,4H),6.16(s,1H),5.24(s,1H),4.70(dd,J=52.7,13.4Hz,4H),4.07-3.90(m,4H),3.87-3.65(m,12H),2.38(s,2H),1.81-1.52(m,16H),1.50–1.26(m,16H),1.16(s,96H),0.93-0.84(m,6H),0.82-0.70(m,18H).13C NMR(126MHz,CDCl3:DMSO-d6=10:1,25℃):δ[ppm]=155.34,152.61,152.46,148.22,135.40,132.20,128.67,126.01,123.62,120.55,119.03,118.65,96.85,90.81,72.86,69.09,68.30,33.48,31.39,31.13,29.78,29.22,29.20,29.17,29.12,28.91,28.85,28.82,25.65,25.61,22.21,22.16,18.80,13.67,13.47.ESI-TOF-HRMS:m/z calcd for[M+H]+C119H193O12N4+,1870.4592;found 1870.4613。
实施例3
在本实施例中,通过与实施例1相同的方法制备得到式II化合物和式III化合物;通过以下方法制备得到式I化合物:
取式III化合物3(800mg,1.5mmol)和N,N-二异丙基乙胺(0.25mL)溶于干燥的二氯甲烷(20mL)中。25℃常温下,通过恒压滴液漏斗将式II化合物(约2.5g,3.75mmol)的干燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加时间30分钟,将得到的混合物再在25℃常温下搅拌12个小时。反应完成后,过滤沉淀并依次用二氯甲烷和甲醇洗涤,得到白色固体2.4g,收率约为85%。
1H NMR(400MHz,CDCl3:DMSO-d6=99:1,25℃):δ[ppm]=8.42(d,J=9.3Hz,2H),7.80(d,J=9.3Hz,2H),7.23(d,J=8.1Hz,2H),7.08(d,J=9.2Hz,2H),6.49(s,4H),6.16(s,1H),5.24(s,1H),4.70(dd,J=52.7,13.4Hz,4H),4.07-3.90(m,4H),3.87-3.65(m,12H),2.38(s,2H),1.81-1.52(m,16H),1.50–1.26(m,16H),1.16(s,96H),0.93-0.84(m,6H),0.82-0.70(m,18H).13C NMR(126MHz,CDCl3:DMSO-d6=10:1,25℃):δ[ppm]=155.34,152.61,152.46,148.22,135.40,132.20,128.67,126.01,123.62,120.55,119.03,118.65,96.85,90.81,72.86,69.09,68.30,33.48,31.39,31.13,29.78,29.22,29.20,29.17,29.12,28.91,28.85,28.82,25.65,25.61,22.21,22.16,18.80,13.67,13.47.ESI-TOF-HRMS:m/z calcd for[M+H]+C119H193O12N4+,1870.4592;found 1870.4615。
实施例4
在本实施例中,通过以下反应式制备如下式I所示相选择性凝胶剂:
其中R为-(CH2)7CH3。
首先通过与实施例(1)中步骤(1)相同的方法来合成式II化合物:在合成过程中将实施例1步骤(1)中溴代十二烷替换成溴代正辛烷,合成得到式II化合物,经核磁氢谱验证了式II化合物结构的正确性。
以与实施例1中步骤(2)相同的方法合成得到式III化合物;
通过以下步骤(3)制备得到式I所示凝胶剂,即取式III化合物(800mg,1.5mmol)和N,N-二异丙基乙胺(0.1mL)溶于干燥的二氯甲烷(20mL)中。常温下,通过恒压滴液漏斗将式II化合物(约1.6g,3.3mmol)的干燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加时间30分钟,将得到的混合物再在20℃下搅拌20个小时。反应完成后,过滤沉淀并依次用二氯甲烷和甲醇洗涤,得到白色固体2.0g,收率约为87%。质谱测试数据如下:ESI-TOF-HRMS:m/z calcd for[M+H]+C95H145O12N4+,1535.1795;found 1535.1788。
实施例5
在本实施例中,通过以下反应式制备如下式I所示相选择性凝胶剂:
