具有提升的酶活性的植酸酶的制作方法
技术领域
本申请涉及一种植酸酶,尤指一种具有提升的酶活性的植酸酶。
现有技术
植酸(phytic acid)又称为肌醇六磷酸(myo-inositol(1,2,3,4,5,6)hexakisphosphate),为植物中储存磷的主要形式,在种子中含量尤其丰富,而种子如谷类及豆类是动物饲料的主要原料。虽然种子中的植酸可成为饲养动物所需磷的重要来源,但只有反刍动物才能代谢植酸以利用其中的磷;对于非反刍动物,不能被消化代谢的植酸反而被视为抗营养物质。而植酸被视为抗营养物质的原因是植酸带有丰富的负电,易与带有正电的离子,如钙离子、镁离子、锌离子、锰离子、铜离子、铁离子螯合,再进一步与蛋白质及淀粉形成复合物,此复合物不仅阻碍金属离子的消化吸收,也影响消化酶作用而阻碍营养物质吸收。
无法代谢植酸的非反刍动物需在饮食中添加无机磷或添加植酸酶(phytase)。以添加无机磷的方式,不仅成本高,无法消除抗营养反应,动物排泄物中未被代谢的植酸还会造成水质污染,破坏生态平衡。而经由植酸酶在饲料中的添加,则可降低磷酸的抗营养现象提高营养吸收,增加动物对饲料中磷的可利用性达到60%之外,还能降低磷的排出达50%。
植酸酶是能够从植酸中水解出磷酸根的酶泛称,在动物、植物、微生物中皆可找到植酸酶基因的存在。植酸酶能够将植酸上的六个磷酸根依序水解出来,不同植酸酶的水解最终程度不一,水解机制也不同,若依此做为分类,植酸酶可分为组氨酸酸性磷酸酶(histidine acid phophatase)、紫色酸性磷酸酶(purple acid phosphatase)、β螺旋桨植酸酶(β-propeller phytase),以及半胱氨酸磷酸酶(cysteine phytase),其中组氨酸酸性磷酸酶因为其最适pH值及作用条件较适合用于饲料添加,因此被广泛的研究。组氨酸酸性磷酸酶催化机制是藉由酸碱反应,将连接肌醇环及磷酸根的磷酸单酯键进行水解作用,释放出磷酸根,其水解步骤分为两个阶段,首先组氨酸酸性磷酸酶会利用活性区的组氨酸攻击植酸中的一个磷酸单酯键,形成磷酸-组氨酸中间体,再由活性区的天冬氨酸利用一个水分子提供质子给磷酸-组氨酸中间体中磷酸根的氧离子,释放出一个磷酸根。
尽管于饲料中添加植酸酶在动物饲养上已显示可增加生产力,且使用500-1000单位活性的植酸酶可代替1克的无机磷添加及减少30-50%磷的排出,但在工业上如何使占有饲料市场60%的植酸酶能具有经济效益,为酶改造持续的目标及需求。植酸酶在饲料工业的制程及应用上,必须符合以下特性:最适酸碱值为pH 2-6.5、抗酸性及抗胃蛋白酶及抗胰蛋白酶分解的特性、产品制粒时的耐热性、产品具有高活性;符合以上条件才能有效降低生产成本,应用于工业上。
适合工业用的酶的取得可由自然界的生物筛选而来,但需要耗费大量时间及人力,往往获得的酶也不符合工业应用条件,需要再加以改造,而运用现有的酶再加以加强改造为较经济的方式。酶改造的策略可分为两种,一是利用定向进化,一是利用理论性的设计。定向进化包括两个步骤,一是制造具有丰富多样性的突变基因库,其次再从基因库中筛选具有特定优化特性的突变株。突变基因库的制造方法常用的有易错聚合酶链式反应(error-prone PCR)及DNA重组反应(DNA shuffling),但庞大的突变基因库需要有大量筛选的方法,否则对于筛选上是相当耗费人力及时间的。近年来,随着可利用的蛋白质结构及序列增加,还有计算机计算软件技术的发展,可识别出蛋白质中与底物结合、活性区域、稳定结构的氨基酸,因此以此理论基础建立的有限突变基因库能更有效地筛选出优化的酶特性。
因此,本申请欲藉由植酸酶的序列比对及结构分析,对一些特定的氨基酸进行基因突变,进而有效提升植酸酶在工业上应用的产业价值。
技术实现要素:
本申请的目的在于改造现有植酸酶,利用序列比对、结构分析及定点突变技术,有效提升植酸酶的作用活性,借以增加植酸酶的工业应用价值。
为达上述目的,本申请的一个较佳实施方式提供一种植酸酶,其氨基酸序列是将SEQ ID NO.2第90个位置的缬氨酸突变成苏氨酸的氨基酸序列。编码该SEQ ID NO.2的基因是从大肠杆菌(Escherichia coli)所筛选出来的EcAppA基因,其所表现的蛋白酶为组氨酸酸性磷酸酶。该植酸酶的氨基酸序列是SEQ ID NO.4的氨基酸序列。
为达上述目的,本申请的一较佳实施方式为提供一种植酸酶,其氨基酸序列是将SEQ ID NO.6第204个位置的天冬酰胺突变成丙氨酸或将第206个位置的丝氨酸突变成丙氨酸的氨基酸序列,以借此移除植酸酶活性区的糖基化位点。编码该SEQ ID NO.6的基因系从大肠杆菌(Escherichia coli)所筛选出来的EcAppA基因进一步经过密码子使用序列优化所得的基因,其所表现的蛋白酶为组氨酸酸性磷酸酶。该植酸酶的氨基酸序列是SEQ ID NO.8或SEQ ID NO.10的氨基酸序列。
附图说明
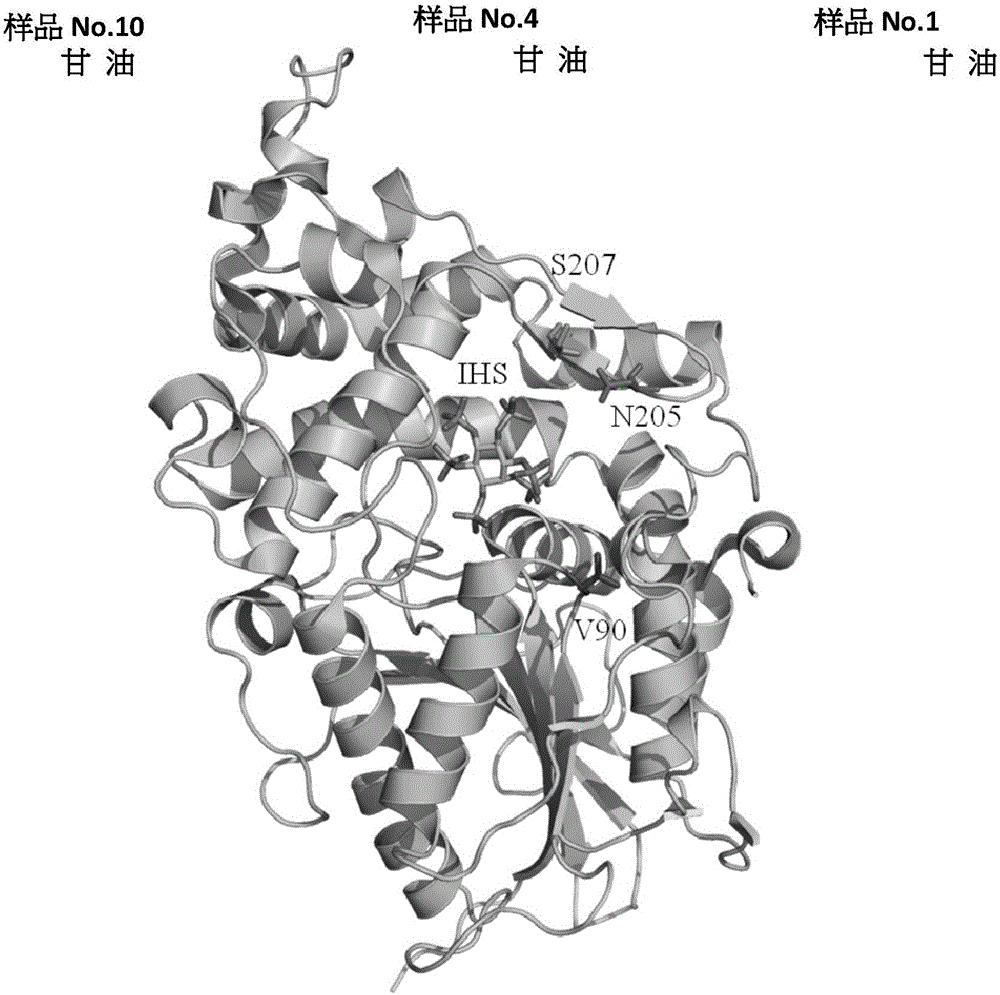
图1显示植酸酶EcAppA蛋白质立体结构及改造点的位置。
图2显示植酸酶EcAppA的基因及氨基酸序列。
图3显示定点突变所采用的引物序列。
图4显示植酸酶EcAppA的Ec-V90T突变株的基因及氨基酸序列。
图5显示植酸酶r-AppA的基因及氨基酸序列。
图6显示植酸酶r-AppA的r-N204A突变株的基因及氨基酸序列。
图7显示植酸酶r-AppA的r-S206A突变株的基因及氨基酸序列。
图8显示野生型Ec-AppA与突变株Ec-V90T的植酸酶活性分析测试。
图9显示野生型r-AppA与突变株r-N204A及r-S206A的植酸酶活性分析测试。
具体实施方式
体现本申请特征与优点的一些典型实施例将在后段的说明中详细叙述。应理解的是本申请能够在不同的实施方式上具有各种的变化,然其均不脱离本申请的范围,且其中的说明及附图在本质上是当作说明的用,而非用以限制本申请。
本申请中的植酸酶基因(EcAppA)是从大肠杆菌(Escherichia coli)所筛选出来的,其所表现的蛋白酶为组氨酸酸性磷酸酶。因为它的高活性及高度底物专一性及最适作用pH范围,均符合饲料的应用,因此具有高产业应用价值,另外,此基因已能成功的在工业生产菌株毕赤酵母(Pichia pastoris)中表达,且此酶已解出蛋白质结构。因此,此植酸酶是相当好的做为进一步酶改良的目标,故本申请欲对此活性已经相当高的植酸酶EcAppA进行改造,以进一步提升其活性。
对于高活性的EcAppA,在改造上要找到增加活性的突变,机率比活性低的酶来的低,因此使用随机点突变大量筛选的成功率相对来的低;再者,即使活性增加也难有乘以倍率的增加,在筛选上不易与野生型植酸酶做区别,因此增加筛选上的困难度,所以本申请使用理智设计(rational design)来提升EcAppA的活性,运用数据库可得的结构及序列加以分析,缩小筛选的目标范围,有效找到活性增加的突变株。