其中R为-(CH2)17CH3。
首先通过与实施例(1)中步骤(1)相同的方法来合成式II化合物:在合成过程中将实施例1步骤(1)中溴代十二烷替换成溴代正十八烷,合成得到式II化合物,经核磁氢谱验证了式II化合物结构的正确性。
以与实施例1中步骤(2)相同的方法合成得到式III化合物;
通过以下步骤(3)制备得到式I所示凝胶剂,即取式III化合物(800mg,1.5mmol)和N,N-二异丙基乙胺(0.16mL)溶于干燥的二氯甲烷(20mL)中。常温下,通过恒压滴液漏斗将式II化合物(约3.2g,3.45mmol)的干燥二氯甲烷(5mL)溶液滴加入上述溶液中,滴加时间30分钟,将得到的混合物再在25℃下搅拌15个小时。反应完成后,过滤沉淀并依次用二氯甲烷和甲醇洗涤,得到白色固体1.74g,收率约为85%。质谱测试数据如下:ESI-TOF-HRMS:m/z calcd for[M+H]+C155H265O12N4+,2376.7743;found 2376.7745。
实施例6
将实施例1制备得到的式I所示凝胶剂粉末放入装有汽油的小瓶中,其用量为小瓶中汽油质量的1.0%,在室温下静置,大约3小时之后,式I所示凝胶剂粉末扩散入汽油中形成透明澄清的溶液,还未形成凝胶。然而,再过3小时,汽油溶液转变成了凝胶,该过程示意图如图4所示。在其他的油类(例如煤油、柴油或机油)以及有机溶剂(例如正己烷、环己烷、苯、甲苯或二甲苯)中也能看到类似的现象,说明该凝胶剂在这些溶液中有很好的溶解性,能够促使这些油类和有机溶剂发生胶凝,测试了在油类和一些有机溶剂中的临界成胶浓度(CGC)和以及在测定浓度下的成胶温度(Tgel),如表1所示。
表1
由表1的结果可以表明,式I所示凝胶剂可以使得油类和有机溶剂发生胶凝,是很好的凝胶剂,形成的凝胶比较稳定。
在该实施例中,对在加入实施例1制备得到的式I所示凝胶剂为汽油质量的5.0%的情况下形成的凝胶的流变性质进行考察,结果如图5A和图5B所示,在旋转模式下(图5A),储能模量(G')一直高于损耗模量(G”),并且这两种模量都随频率的变化而变化,证明是一种较强的凝胶。在震荡模式下(图5B),压力产量(大约100pa)和应力幅(σ0)表明该凝胶强度足以承担自己的重量,说明形成的凝胶具有较好的机械强度,稳定性较好。
为了验证式I所示相选择性凝胶剂分子发生胶凝的时分子中的相互作用,在不同的浓度下测试胶凝剂在混合溶剂氘代氯仿和氘代DMSO(500:1)中的核磁氢谱(1H NMR),如图6所示,可以看出,随着胶凝剂在混合溶剂中浓度的增加,相对的尖锐的信号峰逐渐变宽,表明胶凝剂分子发生了聚集,同时,位于桥连双萘芳香区的质子峰向高场发生位移,表明形成了“从背后拥抱”的类似于“超分子聚合物”的结构,从而发生了屏蔽效应,因此,具有胶凝剂特征。
将式I所示凝胶剂在汽油中形成的凝胶制备成干凝胶,利用扫描电镜(FESEM,ZEISS Merlin)对其进行表征,结果如图7所示,从图中可以看出,形成的干凝胶存在纠缠的网络,说明由式I所示凝胶剂形成的一维纤维进一步缠绕从而形成凝胶。分子模拟(即利用Spartan’14PM6级理论进行计算)表明桥连双萘之间的堆积确实能够与脲基的氢键相互促进。在这些作用下能够形成非常稳定的超分子聚合物。在非极性溶液中,这种效果得到放大,从而形成聚集度很高的超分子聚合物。这些聚合物进一步缠绕从而使这些溶剂形成凝胶。
同样,实施例2-5制备得到的凝胶剂同样能够使得油类(例如汽油、煤油、柴油或机油)以及有机溶剂(例如正己烷、环己烷、苯、甲苯或二甲苯)发生胶凝。
实施例7
在本实施例中,将实施例1-5制备得到的凝胶剂用于油水分离(实施例1-5制备得到的凝胶剂不溶于水并且形成沉淀),其过程示意图如图8所示,即在烧杯中将10mL水和0.5mL汽油的混合得到油水混合物,汽油分散漂浮在水面上,将凝胶剂粉末(图中以1a表示)加入至烧杯中,加入量为汽油质量的2.0wt%,为了促进凝胶过程,将水相进行搅拌,6小时后汽油形成了凝胶,汽油收缩成一大块的凝胶,用勺子很容易就能从水中勺出。通过蒸馏能够收集回收的凝胶中的汽油并且凝胶剂能够再次循环利用。因此,虽然这种凝胶剂可能成本较高,但它是能够回收利用的。因此,本发明制备的凝胶剂可以用于治理油类泄漏。
申请人声明,本发明通过上述实施例来说明本发明的相选择性凝胶剂及其制备方法和应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明所选用原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
- 还没有人留言评论。精彩留言会获得点赞!