首先,将EcAppA与同样具有高植酸酶活性的CaAppA(来自无丙二酸柠檬酸杆菌Citrobacter amalonaticus的植酸酶)及CbAppA(来自布氏柠檬酸杆菌Citrobacter brakii的植酸酶)进行序列比对,比对结果发现,CaAppA及CbAppA分别与EcAPPA有60%的一致度及70%的相似度,且互相有70%的一致度及80%的相似度。利用这三个具有高度相似性的序列且均有高活性的植酸酶,可将所要突变的范围缩小,减少需要筛选的活性优化突变。
接着,将在CaAppA及CbAppA有一致性但在EcAppA不同的氨基酸位点挑出,再利用已知的EcAppA蛋白质立体结构分析这些位点的位置,保留在活性区附近的氨基酸位点,因为在活性区附近位点造成活性改变的机率较大,以此找出活性有增加的突变株。根据前述分析,氨基酸序列上第90个位置的缬氨酸(Valine)即被选择作为进行定点突变的位置。
此外,利用已知的EcAppA蛋白质立体结构分析毕赤酵母表达时可能产生天冬酰胺(Asparagine)糖基化的位点,亦即在序列上天冬酰胺后第二个氨基酸出现丝氨酸(Serine)或酪氨酸(Tyrosine),而天冬酰胺后的氨基酸不为脯氨酸(Proline)时。从序列上来看,EcAppA有三个可能形成糖基化的位置,利用已知的EcAppA蛋白质立体结构分析观察到其中一个糖基化的位置在植酸酶活性区周围,因此可将此糖基化位置的天冬酰胺(原始氨基酸序列第205个位置)或是天冬酰胺后第二个氨基酸位置的丝氨酸(原始氨基酸序列第207个位置)进行点突变,以制造移除糖基化位点的突变株。
请参阅图1,其显示EcAppA蛋白质立体结构及改造点的位置。EcAppA属于组氨酸酸性磷酸酶家族中的植酸酶,此组氨酸酸性磷酸酶家族结构均有类似的结构,大致分为上下两部份,上半部由α-螺旋组成,具有多变性,下半部由α-螺旋及β折叠组成,结构较为固定。IHS为植酸中的磷以硫取代的合成物质,在做植酸酶蛋白质立体结构时用来指示植酸在酶中结合的位置。本申请的目标改造点V90、N205、S207也显示于图1的结构中。
以下将详述本申请改造植酸酶EcAppA的方法,包括定点突变、蛋白表现及活性测试方法,及其所得到的改良植酸酶蛋白。
首先,本申请所使用的植酸酶EcAppA基因序列为去除信号序列(signal peptide)的Escherichia coli K12 AppA序列(GenBank NC_000913.2),即从第67个碱基开始表达,但有几个不影响活性的自然突变位点,包括原始基因的第266个碱基由T变C(使氨基酸由缬氨酸变丙氨酸),第302个碱基由A变T(使氨基酸由谷氨酰胺变成亮氨酸),及第835个碱基由C变T(不改变氨基酸),序列加上启动子ATG后如图2所示,EcAppA基因的全长为1236个碱基(DNA序列以SEQ ID NO.1标示)以及411个氨基酸(氨基酸序列以SEQ ID NO.2标示)。将EcAppA基因利用EcoRI与XhoI限制酶切位构筑到pET22b表达载体中。定点突变(site-directed mutagenesis)方法为利用突变引物进行聚合酶链反应(polymerase chain reaction;PCR),当中所用的突变引物如图3所示,其中Ec-V90T突变株的突变引物是设计为可将植酸酶EcAppA第90个氨基酸由缬氨酸突变成苏氨酸(Threonine)。此突变株的序列列于图4中,其中突变株Ec-V90T的DNA序列以SEQ ID NO.3标示,氨基酸序列则以SEQ ID NO.4标示。
接着在PCR反应产物中加入DpnI于37℃下作用,将原始模板去除。再把含有突变基因的质粒送入大肠杆菌XL1B感受态细胞中,将菌液涂于含有100μg/ml氨苄西林抗生素的LB培养基于37℃下进行培养一天。再挑选菌落,借由测序步骤确认成功突变基因序列。将突变成功的AppA基因转化到大肠杆菌BL21(DE3)内,进行蛋白表达与纯化。
蛋白质表达方法是将转化成功的大肠杆菌BL21(DE3)利用含有100μg/ml氨苄西林的LB培养液进行培养,其中菌株先接种到5ml LB内培养6小时后,再放大体积至200ml LB培养,最终放大到2L的LB培养。在OD值到达0.6-0.8时,利用1mM的IPTG诱导转入的蛋白质大量表达。之后,将菌液以6000g转速离心20分钟收集下来。利用超音波细胞粉碎机(sonicator)破菌,再以15000rpm离心30分钟,并收集上清液,以快速蛋白质液相层析仪(fast protein liquid chromatography;FPLC)纯化,利用DEAE阴离子交换管柱,分离出蛋白纯度达95%以上的植酸酶蛋白。
植酸酶蛋白的活性测试方式是将4ml的7.5mM肌醇六磷酸钠与0.2ml酶蛋白(酶蛋白的缓冲液为0.05%Triton X-100、0.05%BSA、及0.25M乙酸钠,缓冲液为pH5.5)及1.8ml的反应缓冲液0.25M乙酸钠,pH为5.5,在37℃下作用30分钟。接着加入4ml的终止显色液(水:硝酸溶液:10%钼酸胺:0.2M偏钒酸胺=4:2:1:1),在OD415波长测定吸光值,再换算成酶活性单位(unit)。其中酶活性的标准曲线是由磷酸二氢钾标准溶液0-25μmol/ml之间制定,而1单位(unit)的定义为每分钟从浓度5mM植酸钠溶液中释放1μmole无机磷。
而在毕赤酵母表达系统中,首先将AppA序列参考GenBank DQ513832.1的序列合成优化为毕赤酵母易表达的序列(r-AppA),此序列经过密码子使用(codon usage)序列优化,以适合于毕赤酵母中表达,该序列在毕赤酵母中经由分泌表达,于序列N端增加信号序列(signal peptide),且位于原始氨基酸序列第1个位置的甲硫氨酸(methionine)被移至信号序列上,由于信号序列在蛋白质表达过程会被切除,因此在毕赤酵母中分泌出的蛋白质氨基酸序列比原始的序列少了第一个甲硫氨酸。如图5所示,r-AppA基因的全长为1233个碱基(DNA序列以SEQ ID NO.5标示)以及410个氨基酸(氨基酸序列以SEQ ID NO.6标示),并利用EcoRI与NotI限制酶切位构筑到pPICZαA载体中,其中在构筑质粒时发生了不影响活性的自然突变,使r-AppA的第116个氨基酸从丙氨酸变为缬氨酸。再利用定点突变方法以突变引物进行聚合酶链反应,当中所用的突变引物如图3所示,其中r-N204A突变株的突变引物是设计为可将植酸酶r-AppA第204个氨基酸由天冬酰胺突变成丙氨酸(Alanine),此突变株的序列列于图6中,其中突变株r-N204A的DNA序列以SEQ ID NO.7标示,氨基酸序列则以SEQ ID NO.8标示。而r-S206A突变株的突变引物则设计为可将植酸酶r-AppA第206个氨基酸由丝氨酸突变成丙氨酸,此突变株的序列列于图7中,其中突变株r-S206A的DNA序列以SEQ ID NO.9标示,氨基酸序列则以SEQ ID NO.10标示。
接着,使用PmeI把DNA质粒进行线性化之后,借由电转化步骤将DNA送入毕赤酵母内。接着将菌液涂于含有100μg/mlZeocin抗生素的YPD培养皿于30℃下进行培养两天。再挑选菌落并接种到5ml YPD于30℃下培养,再次接种到50ml BMGY中于30℃下培养至隔天。接着,将培养基换成含有0.5%甲醇的20ml BMMY以诱导蛋白表达,同样培养于30℃下。每隔24小时取样并补充0.5%甲醇。此外,将取样的菌液进行离心并收集上清液,进而测定植酸酶活性,其方法如上述。
为了进一步地放大植酸酶生产量,我们先将菌株接种到5mlYPD培养至隔天,接着放大到2L于30℃下培养之后,再次放大到含有19L发酵培养基(FBSM)的50L发酵槽内。发酵过程中,会全程监控温度在30℃并借由氨水控制pH值在5.0左右,而溶氧的调控则借由进气量以及转速维持在40%以上。在批次培养之后,流加50%甘油让酵母生长到一定的菌量。再把甲醇以梯度流加的方式加进发酵槽中,进而诱导酶蛋白表达。定时取样并收集上清液,进而检测植酸酶的表达量与活性。
图8显示野生型Ec-AppA与突变株Ec-V90T的植酸酶活性分析测试,图中酶活性是以野生型EcAppA的酶活性做为100%的基准之下进行比较,统计分析是利用T-test的双尾分析,在P<0.05时判定为显著性差异(*)。由结果得知,将氨基酸序列上第90个位置的缬氨酸(Valine)突变成苏氨酸(Threonine)能有效地提升植酸酶的活性约20%。
图9显示野生型r-AppA与突变株r-N204A及r-S206A的植酸酶活性分析测试,图中酶活性是以野生型r-AppA的酶活性做为100%的基准之下进行比较,统计分析是利用T-test的双尾分析,在P<0.05时判定为显著性差异(*)。由结果得知,利用定点突变制造移除活性区糖基化位点的突变株r-N204A及r-S206A,即第204个位置的天冬酰胺(Asparagine)突变成丙氨酸(Alanine)及第206个位置的丝氨酸(Serine)突变成丙氨酸(Alanine),均能使植酸酶活性上升约10%。
综上所述,为了增加植酸酶的工业应用价值,本申请详细分析及比较EcAppA植酸酶基因与其他活性高的植酸酶序列,加上活性区周遭的结构分析后,针对当中数个氨基酸利用定点突变的技术突变成目标氨基酸,用以提升作用活性。其中,将氨基酸序列上第90个位置的缬氨酸(Valine)突变成苏氨酸(Threonine)所得到的Ec-V90T突变株能有效地提升植酸酶的活性约20%。而考虑到使用毕赤酵母做表达时所造成的糖基化对蛋白质的影响,本申请亦利用定点突变制造移除活性区糖基化位点的突变株r-N204A及r-S206A,即第204个位置的天冬酰胺(Asparagine)突变成丙氨酸(Alanine)及第206个位置的丝氨酸(Serine)突变成丙氨酸(Alanine),结果移除活性区糖基化位点的突变株均能使植酸酶活性上升约10%。
在原本活性就相当高的酶上再找寻增加活性的突变点本属不易,活性能增加的幅度也不大,本申请找到的突变点增加活性的幅度为10~20%,此幅度的增加若用大量筛选的方法也容易在表达量变动下被遗漏,而本申请结合了酶已知序列及结构,利用理性设计,在只筛选少许突变株的情况下,才能将其一一纯化出来,确定其活性,筛选出有效突变株。由于植酸酶占有55亿饲料酶市场的60%,借由本申请提出的突变株进一步提升植酸酶活性后,更能有效的节省生产成本及增加生产价值,故本申请的具有提升的酶活性的植酸酶极具产业价值,于是依法提出申请。
本申请可以由本领域技术人员做出各种改变而不背离本发明的精神,这样的改变也均落入本发明的保护范围权利要求。
- 还没有人留言评论。精彩留言会获得点赞!